
Abstract
A new series of CA4P analogs (5a-g, 6a-g) has been designed and effectively synthesized via a one-pot reaction from Combretastatin A-4/Erianin, commercially available amino acid esters and phenyl dichlorophosphate. To establish new candidates with anticancer activity, the in vitro antiproliferative effect of these compounds was measured by the CCK8 method on different cancer cell lines such as human liver caricinoma (HepG2), cervical cancer (HeLa) and colorectal carcinoma (HCT-116). The structure-activity relationships between CA4P outgrowth-promoting activity and its analogs suggested that the biaryl structure linked with double bond in Part A and the steric effect at the position α-carbon atom in the amino acid ester moiety (Part B) are essential for affecting the in vitro proliferation inhibitory activity of CA4P analogs. Additionally, the results of biological activity and molecular docking simulation showed that the vast majority of these novel Phosphoramidate derivatives exhibited potent anti-cancer activities.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer, a disease that involves the uncontrolled growth of abnormal cells anywhere in a body, has remained one of the leading causes of human death globally [1]. In the last three decades, chemotherapy has been the main modality of treatment for cancer patients. However, its effectiveness remains low, primarily due to limited accessibility of drugs to the tumor tissue, their intolerable toxicity, development of muti-drug resistance, and the dynamic heterogeneous biology of the growing tumors [2]. Therefore, the discovery of new compounds with selective activity against cancerous cells is the center of attention of anticancer drug development.
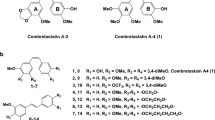
CA4P is a synthetic water-soluble phosphorylated pro-drug of combretastatin A4 (CA-4, Fig. 1), isolated from the South African willow bush, combretum caffrum. What is more, CA4P as a tumor vascular targeting agent is currently in clinical trial [3,4,5]. In animal models, CA4P reversibly binds to the colchicine binding site of endothelial cell tubulin, inhibiting tubulin polymerization and selectively inducing mitotic arrest and apoptosis in cancer cells but not in normal cells [6]. As apoptotic endothelial cells detach from their substrate, the detachment can result in shutdown of blood flow, rapid tumor vascular collapse and tumor necrosis [7]. Due to its reversible effects and the short half-life of about 10–27 min, the adverse effects typical of tubulin-binding agents did not occur in CA4P [8]. CA4P has been demonstrated to disrupt tumor neovasculature and decrease tumor blood flow within both animal and human tumors [9]. The safety and activity profile of CA4P in oncology patients showed that side effects are mild to moderate.

The chemical structure of CA-4, CA4P and AVE-8062
In recent years, a large number structure modification has been made to find more potent CA4 and CA4P analogs [10,11,12,13]. In the search for new bioactive compounds and particularly cytotoxic agents, Phosphornates and phosphoramidates are often used as prodrug moieties to get better restorative potential of the parent drug [14, 15] These derivatives are also used as drugs due to their potential applications as anticancer [16,17,18], anti-HIV [19, 20], inhibitors of hepatitis C virus [21, 22], and anti-malarial agents [23]. Phosphoramidates are the building blocks for the synthesis of phosphate esters in nucleotide chemistry and key structural skeletons in many biologically active natural products such as agrocin 84 [24], phosmidosine and microcin C7 [25]. Therefore, the synthesis of new bioactive phosphoramidate entities has attracted considerable interest.
In continuation of our efforts toward the synthesis of novel potential anticancer active compounds based on the CA-4 scaffold and considering the meaningful biological performance of CA4P [26], we sought to develop a simple one-pot synthesis of novel CA-4 phosphoramidate prodrugs (Fig. 2). In addition, the in vitro anti-proliferative effect of each obtained product was evaluated using the CCK-8 assays on a set of human cancer cells including human hepatocarcinoma (HepG2), Cervix carcinoma (HeLa), and colorectal carcinoma (HCT-116). Besides, for compounds with the highest and lowest in vitro anti-proliferative effect on cancer cells, molecular docking simulations were conducted to investigate the nature of interactions between the compounds and colchicines binding site of tubulin.

The designed structure of CA-4 phosphoramidates
Results and discussion
Chemistry
According to our previous report [26], it was possible to efficiently access the CA-4 derivatives using a modified route that involves a three-step synthetic pathway. In this work, the same synthetic approach was implemented. Once the Combretastatin A-4 and Erianin were synthesized and properly purified, CA-4 and Erianin phosphoramidate derivatives (5a-g and 6a-g) were obtained with good yields (43–90%). The synthetic route of CA-4 phosphoramidates and its analog Erianin phosphoramidates is outlined in Scheme 1. During the experimental design, the method of multistep feeding was replaced by a one-pot reaction and no intermediate treatments were employed on the target compounds. Firstly, various commercially available amino acid esters were treated with phenyl dichlorophosphate in the presence of triethylamine (TEA), after 4 h, the mixture of Combretastatin A-4 or Erianin and TEA was added to the reaction solution. Then the desired compounds were obtained in moderate to good yields after the reaction was stirred for another 4 h. All the HRMS, 1H-NMR and 13C-NMR data of the synthetic compounds were in accordance with the proposed molecular structures.

(1) 2 equiv TEA, CH2Cl2, 1 equiv Phenyl dichlorophosphate (PDP), −70 °C, 0.5 h, then warmed to 0 °C in 3 h; (2) 1 equiv TEA, CH2Cl2, 1 equiv CA-4 or Erianin, 0 °C, 4 h
In vitro anti-proliferative activity
The anti-tumor activities of the target compounds were evaluated in vitro against HepG2, HeLa, and HCT-116 cell lines by the CCK8 method. Meanwhile, CA4P acted as the positive control and the results are summarized in Tables 1 and 2. As we had predicted, all the CA-4 phosphoramidates (5a-g) and most of its analogue Erianin phosphoramidates (6b, 6d, 6e, and 6g) exhibited significant inhibitory effects with more than 50% inhibition rate of all tested cell lines at a low concentration of 1μΜ. In particular, all the tested compounds had a similar inhibition rate to CA4P, displaying >80% inhibition in HeLa cell, showing remarkable anti-tumor activities against HeLa cell line as compared with the other cell lines except 6a, 6c, and 6f.
Structure-activity relationships of all synthetic compounds demonstrated that (1) Comparing the types of bond in Part A of synthetic compounds, the double-bond-linked derivatives (5a-5g) resulted in better inhibitory activity than the single bond linked analogs (6a-6g), revealing that the antiproliferation activity was strongly dependent on the types of linked bond and that the double bond was better than the single bond in Part A of tested compounds for the cytotoxicity in human cancer cells. (2) The comparison of R groups in α-amino acid esters (Part B) indicated that the amino acid ester moiety, especially at the position α-carbon atom has a major steric influence on antitumor activity in vivo. As compounds 5c, 5e, and 5g demonstrated, the R1 methoxy group can be replaced with ethoxy group or Isopropoxy group without substantial loss of activity against HepG2, HeLa and HCT-116 cells. Interestingly, the compounds of the 6 series inhibited different human cancer cells proliferation with varying ability as shown in Table 2. Of the compounds evaluated, 6a, 6c, and 6f exhibited a dramatic reduction in the proliferation inhibitory rate against HepG2 and HCT-116 cells. This result further validated the above conclusion. This is probably due to the fact that the large side chain group at the position of α-carbon atom would sterically hinder the interaction between the amino group and the protein. And the reduction in potency was also observed in compound 5f with 42.20% inhibition rate against HCT-116 cell line.
Molecular docking study
To better understand the discrepancy of anticancer activities between series 5 and 6 for microbubulin polymerization, molecular docking was used to explore the 3D binding modes of the representative CA4P, 5d and 6f at the colchicines binding site of tubulin (PDB ID: 402B from Protein Data Bank) with the help of Surflex-dock, which provided an insight into the binding affinity and hydrogen bond formation of the ligands to the 402B binding sites. Figures 3–5 show the docking results of CA4P, 5d, and 6f respectively. The interacted amino acid residues at the binding site of 402B with Ligands were summarized in Table 3. The initial docking results show that the van der Waals interaction seems to be the key factor for the bioactivity. The hydrogen bond formation was analyzed for each molecule to see the contribution of each molecule to 402B binding sites. CA4P formed three hydrogen bonds with Ala180 (1.92 Å), Lys254 (1.72 Å) and Asn101 (1.91 Å). In 5d, two hydrogen bonds were found to be with Ala250 (1.97 Å) and Thr353 (2.71 Å). In the case of the 6f molecule, only one hydrogen bond formation was analyzed to be with Gln11 (2.69 Å). The formation of hydrogen bonds and van der Waals forces with the active residues by CA4P and 5d when compared to 6f suggests that series 5 are potential inhibitor compounds.

The molecular docking results of CA4P

The molecular docking results of 5d

The molecular docking results of 6f
MD simulation analysis
To gain further detailed insight into the stability of the docked complexes, a 50 ns-long MD simulation and binding free energy calculations of the top 2 docked complexes and CA4P were performed using the GROMACS 5.1.2 package. The overall convergence of MD simulations and system equilibration were monitored in terms of the root-mean-square deviation (RMSD) of tubulin inhibitor backbone atoms (C, Cα, N, and O). The average RMSD value (Å) with respect to simulation time (ns) of three representative inhibitors is represented in Fig. 6. The RMSD value gained till 10 ns and convergence was observed from 20 ns but minor fluctuations remained throughout. The fluctuations in the average RMDS values during the whole simulation are between 1.5 and 2.5 Å. In addition, the initial structure of the 5g-tubulin (PDB ID: 402B) complex and the equilibrium structure after 50 ns MD simulation are superimposed in Fig. 7. Figure 7 shows that there seems to be no significant difference between the initial structure (in green) and the lowest energy structure (in magenta) except for a slight drift and rotation of bonds. It can be inferred that the binding pocket and the conformation of the ligand are stable and the docking results are reliable. The MD simulation results are in good agreement with molecular docking results.

Time dependent RMSD analysis of 5d (blue), 6f (black) and CA4P (red) protein backbone

The superimposed initial structure (green ribbon) and equilibrium structured (magenta ribbon) of 5d-402B complexes
To quantify the docking results, the binding free energy calculations for the three complexes of the last 5 ns trajectory were carried out and are listed in Table 4. The contributions of different energy components are also reported in Table 4. The free energy calculations were studied using g_mmpbsa. The binding free energy (ΔGbind) and the individual components were calculated based on the following formula \(\Delta {\mathrm{G}}_{{\mathrm{bind}}} = {\mathrm{EMM}} + {\mathrm{G}}_{{\mathrm{solv}}} - {\mathrm{T}}\Delta {\mathrm{S}} = {\mathrm{E}}_{{\mathrm{vdw}}} + {\mathrm{E}}_{{\mathrm{elec}}} + {\mathrm{G}}_{{\mathrm{polar}}} + {\mathrm{G}}_{{\mathrm{non - polar}}} - {\mathrm{T}}\Delta {\mathrm{S}}.\)
The four individual energy components were compared for the three ligand complexes to analyze the contribution towards binding affinity. It can be seen that the major contribution to the overall binding free energy is the van der Waals energy. The ΔEvdw component was found to be −234.942 kJ/mol in CA4P complex, −362.206 kJ/mol in 5d complex and −267.283 kJ/mol in 6f complex. The van der Waals energy contribution was found to be significantly higher in 5d complex when compared to the CA4P and 6f complex. ΔEelec changes were also negative and a favorable energetic contribution to the total binding free energy where −126.299 kJ/mol in CA4P complex, −39.299 kJ/mol in 5d complex and −90.568 kJ/mol in 6f complex were found, respectively. When ΔGpolar was analyzed, 238.819 kJ/mol in CA4P complex, 272.943 kJ/mol in 5d complex and 248.887 kJ/mol in 6f complex were determined. The positive polar energy is unfavorable energetic contribution to the binding affinity. In the case of ΔGnon-polar, the energy component was −22.466 kJ/mol in CA4P complex, −38.384 kJ/mol in 5d complex and −27.755 kJ/mol in 6f complex. The lower non-polar energy found in 5d complex with respect to the higher non-polar energies in CA4P and 6f also contributed to the 5d inhibitor higher binding affinity. The binding free energy for the inhibitors CA4P, 5d and 6f via g_mmpbsa was predicted to be −142.888, −166.946, and −136.719 kJ/mol, respectively. The CA4P and 5d complexes with protein had better and similar binding energy when compared to 6f complex with protein.
Conclusion
In summary, a series of novel Phosphoramidate derivatives of Combretastatin A-4 have been synthesized via an environmentally friendly one-pot reaction. According to the results of in vitro proliferation inhibitory activity together with the molecular docking results, the biaryl structure linked with double bond in Part A and the steric effect at the position α-carbon atom in the amino acid ester moiety (Part B) played an important role in activity exhibition. The vast majority of these novel Phosphoramidate derivatives exhibited potent anti-cancer activities. Further research for modifying and enhancing their biological activity potential is undergoing based on the present data.
Experiments
Materials and measurements
All chemicals and reagents used in this study were of analytical grade. All NMR spectra were recorded on a Bruker Avance 500 Spectrometer (resonance frequencies 500 MHz for 1H and 126 MHz for 13C) equipped with a 5 mm inverse broadband probe head with z-gradients at 295.8 K with standard Bruker pulse programs. The samples were dissolved in 0.6 mL CDCl3 (99.8% D.TMS). Chemical shifts were given in values of δH and δC referenced to residual solvent signals (δH 7.26 for 1H, and δC 77.0 for 13C in CDCl3). High resolution mass spectra (HRMS) were recorded on a Bruker solan X70FT-MS (samples were dissolved in CH3OH and the ion source was ESI), and the energy was 22.5 eV at MS/MS. Melting points are uncorrected. TLC was performed on the glassbacked silica gel sheets (Silica Gel 60 GF254) and visualized in UV light (254 nm). Column chromatography was performedusing silica gel (300–400 mesh) eluting with ethyl acetate and petroleum ether.
General procedure for synthesis of target compounds 5a-g, 6a-g
Triethylamine (20 mmol) was dropwise added to a stirring solution of amino acid ester hydrochloride (10 mmol) in CH2Cl2 (250 mL) at −70 °C. After 30 min, phenyl dichlorophosphate (10 mmol) was dropwise added into the reaction mixture. The reaction mixture was under stirring for 30 min at −70 °C before being warmed up to 0 °C in 3 h. Then a solution of CA-4 or erianin (10 mmol) and triethylamine (10 mmol) in CH2Cl2 was dropwise added, and the solution was continually stirred for another 4 h at 0 °C. After the completion of reaction, the white solid (triethylamine hydrochloride) was filtered off and washed with dichloromethane (1 × 25 mL). The solvent of the filtrate was evaporated to obtain the residue. Subsequentially the residue was triturated with methyl tertbutyl ether (TBME, 100 mL), and the triethylamine hydrochloride salt was removed by filtration. The filter cake was washed with TBME (50 mL), and the combined filtrate was concentrated under reduced pressure to give a crude liquid. Then the crude liquid was purified by flash column chromatography using petroleum ether/ethyl acetate (3:1) as eluent to give the mixture of diastereomers 5a-g, 6a-g in 40–88% yields.
(2R)-methyl-2-(((2-methoxy-5-((Z)-3,4,5-trimethoxystyryl)phenoxy)(phenoxy)phosphoryl) amino)-3-methyl butanoate (5a)
Light yellow liquid, yield: 53%. 1H NMR (500 MHz, CDCl3) δ 7.29 (t, J = 7.7 Hz, 2H), 7.22 (t, J = 7.6 Hz, 3H), 7.13 (t, J = 7.2 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.80–6.75 (m, 1H), 6.46 (d, J = 15.8 Hz, 2H), 6.40 (d, J = 12.5 Hz, 2H), 3.99–3.92 (m, 1H), 3.83–3.79 (m, 6H), 3.64 (dd, J = 25.6, 7.2 Hz, 10H), 2.03 (s, 1H), 0.87–0.82 (m, 3H), 0.82–0.77 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 172.78, 172.42, 152.95, 150.91, 149.75, 139.43, 137.34, 132.44, 130.37, 129.64, 129.51, 128.54, 126.43, 124.85, 122.85, 122.62, 120.30, 112.10, 106.09, 60.85, 59.79, 55.95, 51.96, 18.53, 17.40. HRMS-ESI (m/z) calcd. for C30H37NO9P [M + H]+: 586.22004, found: 586.21950.
(2S)-methyl-2-(((2-methoxy-5-((Z)-3,4,5-trimethoxystyryl)phenoxy)(phenoxy)phosphoryl)amino)-4-methylpentanoate (5b)
Light yellow liquid, yield: 47%. 1H NMR (500 MHz, CDCl3) δ 7.30 (dd, J = 9.1, 6.2 Hz, 2H), 7.26–7.18 (m, 3H), 7.14 (t, J = 6.9 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.78 (dd, J = 11.9, 8.6 Hz, 1H), 6.48 (t, J = 7.7 Hz, 2H), 6.42 (t, J = 6.4 Hz, 2H), 4.12 (dd, J = 14.6, 7.5 Hz, 1H), 3.81 (d, J = 17.2 Hz, 6H), 3.65 (dd, J = 28.5, 8.0 Hz, 9H), 1.56–1.42 (m, 2H), 0.91–0.86 (m, 3H), 0.84 (dd, J = 6.2, 4.5 Hz, 3H). 13C NMR(125 MHz, CDCl3) δ 173.84, 152.96, 150.83, 149.74, 139.41, 137.32, 132.47, 130.33, 129.66, 129.53, 128.56, 128.53, 126.40, 124.87, 122.73, 120.30, 112.10, 106.07, 60.87, 55.96, 53.11, 52.12, 43.98, 24.22, 22.64. HRMS-ESI (m/z) calcd. for C31H39NO9P [M + H]+: 600.23569, found: 600.23459.
(2S)-methyl-2-(((2-methoxy-5-((Z)-3,4,5-trimethoxystyryl)phenoxy)(phenoxy)phosphoryl)amino)-3-phenylpropanoate (5c)
Light yellow liquid, yield: 56%. 1H NMR (500 MHz, CDCl3) δ 7.30 (dd, J = 15.5, 7.7 Hz, 2H), 7.21 (dd, J = 15.0, 7.2 Hz, 6H), 7.14 (dd, J = 14.8, 7.4 Hz, 1H), 7.08 (t, J = 7.0 Hz, 1H), 7.00 (dd, J = 9.0, 5.7 Hz, 2H), 6.77 (dd, J = 8.4, 2.7 Hz, 1H), 6.51–6.40 (m, 4H), 4.45 (qd, J = 9.6, 5.2 Hz, 1H), 3.82 (s, 3H), 3.73 (d, J = 9.0 Hz, 3H), 3.65 (s, 6H), 3.58 (d, J = 21.5 Hz, 3H), 3.12–2.90 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 172.18, 152.98, 150.87, 149.71, 139.37, 137.41, 135.46, 132.42, 130.34, 129.72, 129.62, 129.49, 128.51, 128.43, 127.06, 127.00, 126.55, 124.97, 122.60, 120.32, 112.15, 106.15, 60.85, 55.95, 55.86, 55.37, 52.05, 40.44. HRMS-ESI (m/z) calcd. for C34H37NO9P [M + H]+: 634.22004, found: 634.22162.
Methyl N6-(tert-butoxycarbonyl)-N2-((2-methoxy-5-((Z)-3,4,5-trimethoxystyryl)phenoxy)(phenoxy)phosphoryl)-D-lysinate(5d)
Light yellow liquid, yield: 44%. 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.8 Hz, 2H), 7.22 (t, J = 7.2 Hz, 3H), 7.15 (t, J = 7.3 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 6.48 (s, 2H), 6.43 (s, 2H), 4.59 (s, 1H), 3.93 (t, J = 10.4 Hz, 1H), 3.82 (t, J = 8.3 Hz, 6H), 3.70–3.62 (m, 9H), 3.01 (s, 2H), 2.04 (s, 2H), 1.75–1.55 (m, 2H), 1.43 (s, 9H), 1.26 (t, J = 7.1 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 173.15, 155.96, 152.93, 150.75, 149.71, 139.29, 137.23, 132.45, 130.27, 129.67, 129.60, 128.52, 126.54, 124.99, 122.62, 120.29, 112.09, 105.98, 79.05, 60.89, 55.95, 54.29, 52.31, 40.21, 34.24, 28.42, 21.84. HRMS-ESI (m/z) calcd. for C36H48N2O11P [M + H]+: 715.2917, found: 715.2938.
(Z)-ethyl-2-(((2-methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)(phenoxy)phosphoryl)amino) acetate (5e)
Light yellow liquid, yield: 49%. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 3H), 7.16 (t, J = 7.2 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 6.47 (d, J = 9.7 Hz, 2H), 6.43 (d, J = 13.0 Hz, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.83 (d, J = 2.9 Hz, 6H), 3.69 (s, 6H), 2.04 (s, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 170.14, 152.97, 150.77, 149.79, 139.25, 132.45, 130.39, 129.71, 129.61, 128.54, 126.58, 124.98, 122.72, 120.29, 120.25, 112.20, 106.10, 61.53, 60.85, 55.96, 43.06, 14.11. HRMS-ESI (m/z) calcd. for C28H33NO9P [M + H]+: 558.18874, found: 558.18800.
(Z)-ethyl-1-(((2-methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)(phenoxy)phosphoryl)amino)cyclopropanecarboxylate (5f)
Light yellow liquid, yield: 50%. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.8 Hz, 2H), 7.25 (d, J = 6.9 Hz, 3H), 7.16 (t, J = 7.2 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 6.49 (d, J = 13.3 Hz, 2H), 6.44 (t, J = 6.9 Hz, 2H), 4.05 (dd, J = 7.2, 3.4 Hz, 2H), 3.86–3.69 (m, 12H), 1.45–1.37 (m, 2H), 1.26 (ddd, J = 15.6, 13.0, 4.9 Hz, 2H), 1.16 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.11, 152.99, 150.93, 149.81, 139.46, 137.66, 137.52, 132.50, 130.24, 129.62, 129.51, 128.61, 126.47, 124.86, 122.47, 120.26, 112.24, 106.07, 61.43, 60.88, 55.98, 35.06, 18.12, 17.25, 14.09. HRMS-ESI (m/z) calcd. for C30H35NO9P [M + H]+: 584.20440, found: 584.20552.
(2S)-isopropyl-2-(((2-methoxy-5-((Z)-3,4,5trimethoxystyryl)phenoxy)(phenoxy)phosphoryl)amino)propanoate (5g)
Light yellow liquid, yield: 55%. 1H NMR (500 MHz, CDCl3) δ 7.30 (t, J = 7.6 Hz, 2H), 7.22 (d, J = 7.4 Hz, 3H), 7.14 (t, J = 7.2 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.79 (t, J = 8.0 Hz, 1H), 6.50–6.39 (m, 4H), 4.98 (tt, J = 15.8, 6.2 Hz, 1H), 4.00 (dd, J = 20.1, 8.4 Hz, 1H), 3.82 (t, J = 6.1 Hz, 6H), 3.68 (s, 6H), 1.33 (t, J = 7.3 Hz, 3H), 1.22–1.15 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 172.87, 152.93, 150.76, 149.76, 139.31, 137.22, 132.47, 130.25, 129.63, 129.56, 128.57, 126.49, 124.92, 122.81, 122.62, 120.35, 112.10, 105.97, 69.11, 60.88, 55.93, 50.38, 21.67, 21.09. HRMS-ESI (m/z) calcd. for C30H37NO9P [M + H]+: 586.22004, found: 586.21880.
(2R)-methyl-2-(((2-methoxy-5-(3,4,5-trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)amino)-3-methylbutanoate (6a)
Light yellow liquid, yield: 55%. 1H NMR (500 MHz, CDCl3) δ 7.37–7.31 (m, 2H), 7.31–7.25 (m, 2H), 7.22 (d, J = 11.6 Hz, 1H), 7.17 (t, J = 7.1 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.85 (dd, J = 8.3, 4.1 Hz, 1H), 6.39 (d, J = 3.4 Hz, 2H), 3.99 (dtd, J = 15.1, 10.4, 4.7 Hz, 1H), 3.84 (dd, J = 10.8, 7.7 Hz, 12H), 3.63 (d, J = 20.2 Hz, 3H), 2.81 (d, J = 5.0 Hz, 4H), 2.07–1.99 (m, 1H), 0.94–0.87 (m, 4H), 0.80 (t, J = 6.5 Hz, 3H), 0.09 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 172.84, 153.09, 151.02, 148.86, 137.32, 136.28, 134.78, 134.57, 129.54, 125.68, 124.85, 121.90, 120.47, 112.45, 105.47, 60.82, 59.76, 56.08, 51.97, 38.29, 37.03, 32.34, 18.51. HRMS-ESI (m/z) calcd. for C30H39NO9P [M + H]+: 588.23569, found: 588.23430.
(2S)-methyl-2-(((2-methoxy-5-(3,4,5-trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)amino)-4-methylpentanoate (6b)
Light yellow liquid, yield: 40%. 1H NMR (500 MHz, CDCl3) δ 7.36–7.31 (m, 2H), 7.30 (s, 1H), 7.27–7.20 (m, 2H), 7.20–7.15 (m, 1H), 6.94 (d, J = 6.5 Hz, 1H), 6.86 (t, J = 8.3 Hz, 1H), 6.40 (d, J = 5.5 Hz, 2H), 3.84 (q, J = 8.0 Hz, 12H), 3.71 (d, J = 13.1 Hz, 1H), 3.63 (d, J = 21.3 Hz, 3H), 2.85–2.78 (m, 4H), 1.52 (dt, J = 12.3, 6.4 Hz, 2H), 1.28 (t, J = 7.1 Hz, 1H), 0.85–0.81 (m, 3H), 0.09 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 173.88, 153.08, 150.98, 148.83, 139.46, 137.35, 136.23, 134.77, 129.55, 125.63, 124.88, 122.07, 120.43, 112.46, 105.40, 60.86, 56.08, 53.10, 52.10, 43.97, 38.30, 37.09, 24.15, 22.69. HRMS-ESI (m/z) calcd. for C31H41NO9P [M + H]+: 602.25134, found: 602.24958.
(2S,3R)-methyl-2-(((2-methoxy-5-(3,4,5-trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)amino)-3-methylpentanoate (6c)
Light yellow liquid, yield: 62%. 1H NMR (500 MHz, CDCl3) δ 7.33 (td, J = 8.1, 3.0 Hz, 2H), 7.30–7.25 (m, 2H), 7.25–7.19 (m, 1H), 7.16 (t, J = 6.8 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.87–6.82 (m, 1H), 6.39 (d, J = 4.1 Hz, 2H), 4.09–4.00 (m, 1H), 3.87–3.81 (m, 12H), 3.62 (d, J = 20.8 Hz, 3H), 2.80 (dd, J = 8.5, 5.7 Hz, 4H), 1.80–1.67 (m, 1H), 1.27 (dd, J = 15.1, 8.1 Hz, 2H), 0.91–0.75 (m, 7H). 13C NMR (125 MHz, CDCl3) δ 172.74, 153.08, 151.00, 148.85, 137.33, 136.27, 134.75, 134.55, 129.53, 125.61, 124.85, 122.11, 120.43, 112.44, 105.45, 60.82, 58.82, 56.07, 51.91, 39.43, 38.30, 37.04, 24.84, 14.94, 11.51. HRMS-ESI (m/z) calcd. for C31H41NO9P [M + H]+: 602.25135, found: 602.25194.
Methyl N6-(tert-butoxycarbonyl)-N2-((2-methoxy-5-(3,4,5trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)-D-lysinate (6d)
Light yellow liquid, yield: 88%. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.7 Hz, 2H), 7.26 (t, J = 9.1 Hz, 2H), 7.21–7.14 (m, 2H), 6.92 (t, J = 7.4 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 6.37 (d, J = 3.6 Hz, 2H), 4.56 (s, 1H), 3.94 (dt, J = 32.9, 10.3 Hz, 1H), 3.86–3.78 (m, 12H), 3.63 (d, J = 18.2 Hz, 3H), 2.97 (d, J = 30.6 Hz, 2H), 2.79 (s, 4H), 2.28–1.88 (m, 2H), 1.73–1.56 (m, 2H), 1.42 (d, J = 2.3 Hz, 9H), 1.24 (t, J = 7.1 Hz, 2H), 0.07 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 173.21, 155.94, 153.06, 150.93, 148.82, 139.39, 137.28, 136.26, 134.76, 129.58, 125.75, 124.94, 122.14, 120.41, 112.49, 105.46, 79.01, 60.80, 56.06, 54.37, 52.27, 38.21, 36.98, 34.24, 29.42, 28.40, 21.88. HRMS-ESI (m/z) calcd. for C36H50N2O11P [M + H]+: 717.3074, found: 717.3097.
Ethyl-2-(((2-methoxy-5-(3,4,5-trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)amino) acetate (6e)
Light yellow liquid, yield: 53%. 1H NMR (500 MHz, CDCl3) δ 7.35 (t, J = 7.8 Hz, 2H), 7.29 (d, J = 8.6 Hz, 2H), 7.18 (t, J = 7.6 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.86 (d, J = 8.3 Hz, 1H), 6.79 (dd, J = 17.9, 8.1 Hz, 1H), 6.39 (d, J = 4.5 Hz, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.83 (d, J = 4.5 Hz, 14H), 2.82 (d, J = 5.9 Hz, 4H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 170.31, 153.05, 150.82, 148.85, 139.30, 137.36, 134.67, 129.66, 129.37, 125.82, 125.05, 122.05, 120.42, 119.77, 115.42, 112.50, 105.43, 61.56, 60.85, 56.06, 43.08, 38.23, 37.02, 14.12. HRMS-ESI (m/z) calcd. for C28H35NO9P [M + H]+: 560.20439, found: 560.20256.
Ethyl-1-(((2-methoxy-5-(3,4,5-trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)amino)cyclopropanecarboxylate (6f)
Off-white solid, yield: 54%. m.p.: 83.7–84.7 °C. 1H NMR (500 MHz, CDCl3) δ 7.37–7.31 (m, 2H), 7.28 (d, J = 8.5 Hz, 2H), 7.24 (s, 1H), 7.17 (t, J = 7.2 Hz, 1H), 6.93 (d, J = 7.7 Hz, 1H), 6.86 (d, J = 8.3 Hz, 1H), 6.38 (d, J = 11.5 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 3.87–3.72 (m, 12H), 2.82 (d, J = 2.9 Hz, 4H), 1.51–1.25 (m, 4H), 1.13 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.19, 153.08, 150.96, 148.79, 139.56, 137.33, 136.21, 134.63, 129.55, 125.56, 124.87, 121.98, 120.44, 112.57, 105.39, 61.41, 60.85, 56.08, 38.29, 37.06, 35.09, 18.17, 17.59, 14.08. HRMS-ESI (m/z) calcd. for C30H37NO9P [M + H]+: 586.22004, found: 586.21830.
(2S)-isopropyl-2-(((2-methoxy-5-(3,4,5-trimethoxyphenethyl)phenoxy)(phenoxy)phosphoryl)amino)propanoate (6g)
Light yellow liquid, yield: 54%. 1H NMR (500 MHz, CDCl3) δ 7.35 (t, J = 7.1 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.22 (s, 1H), 7.18 (t, J = 7.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.88–6.83 (m, 1H), 6.39 (s, 2H), 5.00 (tt, J = 12.4, 6.2 Hz, 1H), 4.20–4.12 (m, 1H), 3.85 (d, J = 15.3 Hz, 12H), 2.87–2.75 (m, 4H), 1.35 (dd, J = 27.0, 7.0 Hz, 3H), 1.25–1.19 (m, 6H), 0.09 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 172.85, 153.07, 150.95, 148.84, 139.43, 137.35, 136.22, 134.58, 129.58, 125.67, 124.90, 121.97, 120.44, 112.44, 105.41, 69.09, 60.85, 56.07, 50.44, 38.28, 37.06, 21.61, 21.15. HRMS-ESI (m/z) calcd. for C30H39NO9P [M + H]+: 588.23569, found: 588.23571.
In vitro proliferation inhibitory activity assay
The anti-tumor activities of the target compounds were evaluated in vitro against HepG2, HeLa and HCT-116 cell lines respectively by the CCK8 method. Meanwhile, CA4P acted as the positive control. EnoGeneCell™ Counting Kit-8 (CCK-8) and cell lines were available from Nanjing Enogene Biotech. Co., Ltd. Cells were cultured in RPMI1640 medium containing 10% fetal calf serum and only those containing above 90% live cells were be used to carry out the cytotoxicity assessments. Cell proliferation inhibition needed to be tested with EnoGeneCell ™ Kit-8 (CCK-8) Cell Counting Kit. The cell suspension at the concentration of 1 × 105 cells mL−1 was obtained after cell digestion and counting. 100 μL of the cell suspension was added to each well of 96-well culture plates to make sure 1 × 104 cells per well. The subsequent incubation was permitted at 37 °C and 5% CO2 incubator for 24 h. 100 μL of corresponding culture medium containing tested samples in a concentration of 1 μΜ was added to each well. At the same time negative control group, solvent control group and positive control group were established, each group had five replicate wells. After incubation at 37 °C and 5% CO2 atmosphere about 72 h, 10 μL of CCK-8 solution was added in each well. Four hours after incubation, the OD value was measured at the wavelength of 450 nm on an ELISA microplate reader. Then the inhibition rate against HepG2, HeLa, and HCT-116 cells of these compounds were calculated according to the OD values, respectively. The results were summarized in Tables 1 and 2.
Molecule docking
The molecular docking procedure was performed in the Surflex-Dock module in Sybyl-X 2.0. The crystal structure of tubulin (PDBID: 402B) was downloaded from the RSCB Protein Data Bank [27]. The complex with colchicine formed by chains A and B of the tubulin heterodimer was considered for docking. The ligands were docked into the colchicine binding site by an empirical scoring function and a patented search engine in Surflex–Dock. Before the docking process, the colchicine ligand containing the tubulin complex was extracted and water molecules were removed. Subsequently, protein was prepared by using Biopolymer module implemented in Sybyl. Polar hydrogen atoms were added. AMBER7 FF99 charges were assigned to protein atoms [28]. Other parameters were taken by default. Then a protocol was generated based on the original ligand. The molecule was flexibly docked into the active pocket. Surflex-Dock total scores and corresponding conformations were employed for further studies.
Molecule dynamics (MD) simulations
MD simulations were carried out using the GROMACS 5.1.2 package. The general Amber force field (gaff) and the ff99SB force field were used for the ligands and the protein, respectively. The counter-ions Na+ or Cl− were added to neutralize the unbalanced charged in the complexes [29]. Each system was added 10 Å out of the solute using an octahedral TIP3P water box to reduced computational demand.
All the solvated simulations follow the procedure of minimization, heating, density equilibration, and production. Initially, the minimization of the entire system was performed in two steps. In the first step, the atom position of all solute species was minimized with 500 cycles of each steepest descent and conjugate gradient methods by restraining the protein-ligand complex with a force constant of 10 kcal mol−1 Å−2. In the second step, the whole system was minimized without any restraints by 1000 cycles of steepest descent and 5000 cycles of conjugate gradient [30]. Then the minimized system was gradually heated under and NVT ensemble from 0 to 300 K over a period of 20 ps, followed by density equilibration under and NPT ensemble at 300 K over 100 ps with a weak restraint of 2 kcal mol−1 Å−2 of constant pressure (1 atm). In this method the SHAKE algorithm was applied to all bonds involving hydrogen atoms, and the Langevin dynamics was used for temperature control [31]. Finally, a 50 ns production run was performed under an NPT (1 atm) ensemble with a cut-off distance of 10 Å at 300 K. a periodic boundary condition was used for the whole simulation run in order to counter the bulk effect. The integration time step was set to 2 fs and coordinate trajectories were recorded at every 2 ps in the whole simulation. Visualization and analysis were carried out by VMD. The figure of root mean square deviation (RMSD) and cluster analysis were performed using the Xmgrace program. The free energy of binding was calculated using g_mmpbsa (Molecular Mechanics Poison-Boltzamann Surface Area) Gromace tool [32].
References
Bernal CC, Vesga LC, Mendes-Sánchesz SC Synthesis and anticancer activity of new tetrahydroquinoline hybrid derivatives tethered to isoxazoline moiety. Med Chem Res. 2020;29:675–89.
Vasir JK, Labhasetwar V Targeted drug delivery in cancer therapy. Technol Cancer Res T. 2005;4:363–74.
Izumi Y, Aoshima K, Hoshino Y, Takagi S. Effects of combretastatin A-4 phosphate on canine normal and tumor tissue-derived endothelial cells. Res Vet Sci. 2017;112:222–8.
Lin CM, Ho HHPG, Hame L Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6911.
Chaplin D, Pettit G, Hill S Anti-vascular approaches to solid tumour therapy: evaluation of combretastatin A4 phosphate. Anticancer res. 1998;19:189–95.
Siemann DW, Chaplin DJ, Walicke PA A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin Investig Drugs. 2009;18:189–97.
Ibrahim MA, Do DV, Sepah YJ, Shah SM, Anden EV, Hafiz G, et al. Vascular disrupting agent for neovascular age related macular degeneration: a pilot study of the safety and efficacy of intravenous combretastatina-4 phosphate. BMC Pharm Toxicol. 2013;14:1–10.
Hu E, Ko R, Koda R, Rosen P, Jeffers S, Scholtz M, et al. Phase I toxicity and pharmacology study of trimethylcolchicinic acid in patients with advanced malignancies. Cancer Chemother Pharm. 1990;26:359–64.
Duncan DD, Lemaillet P, Ibrahim M, Nguyen QD, Hiller M, Roman JR. Absolute blood velocity measured with a modified fundus camera. J Biomed Opt. 2010;15:056014.
Ohsumi K, Nakagawa R, Fukuda Y, Hatanaka T, Morinaga Y, Nihei Y, et al. Novel combretastatin analogues effective against murine solid tumors: design and structure–activity relation- ships. J Med Chem. 1998;41:3022–32.
Pettit GR, Toki BE, Herald DL, Boyd MR, Hamel E, Pettit RK, et al. Antineoplastic agents. 410. Asymmetric hydroxylation of transcombretastatin A-4. J Med Chem. 1999;42:1459–65.
Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J Med Chem. 2006;49:3033–44.
Hori K, Saito S Microvascular mechanisms by which the combretastatin A-4 derivative AC7700 (AVE8062) induces tumour blood flow stasis. Br J Cancer. 2003;89:1334–44.
Rasool SN, Subramanyam C, Janakiramudu DB, Supraja P, Usha R, Raju CN. Convenient one-pot synthesis and biological evaluation of phosphoramidates and phosphonates containing heterocycles. Phosphorus Sulfur Silicon Relat Elem. 2018;193:470–4.
Wang G, Dyatkina N, Prhavc M, Williams C, Serebryany V, Hu Y, et al. Synthesis and anti-HCV activities of 4’-Fluoro-2’-substituted uridine triphosphates and nucleotide prodrugs: discovery of 4’-Fluoro-2’- C-methyluridine 5’-Phosphoramidate Prodrug (AL-335) for the Treatment of Hepatitis C Infection. J Med Chem. 2019;62:4555–70.
Congiatu C, McGuigan C, Jiang WG, Davies G, Mason MD. Naphthyl phosphoramidate derivatives of bvdu as potential anticancer agents: design, synthesis and biological evaluation. Nucleotides Nucleic Acids. 2006;24:485–9.
Wang ZW, Guo CC, Xie WZ, Liu CZ, Xiao CG, Tan Z. Novel phosphoramidates with porphine and nitrogenous drug: one-pot synthesis and orientation to cancer cells. Eur J Med Chem. 2010;45:890–5.
Duan JX, Jiao H, Kaizerman J, Stanton T, Evans JW, Lan L, et al. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J Med Chem. 2008;51:2412–20.
Mehellou Y, Balzarini J, McGuigan C An investigation into the anti-HIV activity of 2’,3’ -Didehydro-2’,3’ Dideoxyuridine (d4U) and 2’, 3’-Dideoxyuridine (ddU) Phospho-Ramidate ‘ProTide’ Derivatives. Org Biomol Chem. 2009;7:2548–53.
Zakirova NF, Shipitsyn AV, Jasko MV, Prokofjeva MM, Andronova VL, Galegov GA, et al. Phosphoramidate derivatives of acyclovir: synthesis and antiviral activity in HIV-1 and HSV-1 models in vitro. Bioorg Med Chem. 2012;20:5802–9.
McGuigan C, Kelleher MR, Perrone P, Mulready S, Luoni G, Daverio F, et al. The application of phosphoramidate ProTide technology to the potent anti-HCV compound 4’ -azidocytidine (R1479). Bioorg Med Chem Lett. 2009;19:4250–4.
Perrone P, Luoni GM, Kelleher MR, Daverio F, Angell A, Mulready S, et al. Application of the phosphoramidate ProTide approach to 4’-Azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive nucleoside. J Med Chem. 2007;50:1840–9.
Mara C, Dempsey E, Bell A, Barlow JW. Synthesis and evaluation of phosphoramidate and phosphorothioamidate analogues of amiprophos methyl as potential antimalarial agents. Bioorg Med Chem Lett. 2011;21:6180–3.
Sahili AE, Li SZ, Lang J, Virus C, Planamente S, Ahmar M, et al. A oyranose-2-phosphate Motif is responsible for both antibiotic import and quorum-sensing regulation in agrobacterium tumefaciens. PLoS Pathog. 2015;11:1–24.
Ran R, Zeng H, Zhao D, Liu R, Xu X. The novel property of heptapeptide of microcin C7 in affecting the cell growth of Escherichia coli. Molecules 2017;22:432–45.
Zhao L, Zhou JJ, Huang XY, Cheng LP, Pang W, Kai ZP, et al. Design, synthesis and anti-proliferative effects in tumor cells of new combretastatin A-4 analogs. Chin Chem Lett. 2015;26:993–9.
Prota AE, Danel F, Bachmann F, Bargsten K, Buey RM, Pohlmann J, et al. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J Mol Biol. 2014;426:1848–60.
Jain A, Jay N Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511.
Sun HY, Li YY, Shen MY, Tian S, Xu L, Pan PC, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys. 2014;16:22035–45.
Sanghai N, Jain V, Preet R, Kandekar S, Das S, Trivedi N, et al. Combretastatin A-4 inspired novel 2-aryl-3-arylamino-imidazo-pyridines/pyrazines as tubulin polymerization inhibitors, antimitotic and anticancer agents. MedChemcomm 2014;5:766–82.
Miyamoto S, Kollman PA SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comp. Chem. 1992;13:952–62.
Pettit G, Rhodes MR, Herald DL, Chaplin DJ, Stratford MRL, Hamel E, et al. Antineoplastic agents 393. Synthesis of the trans-isomer of combretastatin A-4 prodrug. AntiCancer Drug Des. 1999;13:981–93.
Acknowledgements
The authors are grateful for financial supports from National Natural Science Foundation of China (Grant No. 21672151, 21602135).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that: (i) except for NNSFC, no support, financial or otherwise, has been received from any other organization that may have an interest in the submitted work; and (ii) there are no other relationships or activities that could appear to have influenced the submitted work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, S., Li, T., Pang, W. et al. Synthesis, biological evaluation and molecular docking studies of Combretastatin A-4 phosphoramidates as novel anticancer prodrugs. Med Chem Res 29, 2192–2202 (2020). https://doi.org/10.1007/s00044-020-02632-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02632-2