
Abstract
Carboxylated chalcones and other related flavonoids were synthesized and evaluated as inhibitors of xanthine oxidase, which is a known target for synthetic and herbal drugs used against hyperuricemia, gout, and other diseases. The 4-carboxylated chalcones with hydroxy, methoxy, and ethoxy groups at ring A were found to exhibit in vitro inhibitory activities with IC50 values in the range of 0.057 to 0.26 μM, being 10–60-fold more potent than allopurinol. Structurally related carboxylic acids with Δ3,9-homoisoflavonoid and flavone scaffolds also showed micromolar activity towards xanthine oxidase. At the same time, dihydrochalcone and Δ2,3-homoisoflavonoid carboxylic acids as well as their oxa-analogues were more than two orders of magnitude less effective inhibitors. Kinetic and molecular docking studies indicated that the carboxylated chalcones and Δ3,9-homoisoflavonoids are mixed-type inhibitors, which mostly bind to free enzyme occupying the active site of xanthine oxidase.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Naturally occurring chalcones as well as flavones, flavonols, flavanones, flavanonols, anthocyanidins, isoflavones, and homoisoflavonoids form large groups of polyphenolic compounds belonging to the flavonoid family [1]. Biosynthesis of flavones as precursors of most flavonoids occurs through the transformation of 2'-hydroxychalcones [2, 3], while biosynthesis of various types of homoisoflavonoids is carried out by involving of methoxy group of 2'-methoxychalcones into the formation of C6-C4-C6 homoisoflavonoid skeleton [4]. The members of the flavonoid family (Fig. 1) as well as their synthetic structural analogues are considered promising starting points for drug design, demonstrating anticancer, antimicrobial, antiviral, anti-diabetic, anti-inflammatory, and other activities [1,2,3,4,5]. Some chalcone derivatives such as metochalcone and sofalcone were approved to use as choleretic and gastrointestinal drugs [3], while chalcone analog ilepcimide is considered a potential antiepileptic medication [6]. Mechanisms of bioactivity of chalcone derivatives can be related to reversible and irreversible inhibition of enzymes and proteins, as well as to scavenging ROS and other free radicals [7, 8]. As an example, the chalcones with anti-cancer activity can be directed to 5α-reductase, VEGFR-2 kinase, tubulin, cathepsin-K, topoisomerase II, and mTOR [9,10,11].

Basic structures of chalcones, dihydrochalcones, flavones, and homoisoflavonoids
Xanthine oxidase (XO) is an enzyme of the terminal step of purine metabolism catalyzing the oxidative transformation of hypoxanthine and xanthine to uric acid with the generation of superoxide radical. Overproduction of urates is the reason for hyperuricemia which can be responsible for other diseases such as gout, renal failure, and cardiovascular disorders [12]. In addition, the high level of serum uric acid may lead to cancer [13]. This enzyme is known to be a target for many synthetic [14,15,16,17] and natural compounds including chalcones [18,19,20,21], and other flavonoids [22]. Purine analog allopurinol and non-purine XO inhibitor febuxostat are used for the treatment of hyperuricemia and gout [23, 24]. However, there is a current interest in further searching for new inhibitors of this enzyme.
Mono- and polyhydroxylated chalcones were established to inhibit XO activity and scavenge free radicals. The number and position of hydroxyl groups of monohydroxy-, dihydroxy-, and trihydroxy-substituted chalcones influenced their inhibitory potency against human XO [18,19,20,21]. Structure-activity relationship of a series of mono- and polyhydroxylated chalcone derivatives with dual properties showed that the most active XO inhibitors had a minimum of three hydroxyl groups, while the most effective radical scavengers carried two neighboring hydroxyl groups on at least one phenyl ring [18]. It was shown that replacing the hydroxyls with the carboxyl group yielded effective A-ring carboxylated chalcones with higher aqueous solubility and activities against the Gram-positive bacterium S. aureus [25]. The database consisting of 4'-carboxylated chalcones was used for virtual screening and biological evaluation of new compounds with moderate to good CysLT1 antagonistic activities [26]. Chalcone derivatives with A-ring modification by carboxylic and hydroxyl groups were reported as a new class of HIV-1 integrase inhibitors [27]. The introduction of the carboxylic group on ring B significantly increased the chalcones activity in antinociceptive pain models [28]. Chalcones bearing 4-carboxy groups were studied as allosteric inhibitors of YopH [29].
In the present paper, a series of new chalcone-4-carboxylic acids were synthesized and studied in vitro as inhibitors of XO. The inhibitory effects of the 4-carboxylated chalcones were compared with the inhibitory properties of related carboxylic acids bearing flavone, Δ3,9-homoisoflavonoid, Δ2,3-homoisoflavonoid, and dihydrochalcone scaffolds, as well as with the properties of some oxa-analogues of these compounds.
Results and discussion
Chemistry
Chalcones 2a-2i were synthesized by Claisen–Schmidt condensation of acetophenones 1a-1i with 4-formylbenzoic acid in alcohol in the presence of KOH according to the standard procedure [28, 29]. The two-step process which includes the reaction of non-hydroxylated acetophenones with methyl 4-formylbenzoate followed by saponification affords the chalcones with comparable yields (Scheme 1). In the case of 2-hydroxyacetophenones, this method gave poor results due to the formation of flavanones as by-products whose quantities depended on substituents in acetophenones 1. The carboxylated chalcones 2a-2i were identified as E-isomers due to the splitting of alkene protons with a J value of 15.5–16.0 Hz.

Synthesis of chalcones. Reagents and conditions: a) i, 4-formylbenzoic acid, EtOH, KOH, 60–70 °С, ii, HCl, H2O; b) i, methyl 4-formylbenzoate, EtOH, KOH, r. t., ii, KOH, H2O, 60–70 °С, than HCl
The series of chalcone derivatives 2a-2i was also supplemented by the synthesis of 4′-carboxy-7-methoxyflavone (3), obtained by I2-DMSO oxidative cyclization of carboxylated chalcone 2d (Scheme 2).

Synthesis of flavone 3. Reagents and conditions: a) I2, DMSO, 130–140 °С, 6 h
Sappanin-like Δ3,9-homoisoflavonoids 5a and 5b were synthesized by condensation of chromane-4-ones 4a and 4b, respectively, in acetic acid in the presence of H2SO4 (Scheme 3). It should be mentioned that condensation of chromanones 4 with 4-formylbenzoic acid was not possible in the basic medium.

Synthesis of homoisoflavonoids 5a and 5b. Reagents and conditions: a) 4-formylbenzoic acid, CH3COOH, H2SO4, reflux, 8 h
The stereochemical structure of homoisoflavonoid 5b, a conformationally constrained analog of chalcone 2f, was elucidated using COSY and 2D NOESY NMR spectra (Fig. 2). Cross-peaks between H-6 and H-5 protons as well CH2-2 and H-9 were found in the COSY spectrum. In addition, cross-peaks between H-6 and H-5 protons as well as H-2′ and CH2-2 protons were present in the 2D NOESY spectrum. Cross-peak between CH2-2 and H-9 protons has not been observed. These data suggest that compound 5b exists in the E-isomeric form assigned for similar homoisoflavonoids [30, 31].

Main correlations in COSY and 2D NOESY spectra for compound 5b
4-Carboxydihydrochalcone and 4′-carboxyhomoisoflavonoids as well as their oxa-analogues that can be considered similar to 4-carboxychalcone compounds have also been synthesized. Condensation of resorcinol (6a) or its monomethyl ether (6b) with 3-[4-(methoxycarbonyl)phenyl]propanoic acid in boron trifluoride etherate led to methyl 4-[3-(2-hydroxyphenyl)-3-oxopropyl]benzoates 7a and 7b. Subsequent saponification of compound 7b affords target dihydrochalcone acid 8a. Related oxa-dihydrochalcone 7c was synthesized by the Hoesh procedure as we reported early [32]. Further selective and exhaustive alkylation of compound 7c led to esters 7d and 7e, which saponification gave corresponding acids 8b and 8c. Under Vilsmeier-Haack reaction conditions, compounds 7a and 7b were converted to substituted Δ2,3-homoisoflavonoids 9a and 9b which were further transformed into corresponding acids 10a and 10b. 9-Oxa-homoisoflavonoid acid 10c was synthesized by ring-closure reaction of acid 7c under the Vilsmeier-Haack reaction (Scheme 4).

Reagents and conditions: a) 4-MeOOCC6H4CH2CH2COOH or 4-MeOOCC6H4CH2COOH, BF3·EtO2, 80–90 °C, 2 h; b) i, 4-MeOOCC6H4OCH2CN, BF3·EtO2, HCl, rt, 6 h, ii, H2SO4, H2O, reflux, 2 h; c) Me2SO4, K2CO3, acetone, 50–60 °С, 4–8 h; d) KOH, EtOH, 50 °С, 4 h; e) i, BF3·EtO2, POCl3, DMF, 55–60 °С, 2 h, ii, H2O, 80 °С, 0.5 h; f) H2SO4, AcOH, reflux, 8 h
Xanthine oxidase inhibition and antioxidant properties
The studies of XO inhibition by chalcone-4-carboxylic acids 2a-2i as well as related to them flavonoids were performed using the enzyme from bovine milk, which shared 90% identity to human liver XO [33]. The dose-dependent curves (Fig. 3) demonstrate differences in the inhibitory properties of chalcone 2f, Δ3,9-homoisoflavonoid 5b, and Δ2,3-homoisoflavonoid 10b.

Dose-dependent curves of XO inhibition by compounds 2f (○), 5b (□), and 10b (∆)
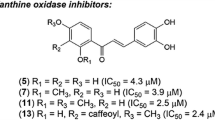
Control experiments showed that the carboxylic acid group at the B-ring of the chalcone is necessary for the inhibition of XO. As an example, 4-carboxy-4′-methoxychalcone (2b) has an IC50 value of 0.10 μM, while 4-hydroxy-4′-methoxychalcone at a concentration of 10 µM inhibited the XO activity by only ten percent. The carboxylated chalcone 2a containing hydroxy group at 4ʹ-position of A-ring had an IC50 value of 0.26 µM. Replacement of the hydroxyl by the ethoxy group increased the inhibitory potential of compound 2c (IC50 = 0.057 µM). The presence of hydroxyl substituent at the 2ʹ-position of the A-ring did not change significantly the IC50 value for compound 2d. Chalcone 2f with two methoxy groups showed a somewhat increased inhibition effect in comparison with compound 2d. A further variation of substituents at the A-ring of the chalcone scaffold gave derivatives 2g and 2h, which have similar IC50 values as compared to compound 2d. The introduction of halogen atoms at the A-ring did not improve the inhibitory properties of compounds 2e and 2i. It was interesting that the inhibitory activity of 4′-carboxy-7-methoxyflavone (3) was the same as that of chalcone 2d (Table 1). As compared to compound 2c, a similar binding affinity to XO was observed previously for 4′-carboxylated aurones [34].
At the same time, dihydrochalcone-4-carboxylic acid 8a and oxa-dihydrochalcone-4-carboxylic acids 8b and 8c exhibited much lower inhibitory effects than chalcones 2a-i, probably indicating an important role of alkene fragment in the inhibitor structure. Taking this into account, the activities of carboxylated ∆3,9-homoisoflavonoids and ∆2,3-homoisoflavonoids were compared. The conformationally constrained ∆3,9-homoisoflavonoid-4′-carboxylic acids 5a and 5b turned out to be micromolar inhibitors of XO. However, the presence of an endocyclic double bond led to a significant decrease in inhibition of XO by ∆2,3-homoisoflavonoid-4′-carboxylic acids 10a and 10b, as well as oxa-homoisoflavonoid-4′-carboxylic acid 10c.
It can be summarized that the most active inhibitors among the compounds studied were derivatives of carboxylated chalcones. Carboxylated dihydrochalcones and oxa-dihydrochalcones exhibited much lower inhibitory effects than the chalcones. The activity of carboxylated ∆3,9-homoisoflavonoids significantly exceeded the activity of ∆2,3-homoisoflavonoids.
Antioxidant properties of carboxylated chalcones and related flavonoids were determined by measuring the effect of the compounds on the degradation of deoxyribose by hydroxyl radicals generated by the Fenton reaction [35]. According to the data obtained, chalcone-4-carboxylic acids 2a, 2b, and 2g exhibited the best activity. Some lower effects were observed in the case of carboxylated chalcones 2f, 2h, and 2i. Oxa-dihydrochalcone-4-carboxylic acid 8c, ∆2,3-homoisoflavonoid-4′-carboxylic acid 10a, and 4′-carboxy-7-methoxyflavone 3 possessed antioxidant properties similar to those of chalcone-4-carboxylic acids 2f, 2h, and 2i. The activities of all compounds were higher than that of Trolox as a reference antioxidant. These data indicated that carboxylated chalcones can combine the XO inhibition effect with scavenging ability towards free hydroxyl radicals.
Kinetic studies of xanthine oxidase inhibition
The double reciprocal Lineweaver-Burk plots (Fig. 4) showed that inhibition of XO by compounds 2f and 5b is characterized by increasing Km and decreasing Vmax values, indicating a mixed-type mechanism. The inhibition constants Ki and Ki’ were 21 ± 1 nM and 195 ± 16 nM for chalcone-4-carboxylic acid 2f and 72 ± 13 nM and 822 ± 167 nM for ∆3,9-homoisoflavonoid-4′-carboxylic acid 5b. The values of Ki and Ki’ for both inhibitors indicate that their affinity for the free enzyme is much higher than for the enzyme-substrate complex.

Lineweaver-Burk plots for inhibition of XO by compounds 2f (a) and 5b (b). The concentrations were: a) 0 (○), 35 nM (□), 70 nM (∆), and 140 nM (◊); b) 0 (○), 150 nM (□), 300 nM (∆), and 600 nM (◊)
Molecular docking
Molecular docking by AutoDock Vina [36] was performed to predict the binding modes of the inhibitors to the active site of XO. The docking results suggest that the carboxylated B-ring of all compounds is located near residues Arg880, Thr1010, and Phe914. The calculated docking energies for compounds 2f and 5b were −9.6 kcal/mol and −9.2 kcal/mol, respectively. The carboxyl groups of chalcone-4-carboxylic acid 2f and Δ3,9-homoisoflavonoid-4′-carboxylic acid 5b (E-isomers) form hydrogen bonds with amino acids residues of Arg880 and Thr1010 (Fig. 5). Additional hydrogen bonds were between these carboxyl groups and Glu1261 through water molecule 1457, which is sufficient for the catalysis [37]. The positions of B-rings of compounds 2f and 5b are stabilized by π-π stacking interactions with amino acids residue of Phe914. The oxygen atoms of carbonyl groups of chalcone and Δ3,9-homoisoflavonoid have similar orientations, but only compound 2f interacts with Asn768. The carbonyl group of Δ2,3-homoisoflavonoid derivatives is more distant from Asn768 as compared with that of Δ3,9-homoisoflavonoids. The A-rings of chalcone 2f and Δ3,9-homoisoflavonoid 5b are located in a hydrophobic region which is formed by amino acid residues of Leu648, Met770, Lys771, Leu873, Phe1013, and Leu1014. The data obtained suggest that the differences between binding poses of chalcones and other compounds studied can be related to cycle A and carbonyl fragment of the inhibitors.

Possible binding modes of compounds 2f (a) and 5b (b) at the active site of XO
Conclusions
A series of carboxylated chalcones and related compounds were synthesized and studied as inhibitors of XO. The carboxylated chalcones bearing hydroxy, methoxy, and ethoxy groups at ring A (compounds 2a-2i), Δ3,9-homoisoflavonoids (5a, 5b), and flavone (3) were found to be submicromolar inhibitors of the enzyme, more potent than carboxylated Δ2,3-homoisoflavonoids (10a, 10b), dihydrochalcone (7a), oxa-dihydrochalcones (8b, 8c), and oxa-homoisoflavonoid (10c). These data indicate that the carboxylated ring B, linked to the alkene fragment, can be crucial for activity of chalcone inhibitor against XO. The results of the study showed also that the compounds can be capable of scavenging free radicals. Kinetic data suggest the mixed-type inhibition of XO by carboxylated chalcones and Δ3,9-homoisoflavonoids. Docking studies revealed that the B-ring of carboxylated inhibitor fits deep in the active site, while A-ring is located at the periphery of the binding site. The in vitro and in silico results demonstrate the potential of carboxylated chalcones and some other carboxylated flavonoids for designing XO inhibitors.
Experimental Section
Chemistry
1H and 13C, and 2D NMR spectra were recorded on Varian 500 (500/125 MHz) or Varian 400 (400/100 MHz) spectrometers in CDCl3 [residual CHCl3 (δH = 7.26 ppm) or CDCl3 (δC = 77.16 ppm) as internal standard] or DMSO-d6 [residual SO(CD3)(CD2H) (δH = 2.50 ppm) or SO(CD3)2 (δC = 39.52 ppm) as internal standard]. The spectra are represented in Supplementary data. Melting points were determined in open capillary tubes using the Buchi B-535 apparatus and were uncorrected. Mass spectra were obtained using an Agilent 1100 spectrometer using APCI (atmospheric-pressure chemical ionization). Elemental analysis was performed on a vario MICRO cube automated CHNS analyser. Column chromatography was performed using Macherey-Nagel Silica 60 0.04–0.063 mm silica gel.
4-Hydroxy-4′-methoxychalcone was synthesized as described previously [38].
General procedure for the synthesis of chalcones 2a-2i
To a solution of 2 mmol of acetophenone and 2 mmol of 4-formylbenzoic acid in 15 mL of EtOH was added 1 mL of 50% aqueous KOH. The reaction mixture was stirred for 5 h at 60–70 °C. It was poured into 70 mL of water with vigorous stirring and neutralized with concentrated HCl to pH 3–4. After cooling, the precipitate was filtered off, washed with water, and crystallized from the appropriate solvent with a yield of chalcones 2a-2i.
4-[(1E)-3-(4-Hydroxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2a)
Yellow solid (55% yield); mp 278–280 °C; 1H NMR (400 MHz, DMSO-d6): δ 6.91 (2H, d, J = 8.4 Hz), 7.71 (1H, d, J = 15.6 Hz), 7.92–8.05 (5H, m), 8.09 (2H, d, J = 8.4 Hz), 10.48 (1H, s), 13.12 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 115.5, 124.4, 128.7, 129.0, 129.8, 131.4, 131.9, 139.1, 141.3, 162.4, 166.9, 187.1 ppm; MS (ACPI) m/z (%): 269.1 (100) [M + H]+. Anal. calcd. for C16H12O4: C, 71.64; H, 4.51. Found: C, 71.78; H, 4.68.
4-[(1E)-3-(4-Methoxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2b)
White solid (68% yield); mp 232–234 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.83 (3H, s), 7.05 (2H, d, J = 8.5 Hz), 7.72 (1H, d, J = 15.6 Hz), 7.91–8.06 (5H, m), 8.15 (2H, d, J = 8.5 Hz), 13.14 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 55.6, 114.1, 124.2, 128.8, 129.8, 130.3, 131.1, 132.0, 139.0, 141.7, 163.4, 166.9, 187.3 ppm; MS (ACPI) m/z (%): 283.0 (100) [M + H]+. Anal. calcd. for C17H14O4: C, 72.33; H, 5.00. Found: C, 72.24; H, 5.14.
4-[(1E)-3-(4-Ethoxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2c)
Yellow solid (59% yield); mp 257–259 °C; 1H NMR (400 MHz, DMSO-d6): δ 1.36 (3H, t, J = 6.9 Hz), 4.15 (2H, q, J = 6.9 Hz), 7.07 (2H, d, J = 8.8 Hz), 7.73 (1H, d, J = 15.6 Hz), 7.96–8.02 (4H, m), 8.05 (1H, d, J = 15.6 Hz), 8.17 (2H, d, J = 8.8 Hz), 13.11 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 14.5, 63.6, 114.4, 124.2, 128.8, 129.7, 130.1, 131.0, 131.9, 139.0, 141.6, 162.7, 166.9, 187.2 ppm; MS (ACPI) m/z (%): 297.0 (100) [M + H]+. Anal. calcd. for C18H16O4: C, 72.96; H, 5.44. Found: C, 72.77; H, 5.32.
4-[(1E)-3-(2-Hydroxy-4-methoxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2d)
Yellow solid (56% yield); mp 252–254 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.85 (3H, s), 6.52 (1H, d, J = 2.1 Hz), 6.57 (1H, dd, J = 9.0, 2.1 Hz), 7.84 (1H, d, J = 15.5 Hz), 8.11 (1H, d, J = 15.5 Hz), 8.28 (1H, d, J = 9.0 Hz), 13.13 (1H, s), 13.31 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 55.8, 100.9, 107.5, 113.9, 123.5, 129.0, 129.7, 132.2, 132.8, 138.6, 142.5, 165.7, 166.2, 166.8, 191.6 ppm; MS (ACPI) m/z (%): 299.0 (100) [M + H]+. Anal. calcd. for C17H14O5: C, 68.45; H, 4.73. Found: C, 68.26; H, 4.85.
4-[(1E)-3-(3,5-Dichloro-2-hydroxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2e)
Yellow solid (34% yield); mp 257–259 °C; 1H NMR (400 MHz, DMSO-d6): δ 7.87–7.95 (2H, m), 8.00 (2H, d, J = 8.2 Hz), 8.07 (2H, d, J = 8.2 Hz), 8.14 (1H, d, J = 15.5 Hz), 8.42 (1H, d, J = 2.2 Hz), 13.02 ppm (2H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 121.9, 122.5, 122.8, 122.9, 129.0, 129.5, 129.6, 132.7, 135.3, 138.1, 145.1, 156.6, 166.7, 192.7 ppm; MS (ACPI) m/z (%): 337.0 (100) [M + H]+. Anal. calcd. for C16H10Cl2O4: C, 57.00; H, 2.99. Found: C, 56.79; H, 3.12.
4-[(1E)-3-(2,4-Dimethoxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2f)
Yellow solid (52% yield); mp 200–202 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.83 (3H, s), 3.90 (3H, s), 6.58–6.72 (2H, m), 7.51–7.69 (3H, m), 7.80 (2H, d, J = 7.3 Hz), 7.97 (2H, d, J = 7.3 Hz), 13.07 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 55.6, 56.0, 98.6, 106.1, 121.2, 128.3, 129.2, 129.9, 131.8, 132.3, 139.1, 139.6, 160.5, 164.3, 166.9, 189.0 ppm; MS (ACPI) m/z (%): 313.0 (100) [M + H]+. Anal. calcd. for C18H16O5: C, 69.22; H, 5.16. Found: C, 69.39; H, 5.37.
4-[(1E)-3-(2,5-Dimethoxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2 g)
Yellow solid (64% yield); mp 181–183 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.74 (3H, s), 3.82 (3H, s), 7.03–7.16 (3H, m), 7.51 (1H, d, J = 16.0 Hz), 7.57 (1H, d, J = 16.0 Hz), 7.82 (2H, d, J = 8.0 Hz), 7.97 (2H, d, J = 8.0 Hz), 13.12 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 55.6, 56.4, 113.9, 114.0, 119.0, 128.6, 128.8, 129.0, 129.9, 132.0, 138.8, 141.0, 152.2, 153.1, 166.9, 191.4 ppm; MS (ACPI) m/z (%): 313.0 (100) [M + H]+. Anal. calcd. for C18H16O5: C, 69.22; H, 5.16. Found: C, 69.12; H, 4.99.
4-[(1E)-3-(2-Ethoxy-4-methoxyphenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2h)
Yellow solid (65 % yield); mp 224–226 °C; 1H NMR (400 MHz, DMSO-d6): δ 1.35 (3H, t, J = 6.8 Hz), 3.84 (3H, s), 4.17 (2H, q, J = 6.8 Hz), 6.58–6.69 (2H, m), 7.56 (1H, d, J = 15.8 Hz), 7.65 (1H, d, J = 8.4 Hz), 7.73 (1H, d, J = 15.8 Hz), 7.81 (2H, d, J = 8.0 Hz), 7.98 (2H, d, J = 8.0 Hz), 13.05 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 14.5, 55.6, 64.1, 99.1, 106.2, 121.1, 128.1, 129.5, 129.9, 131.7, 132.3, 138.9, 139.2, 159.9, 164.3, 166.9, 188.8 ppm; MS (ACPI) m/z (%): 327.0 (100) [M + H]+. Anal. calcd. for C19H18O5: C, 69.93; H, 5.56. Found: C, 70.05; H, 5.73.
4-[(1E)-3-(2-Ethoxy-5-fluorophenyl)-3-oxoprop-1-en-1-yl]benzoic acid (2i)
Yellow solid (46% yield); mp 202–204 °C; 1H NMR (400 MHz, DMSO-d6): δ 1.28 (3H, t, J = 6.7 Hz), 4.11 (2H, d, J = 6.7 Hz), 7.13–7.22 (1H, m), 7.27–7.45 (2H, m), 7.53–7.63 (2H, m), 7.82 (2H, d, J = 8.0 Hz), 7.97 (2H, d, J = 8.0 Hz), 13.11 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 14.6, 64.7, 115.1 (d, JC-F = 7.6 Hz), 115.8 (d, JC-F = 24.0 Hz), 119.8 (d, JC-F = 23.0 Hz), 128.5, 128.6, 129.3 (d, JC-F = 6.0 Hz), 129.9, 132.1, 138.7, 141.0, 153.8, 156.0 (d, JC-F = 238.0 Hz), 166.8, 190.4 ppm; 19F NMR (470 MHz, DMSO-d6): δ −123.3 ppm; MS (ACPI) m/z (%): 315.0 (100) [M + H]+. Anal. calcd. for C18H15FO4: C, 68.78; H, 4.81. Found: C, 68.98; H, 4.69.
4-(7-Methoxy-4-oxo-4H-chromen-2-yl)benzoic acid (3)
A solution of 596 mg (2 mmol) of chalcone 2d and 10 mg of I2 in 5 mL of DMSO was heated at 130–140 °С for 6 h. The reaction mixture was diluted with 10 mL of i-PrOH, formed solid was filtered off and recrystallized from the DMF-MeOH mixture. Beige solid (66% yield); mp 290–292 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.93 (3H, s), 7.01–7.12 (2H, m), 7.33 (1H, s), 7.95 (1H, d, J = 8.8 Hz), 8.09 (3H, d, J = 8.2 Hz), 8.21 (3H, d, J = 8.2 Hz), 13.29 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 56.0, 100.8, 107.9, 114.8, 117.1, 126.1, 126.2, 129.7, 133.1, 134.9, 157.4, 160.9, 163.9, 166.6, 176.3 ppm; MS (ACPI) m/z (%): 297.0 (100) [M + H]+. Anal. calcd. for C17H12O5: C, 68.92; H, 4.08. Found: C, 69.07; H, 4.27.
General synthesis of homoisoflavonoids 5a, 5b
Concentrated sulfuric acid (0.1 mL) was added to a solution of 2 mmol of chromane-4-ones 4a or 4b and 2 mmol of 4-formyl benzoic acid in 10 mL of acetic acid, and then the mixture was refluxed for 8 h. The reaction mixture was diluted with water; the precipitate was filtered and washed with water. Recrystallization from ethanol affords Δ3,9-homoisoflavonoids 5a, 5b.
4-[(E)-(4-Oxo-2H-chromen-3(4H)-ylidene)methyl]benzoic acid (5a)
Beige solid (75% yield); mp 277–279 °C; 1H NMR (400 MHz, DMSO-d6): δ 5.42 (2H, s), 7.06 (1H, d, J = 8.3 Hz), 7.14 (1H, t, J = 7.5 Hz), 7.51–7.65 (3H, m), 7.78 (1H, s), 7.89 (1H, d, J = 7.8 Hz), 8.03 (2H, d, J = 7.9 Hz), 13.17 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 67.3, 118.0, 121.4, 122.0, 127.3, 129.5, 130.3, 131.3, 132.3, 135.3, 136.4, 137.9, 160.7, 166.8, 181.0 ppm; MS (ACPI) m/z (%): 281.0 (100) [M + H]+. Anal. calcd. for C17H12O4: C, 72.85; H, 4.32. Found: C, 72.72; H, 4.22.
4-[(E)-(7-Methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl]benzoic acid (5b)
Beige solid (59% yield); mp 308–310 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.83 (3H, s), 5.30–5.51 (2H, m), 6.57 (1H, d, J = 2.1 Hz), 6.71 (1H, dd, J = 8.8, 2.1 Hz), 7.55 (2H, d, J = 8.2 Hz), 7.71–7.76 (1H, m), 7.82 (1H, d, J = 8.8 Hz), 8.02 (2H, d, J = 8.2 Hz), 13.05 ppm (1H, s); 13C{1H} NMR (100 MHz, DMSO-d6): δ 55.9, 67.5, 100.9, 110.7, 115.0, 129.1, 129.5, 130.3, 131.1, 132.4, 134.5, 138.1, 162.8, 165.9, 166.8, 179.6 ppm; MS (ACPI) m/z (%): 311.0 (100) [M + H]+. Anal. calcd. for C18H14O5: C, 69.67; H, 4.55. Found: C, 69.84; H, 4.68.
Methyl 4-[3-(2-hydroxy-4-methoxyphenyl)-3-oxopropyl]benzoate (7b)
A solution of 1.24 mg (10 mmol) of resorcinol monomethyl ether (6b) and 2.08 g (10 mmol) of the 3-[4-(methoxycarbonyl)phenyl]propanoic acid in 10 mL of boron trifluoride etherate was heated at 80–90 °C for 2 h. The mixture was carefully poured into 100 mL of chilled water. The resulting solid was filtered off, dried, and purified by column chromatography using a CH2Cl2-MeOH mixture (50:1) as eluent. Yellow solid (25% yield); mp 115–117 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.01 (2H, d, J = 7.5 Hz), 3.38 (2H, t, J = 7.5 Hz), 3.80 (3H, s), 3.83 (3H, s), 6.46 (1H, d, J = 2.2 Hz), 6.50 (1H, dd, J = 9.0, 2.2 Hz), 7.43 (2H, d, J = 8.2 Hz), 7.83–7.93 (3H, m), 12.53 (1H, d, J = 2.1 Hz) ppm; 13C{1H} NMR (125 MHz, DMSO-d6): δ 29.4, 38.6, 52.0, 55.7, 100.8, 107.3, 113.4, 127.4, 128.8, 129.2, 132.5, 146.9, 164.0, 165.6, 166.1, 203.4 ppm; MS (ACPI) m/z (%): 315.1 (100) [M + H]+. Anal. calcd. for C18H18O5: C, 68.78; H, 5.77. Found: C, 68.93; H, 5.60.
Ethyl 4-[2-(2-hydroxy-4-methoxyphenyl)-2-oxoethoxy]benzoate (7d)
To a stirred solution of 2 mmol of compound 7c in 30 mL of acetone were added 272 mg (2 mmol) of K2CO3 and 0.20 mL (2.05 mmol) of Me2SO4. The reaction mixture was stirred at 50–60 °C for 6 h, poured into 100 mL of water, and acidified with HCl to pH 4–5. The precipitate was filtered off, dried, and crystallized from methanol. White solid (72% yield); mp 125–127 °C; 1H NMR (400 MHz, DMSO-d6): δ 1.29 (3H, t, J = 7.1 Hz), 3.82 (3H, s), 4.27 (2H, q, J = 7.1 Hz), 5.55 (2H, s), 6.52 (1H, d, J = 2.3 Hz), 6.57 (1H, dd, J = 8.9, 2.3 Hz), 7.03 (2H, d, J = 8.8 Hz), 7.84 (1H, d, J = 8.9 Hz), 7.89 (2H, d, J = 8.8 Hz), 11.70 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 14.2, 55.6, 60.3, 70.5, 101.0, 107.3, 112.9, 114.5, 122.4, 131.1, 131.7, 161.9, 162.8, 165.3, 165.6, 195.6 ppm; MS (ACPI) m/z (%): 331.2 (100) [M + H]+. Anal. calcd. for C18H18O6: C, 65.45; H, 5.49. Found: C, 65.69; H, 5.63.
Ethyl 4-[2-(2,4-dimethoxyphenyl)-2-oxoethoxy]benzoate (7e)
To a stirred solution of 2 mmol of compound 7c in 30 mL of acetone were added 816 mg (6 mmol) of K2CO3 and 0.57 mL (6 mmol) of Me2SO4. The reaction mixture was stirred at 50–60 °C for 6 h, poured into 100 mL of water, and acidified with HCl to pH 4–5. The precipitate was filtered off, dried, and crystallized from methanol. Beige solid (85% yield); mp 110–112 °C; 1H NMR (400 MHz, DMSO-d6): δ 1.29 (3H, t, J = 7.1 Hz), 3.87 (3H, s), 3.96 (3H, s), 4.27 (2H, q, J = 7.1 Hz), 5.34 (2H, s), 6.67 (2H, dd, J = 8.8, 2.1 Hz), 6.71 (1H, d, J = 2.1 Hz), 6.96 (2H, d, J = 8.8 Hz), 7.78 (1H, d, J = 8.8 Hz), 7.87 ppm (2H, d, J = 8.8 Hz); 13C{1H} NMR (125 MHz, DMSO-d6): δ 14.2, 55.7, 56.0, 60.2, 73.2, 98.2, 106.7, 114.4, 117.2, 122.2, 131.0, 131.9, 161.5, 162.1, 165.2, 165.3, 191.9 ppm; MS (ACPI) m/z (%): 345.0 (100) [M + H]+. Anal. calcd. for C19H20O6: C, 66.27; H, 5.85. Found: C, 66.45; H, 5.73.
General procedures for the synthesis of compounds 8a-8c
To a solution of the corresponding ester 7b, 7d, or 7e (2 mmol) in 10 mL of EtOH was added 2 ml of 50% aqueous KOH. The reaction mixture was heated at 50 °C for 4 h, diluted with 50 mL of water, and acidified with 1 N HCl solution to pH 4–5. The formed precipitate of acids 8a-8c was filtered and re-crystallized from the MeOH-H2O mixture (1:1).
4-[3-(2-Hydroxy-4-methoxyphenyl)-3-oxopropyl]benzoic acid (8a)
Beige solid (77% yield); mp 175–177 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.00 (2H, t, J = 7.2 Hz), 3.38 (2H, t, J = 7.3 Hz), 3.81 (3H, s), 6.44–6.48 (1H, m), 6.48–6.54 (1H, m), 7.41 (2H, d, J = 8.0 Hz), 7.86 (2H, d, J = 8.0 Hz), 7.90 (1H, d, J = 9.0 Hz), 12.54 (1H, s), 12.79 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 29.4, 38.7, 55.7, 100.8, 107.3, 113.5, 128.5, 128.6, 129.3, 132.6, 146.4, 164.0, 165.6, 167.2, 203.5 ppm; MS (ACPI) m/z (%): 301.0 (100) [M + H]+. Anal. calcd. for C17H16O5: C, 67.99; H, 5.37. Found: C, 67.83; H, 5.25.
4-[2-(2-Hydroxy-4-methoxyphenyl)-2-oxoethoxy]benzoic acid (8b)
Beige solid (65% yield); mp 260–262 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.82 (3H, s), 5.54 (2H, s), 6.52 (1H, d, J = 2.2 Hz), 6.57 (2H, dd, J = 8.9, 2.2 Hz), 7.01 (2H, d, J = 8.8 Hz), 7.81–7.92 (3H, m), 11.71 (1H, s), 12.62 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 55.7, 70.5, 101.0, 107.4, 113.0, 114.4, 123.3, 131.3, 131.7, 161.7, 162.9, 165.6, 167.0, 195.7 ppm; MS (ACPI) m/z (%): 303.0 (100) [M + H]+. Anal. calcd. for C16H14O6: C, 63.57; H, 4.67. Found: C, 63.46; H, 4.79.
4-[2-(2,4-Dimethoxyphenyl)-2-oxoethoxy]benzoic acid (8c)
Beige solid (90% yield); mp 208–210 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.87 (3H, s), 3.96 (3H, s), 5.34 (2H, s), 6.64–6.70 (1H, m), 6.70–6.73 (1H, m), 6.93 (2H, d, J = 8.7 Hz), 7.79 (1H, d, J = 8.7 Hz), 7.86 (2H, d, J = 8.7 Hz), 12.61 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 55.8, 56.1, 73.2, 98.3, 106.7, 114.3, 117.3, 123.0, 131.2, 131.9, 161.6, 161.8, 165.2, 167.0, 192.1 ppm; MS (ACPI) m/z (%): 317.2 (100) [M + H]+. Anal. calcd. for C17H16O6: C, 64.55; H, 5.10. Found: C, 64.72; H, 4.96.
Methyl 4-[(7-hydroxy-4-oxo-4H-chromen-3-yl)methyl]benzoate (9a)
A solution of 1.1 g (10 mmol) of resorcinol and 2.08 g (10 mmol) of 3-[4-(methoxycarbonyl)phenyl]propanoic acid in 10 mL of boron trifluoride etherate was stirred at 90 °C for 2 h. The reaction mixture was cooled to room temperature, and then DMF (10 mL) and POCl3 (1.86 mL, 20 mmol) were added. The mixture was heated at 55–60 °C for 2 h and poured into 50 mL of hot water with vigorous stirring and then cooled. A precipitate was collected and washed with water. Recrystallization from MeOH gave homoisoflavonoid 9a as a white powder with 46% yield; mp 238–240 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.74 (2H, s), 3.82 (3H, s), 6.83 (1H, d, J = 2.0 Hz), 6.89 (1H, dd, J = 8.8, 2.0 Hz), 7.42 (2H, d, J = 8.1 Hz), 7.79–7.94 (3H, m), 8.27 (1H, s), 10.77 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 30.8, 52.0, 102.2, 115.1, 116.2, 122.1, 126.8, 127.5, 128.8, 129.1, 145.6, 153.7, 157.8, 162.5, 166.1, 175.4 ppm; MS (ACPI) m/z (%): 311.0 (100) [M + H]+. Anal. calcd. for C18H14O5: C, 69.67; H, 4.55. Found: C, 69.48; H, 4.42.
Methyl 4-[(7-methoxy-4-oxo-4H-chromen-3-yl)methyl]benzoate (9b)
To a solution of 1.57 g (5 mmol) of dihydrochalcone 7b in 5 mL of N,N-dimethylformamide at 30–40 °C was added boron trifluoride etherate (1.92 mL, 15 mmol). The mixture was stirred for 0.5 h and POCl3 (0.93 mL, 10 mmol) was added at the same temperature. The mixture was heated at 55–60 °C for 2 h and poured into 100 mL of hot water with vigorous stirring and then cooled. A precipitate was collected and washed with water. Recrystallization from methanol gave homoisoflavonoid 9b as a white powder with 67% yield; mp 172–174 °C; 1H NMR (400 MHz, CDCl3): δ 3.83 (2H, s), 3.88 (3H, s), 3.89 (3H, s), 6.75–6.83 (1H, m), 6.89–7.00 (1H, m), 7.36 (2H, d, J = 7.9 Hz), 7.58 (1H, s), 7.96 (2H, d, J = 7.9 Hz), 8.11 ppm (1H, d, J = 8.9 Hz); 13C{1H} NMR (125 MHz, CDCl3): δ 31.8, 52.1, 55.9, 100.2, 114.7, 117.9, 123.8, 127.5, 128.6, 129.1, 130.0, 144.5, 152.7, 158.4, 164.1, 167.1, 176.7 ppm; MS (ACPI) m/z (%): 325.2 (100) [M + H]+. Anal. calcd. for C19H16O5: C, 70.36; H, 4.97. Found: C, 70.25; H, 5.15.
General procedures for the synthesis of compounds 10a, 10b
To a solution of the corresponding ester 9a and 9b (2 mmol) in 10 mL of acetic acid was added concentrated sulfuric acid (0.1 mL) and refluxed for 8 h. To the reaction mixture was added water, and the precipitate was collected, washed with water, and dissolved in 5% NaHCO3. The resulting solution was added concentrated HCl to pH 7. The precipitates of each of acids 10a and 10b were filtered and re-crystallized from an appropriate solvent.
4-[(7-Hydroxy-4-oxo-4H-chromen-3-yl)methyl]benzoic acid (10a)
Beige solid (90% yield); mp 313–315 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.74 (2H, s), 6.83 (1H, d, J = 2.0 Hz), 6.90 (1H, dd, J = 8.8, 2.0 Hz), 7.39 (2H, d, J = 8.2 Hz), 7.78–7.89 (3H, m), 8.25 (1H, s), 10.74 (1H, s), 12.76 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 30.8, 102.2, 115.1, 116.2, 122.2, 126.8, 128.7, 129.3, 145.1, 153.7, 157.9, 162.5, 167.3, 175.4 ppm; MS (ACPI) m/z (%): 297.0 (100) [M + H]+. Anal. calcd. for C17H12O5: C, 68.92; H, 4.08. Found: C, 68.83; H, 4.23.
4-[(7-Methoxy-4-oxo-4H-chromen-3-yl)methyl]benzoic acid (10b)
Beige solid (92% yield); mp 289–291 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.74 (2H, s), 3.86 (3H, s), 6.95–7.04 (1H, m), 7.05–7.11 (1H, m), 7.40 (2H, d, J = 8.0 Hz), 7.84 (2H, d, J = 8.0 Hz), 7.90 (1H, d, J = 8.9 Hz), 8.30 (1H, s), 12.81 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 30.8, 56.0, 100.6, 114.7, 117.2, 122.5, 126.4, 128.7, 129.3, 144.9, 153.9, 157.8, 163.7, 167.2, 175.4 ppm; MS (ACPI) m/z (%): 311.0 (100) [M + H]+. Anal. calcd. for C18H14O5: C, 69.67; H, 4.55. Found: C, 69.82; H, 4.72.
4-[(7-Methoxy-4-oxo-4H-chromen-3-yl)oxy]benzoic acid (10c)
It was obtained similarly to the Vilsmeier-Haack procedure from acid 8c and purified by recrystallization from a DMF-MeOH mixture. Yellow solid (83% yield); mp 294–296 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.92 (3H, s), 7.04–7.14 (3H, m), 7.25 (1H, d, J = 2.2 Hz), 7.89 (2H, d, J = 8.8 Hz), 7.98 (1H, d, J = 8.9 Hz), 8.75 (1H, s), 12.78 ppm (1H, s); 13C{1H} NMR (125 MHz, DMSO-d6): δ 56.2, 100.9, 115.0, 115.1, 117.9, 124.8, 126.6, 131.3, 138.5, 150.5, 157.6, 161.0, 164.1, 166.8, 171.1 ppm; MS (ACPI) m/z (%): 313.0 (100) [M + H]+. Anal. calcd. for C17H12O6: C, 65.39; H, 3.87. Found: C, 65.62; H, 4.02.
In vitro study of xanthine oxidase inhibition and molecular docking calculation
Xanthine oxidase inhibition assay
XO from bovine milk and xanthine as substrate were purchased from Sigma-Aldrich. The inhibitor activities of compounds were studied in the system containing sodium-phosphate buffer (50 mM, pH 7.4), xanthine (50 µM), EDTA (0.1 mM), and DMSO (1%). After incubation of the mixture at 25 °С for 5 min, the enzymatic reaction was started by the addition of XO. The enzyme activity was detected spectrophotometrically at 293 nm. The IC50 values represented the inhibitor concentration required to reduce enzyme activity by 50% and were calculated from a linear regression equation. The values are the mean of 2–3 experiments. The Km value obtained from Lineweaver-Burk plots (Fig. 4) was 2.7 ± 0.1 µM.
Docking study
The docking was performed into the active site of the C chain of the XO crystal structure with PDB code 1FIQ [33] as described previously [34]. Before the calculation, the chains A and B, cofactors, ligands, and water molecules were removed from the file, which was downloaded from RCSB Protein Data Bank (RCSB PDB, rcsb.org) [39]. However, the water molecule HOH1457, which plays a role in the enzyme catalytic mechanism [33, 37], was not removed. In addition, the catalytically important hydroxyl group of the molybdopterin cofactor was replaced by a water molecule. The structures of ligands (carboxylic group in ionized form) were prepared by the MarvinSketch program [40] and optimized using MMFF94s force field in Avogadro software [41]. AutoDockTools 1.5.6 was used to prepare the docking files [42]. Possible binding modes of ligands at the active site of the C chain of XO were predicted by the AutoDock Vina program [36]. Discovery Studio 3.5 visualizer (Accelrys, San Diego, USA) was used for the analysis of the model complexes.
Antioxidant activity study
The ability of compounds to scavenge hydroxyl radicals was estimated using the deoxyribose degradation method [35]. The 2 mL system containing iron (II) chloride (50 µM), EDTA (100 µM), phosphate buffer (50 mM, pH 7.4), 2-deoxyribose (2.8 mM), hydrogen peroxide (2.8 mM), and compound (0.3 mM) was incubated during 1 h at 37 °С. After adding 1 mL of 2.8% aqueous solution of trichloroacetic acid and 1 mL of 1% solution of thiobarbituric acid in 50 mM of sodium hydroxide, the system was incubated in a water bath for 20 min at 80–100 °С. The activity of compounds was determined by measuring the decrease in absorption at 532 nm.
References
Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. 2016;5:e47. https://doi.org/10.1017/jns.2016.41
Rozmer Z, Perjési P. Naturally occurring chalcones and their biological activities. Phytochem Rev. 2016;15:87–120. https://doi.org/10.1007/s11101-014-9387-8
Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem Rew. 2017;117:7762–810. https://doi.org/10.1021/acs.chemrev.7b00020
Castelli MV, López SN Homoisoflavonoids: occurrence, biosynthesis, and biological activity. In: Atta-ur-Rahman, editor. Studies in Natural Products Chemistry. Amsterdam: Elsevier. 2017. p. 315–354. https://doi.org/10.1016/B978-0-444-63929-5.00009-7
Rocha S, Ribeiro D, Fernandes E, Freitas M. A systematic review on anti-diabetic properties of chalcones. Curr Med Chem. 2020;27:2257–321. https://doi.org/10.2174/0929867325666181001112226
Aboul-Enein NM, El-Azzouny AA, Saleh AO, Maklad AY. On chemical structures with potent antiepileptic/anticonvulsant profile. Mini-Rev Med Chem. 2012;12:671–700. https://doi.org/10.2174/138955712800626665
Zhou B, Xing C. Diverse molecular targets for chalcones with varied bioactivities. Med Chem (Los Angeles). 2015;5:388–404. https://doi.org/10.4172/2161-0444.1000291
Nile SH, Keum YS, Nile AS, Jalde SS, Patel RV. Antioxidant, anti-inflammatory, and enzyme inhibitory activity of natural plant flavonoids and their synthesized derivatives. J Biochem Mol Toxicol. 2018;32:e22002. https://doi.org/10.1002/jbt.22002
Mahapatra DK, Bharti SK, Asati V. Anti-cancer chalcones: structural and molecular target perspectives. Eur J Med Chem. 2015;98:69–114. https://doi.org/10.1016/j.ejmech.2015.05.004
Ouyang Y, Li J, Chen X, Fu X, Sun S, Wu Q. Chalcone derivatives: role in anticancer therapy. Biomolecules. 2021;11:894 https://doi.org/10.3390/biom11060894
Liu W, He M, Li Y, Peng Z, Wang G. A review on synthetic chalcone derivatives as tubulin polymerisation inhibitors. J Enzyme Inhib Med Chem. 2022;37:9–38. https://doi.org/10.1080/14756366.2021.1976772
Gliozzi M, Malara N, Muscoli S, Mollace V. The treatment of hyperuricemia. Int J Cardiol. 2016;213:23–27. https://doi.org/10.1016/j.ijcard.2015.08.087
Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med. 2016;5:546–57. https://doi.org/10.1002/cam4.601
Luna G, Dolzhenko AV, Mancera RL. Inhibitors of xanthine oxidase: scaffold diversity and structure-based drug design. ChemMedChem. 2019;14:714–43. https://doi.org/10.1002/cmdc.201900034
Rashad AY, Kassab SE, Daabees HG, Moneim AEA, Rostom SAF. Febuxostat-based amides and some derived heterocycles targeting xanthine oxidase and COX inhibition. Synthesis, in vitro and in vivo biological evaluation, molecular modeling and in silico ADMET studies. Bioorg Chem. 2021;113:104948 https://doi.org/10.1016/j.bioorg.2021.104948
Song JU, Choi SP, Kim TH, Jung C-K, Lee J-Y, Jung S-H, et al. Design and synthesis of novel 2-(indol-5-yl)thiazole derivatives as xanthine oxidase inhibitors. Bioorg Med Chem Lett. 2015;25:1254–8. https://doi.org/10.1016/j.bmcl.2015.01.055
Guan Q, Cheng Z, Ma X, Wang L, Feng D, Cui Y, et al. Synthesis and bioevaluation of 2-phenyl-4-methyl-1,3-selenazole-5-carboxylic acids as potent xanthine oxidase inhibitors. Eur J Med Chem. 2014;85:508–16. https://doi.org/10.1016/j.ejmech.2014.08.014
Hofmann E, Webster J, Do T, Kline R, Snider L, Hauser Q, et al. Hydroxylated chalcones with dual properties: xanthine oxidase inhibitors and radical scavengers. Bioorg Med Chem. 2016;24:578–87. https://doi.org/10.1016/j.bmc.2015.12.024
Bui TH, Nguyen NT, Dang PH, Hguyen HX, Nguyen MTT. Design and synthesis of chalcone derivatives as potential non-purine xanthine oxidase inhibitors. SpringerPlus. 2016;5:1–8. https://doi.org/10.1186/s40064-016-3485-6
Xie Z, Luo X, Zou Z, Zhang X, Huang F, Li R, et al. Synthesis and evaluation of hydroxychalcones as multifunctional non-purine xanthine oxidase inhibitors for the treatment of hyperuricemia. Bioorg Med Chem Lett. 2017;27:3602–6. https://doi.org/10.1016/j.bmcl.2017.01.053
Yang C, Liu Y, Tu Y, Li L, Du J, Yu D, et al. Chalcone derivatives as xanthine oxidase inhibitors: synthesis, binding mode investigation, biological evaluation, and ADMET prediction. Bioorg Chem. 2023;131:106320. https://doi.org/10.1016/j.bioorg.2022.106320
Mehmood A, Ishaq M, Zhao L, Safdar B, Rehman A, Munir M, et al. Natural compounds with xanthine oxidase inhibitory activity: a review. Chem Biol Drug Des. 2019;93:387–418. https://doi.org/10.1111/cbdd.13437
Finch A, Kubler P. The management of gout. Aust Prescr. 2016;39:119–22. https://doi.org/10.18773/austprescr.2016.047
White WB, Saag KG, Becker MA, Borer JS, Gorelick PB, Whelton A, et al. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N Engl J Med. 2018;387:1200–10. https://doi.org/10.1056/NEJMoa1710895
Nielsen SF, Boesen T, Larsen M, Schønning K, Kromann H. Antibacterial chalcones–bioisosteric replacement of the 4′-hydroxy group. Bioorg Med Chem. 2004;12:3047–54. https://doi.org/10.1016/j.bmc.2004.03.071
Dong X, Wang L, Huang X, Liu T, Wei E, Du L, et al. Pharmacophore identification, synthesis, and biological evaluation of carboxylated chalcone derivatives as CysLT1 antagonists. Bioorg Med Chem. 2010;18:5519–27. https://doi.org/10.1016/j.bmc.2010.06.047
Sharma H, Patil S, Sanchez TW, Neamati N, Schinazi RF, Buolamwini JK. Synthesis, biological evaluation and 3D-QSAR studies of 3-keto salicylic acid chalcones and related amides as novel HIV-1 integrase inhibitors. Bioorg Med Chem. 2011;19:2030–45. https://doi.org/10.1016/j.bmc.2011.01.047
Begum S, Begum SKA, Mallika A, Bharathi K. Synthesis, evaluation and in silico studies of 4-N,N-dimethylamino and 4-carboxy chalcones as promising antinociceptive agents. In: Jyothi S, Mamatha D, Satapathy S, Raju K, Favorskaya M, editors. International conference on computational and bio-engineering. CBE 2019. Learning and analytics in intelligent systems. Springer, Cham; 2020;15. p. 481–90. https://doi.org/10.1007/978-3-030-46939-9_42
De Souza ACA, Mori M, Sens L, Rocha RF, Tizziani T, de Souza LFS, et al. A chalcone derivative binds a putative allosteric site of YopH: inhibition of a virulence factor of Yersinia. Bioorg Med Chem Lett. 2020;30:127350. https://doi.org/10.1016/j.bmcl.2020.127350
Siddaiah V, Rao CV, Venkateswarlu S, Krishnaraju AV, Subbaraju GV. Synthesis, stereochemical assignments, and biological activities of homoisoflavonoids. Bioorg Med Chem. 2006;14:2545–51. https://doi.org/10.1016/j.bmc.2005.11.031
Regenass P, Abboud D, Daubeuf F, Lehalle C, Gizzi P, Riché PS, et al. Discovery of a locally and orally active CXCL12 neutraligand (LIT-927) with anti-inflammatory effect in a murine model of allergic airway hypereosinophilia. J Med Chem. 2018;61:7671–86. https://doi.org/10.1021/acs.jmedchem.8b00657
Frasinyuk MS. Synthesis and aminomethylation of 3-substituted 6-hydroxy-1,2-benzisoxazoles. Chem Heterocycl Compd. 2014;50:1616–23. https://doi.org/10.1007/s10593-014-1631-z
Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proc Natl Acad Sci USA. 2000;97:10723–8. https://doi.org/10.1073/pnas.97.20.10723
Muzychka OV, Kobzar OL, Popova AV, Frasinyuk MS, Vovk AI. Carboxylated aurone derivatives as potent inhibitors of xanthine oxidase. Bioorg Med Chem. 2017;25:3606–13. https://doi.org/10.1016/j.bmc.2017.04.048
Aruoma OI. Deoxyribose assay for detecting hydroxyl radicals. Methods Enzymol. 1994;233:57–66. https://doi.org/10.1016/S0076-6879(94)33008-5
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–61. https://doi.org/10.1002/jcc.21334
Huber R, Hof P, Duarte RO, Moura JJ, Moura I, Liu MY, et al. A structure-based catalytic mechanism for he xanthine oxidase family of molybdenum enzymes. Proc Natl Acad Sci. 1996;93:8846–51. https://doi.org/10.1073/pnas.93.17.8846
Gowthaman R, Miller SA, Rogers S, Khowsathit J, Lan L, Bai N, et al. DARC: Mapping surface topography by Ray-Casting for effective virtual screening at protein interaction sites. J Med Chem. 2016;59:4152–70. https://doi.org/10.1021/acs.jmedchem.5b00150
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. https://doi.org/10.1093/nar/28.1.235
MarvinSketch was used for drawing chemical structures, MarvinSketch version 5.2.4, ChemAxon (https://www.chemaxon.com)
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4:1–17. https://doi.org/10.1186/1758-2946-4-17
Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17:57–61
Acknowledgements
This work was supported by the National Academy of Sciences of Ukraine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kobzar, O.L., Tatarchuk, A.V., Mrug, G.P. et al. Carboxylated chalcones and related flavonoids as inhibitors of xanthine oxidase. Med Chem Res 32, 1804–1815 (2023). https://doi.org/10.1007/s00044-023-03109-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03109-8