
Abstract
In our continued efforts to develop targeted prodrugs activated by prostate-specific antigen (PSA), we designed and synthesized novel phosphoramide mustard peptide conjugates using previously optimized PSA substrates. Initial Nu/Nu mouse PK studies indicated that prodrug I (glutaryl-Hyp-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard) exhibits high clearance with significant extrahepatic metabolism in vivo. Substrate optimization studies were thus carried out to further improve PSA specificity and enable the design of prodrugs with reduced in vivo clearance and enhanced tumor selectivity. To assess the utility of the newly optimized sequences as promoieties, they were coupled to phosphoramide mustard using a 4-amino-2-fluorobenzyl alcohol linker akin to prodrug I. In the presence of human PSA, prodrug I was rapidly cleaved with a half-life (t1/2) of 35 min. Prodrugs II (glutaryl-Ser-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard) and III (GABA ← mGly-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard) were hydrolyzed at slower rates with t1/2 values of 80 and 107 min, respectively. These results we observed here are different from our previously reported data but may be explained by the fact that PSA-activated release of phosphoramide mustard and reactive quinonimine methides resulted in mechanism-based inhibition of PSA, thereby preventing further hydrolysis of prodrugs I–III. Prodrug I was cytotoxic to PSA-producing LNCaP cells with an IC50 value of 7.3 μM and demonstrated 14-fold selectivity over the non-PSA-producing DU145. Despite its poor in vitro antiproliferative activity (IC50 = 30 µM), prodrug III was found to be more stable against non-PSA-mediated hydrolysis compared with prodrug I as revealed by metabolite profiling studies, which was in agreement with its improved stability in human hepatocyte cultures. These results suggested that a combination of the peptide sequence GABA ← mGly-Ala-Ser-Chg-Gln with optimal linkers and/or other cytotoxic agents can help achieve an adequate balance between PSA cleavage rate and enhanced resistance to non-PSA-mediated hydrolysis.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The feasibility of targeted prodrugs activated by prostate-specific antigen (PSA) has previously been demonstrated with various cytotoxic agents (Aloysius and Hu 2015b; Yang et al. 2011; Giang et al. 2014; Choi et al. 2012). Prostate disease and cancer progression is characterized by high systemic PSA levels (McDonald et al. 2014; Herschman et al. 1997; Nadler et al. 1995; O’Malley et al. 2014) although low serum concentrations of the marker are also detectable in healthy males (Balk et al. 2003; Catalona et al. 1991; Diamandis 1995; Levesque et al. 1995; Ayyıldız and Ayyıldız 2014). PSA is a kallikrein serine protease with high substrate specificity and a unique ability to cleave the peptide bond after Gln. In addition, the proteolytic activity of PSA is confined to the microenvironment of prostate tumors due to α1-antichymotrypsin and α2-macroglobulin inhibition in the blood (Leinonen et al. 1996; Otto et al. 1998; De Angelis et al. 2007; Nadler et al. 1995) making it an attractive target for tumor-specific activation of peptide prodrugs. Based on the cleavage maps for its natural substrates, semenogelins I and II, several short peptides were engineered as promoieties for prodrugs activated by PSA (Aloysius and Hu 2015a; DeFeo-Jones et al. 2000; Denmeade et al. 1997; Doan et al. 2015; Garsky et al. 2001; Kwiatkowska et al. 2019; Lee et al. 2015; Tarvainen et al. 2020; Wong et al. 2001; Wu and Hu 2016; DiPaola et al. 2002; Jiang and Hu 2007; Jiang et al. 2009).
In this study, phosphoramide mustard was selected as a candidate for peptide conjugation due to its cytotoxic activity against both cycling and non-cycling cells (Kres 1995; Ganesan and Keating 2015), its small molecular weight and size as well as the non-peptidic nature of the self-immolative linker (anticipated to resist proteolysis) required for its attachment to various promoieties (Hu and Wu 2007; Wu and Hu 2016). We previously reported that the prodrug 4-nitrobenzyl phosphoramide mustard demonstrates improved efficacy in SKOV3 ovarian cells expressing E. coli nitroreductase (NTR) and a toxicity profile comparable to that of CB1954 (Wu et al. 2011). The cytotoxic agent released upon NTR activation of 4-nitrobenzyl phosphoramide mustard is phosphoramide mustard, an active metabolite of the DNA-alkylating agent cyclophosphamide (Korkmaz et al. 2007). In combination with other anticancer agents, cyclophosphamide is used for the treatment of lymphomas and certain types of leukemia (Shanafelt et al. 2007; Tabchi et al. 2019). The administration of cyclophosphamide is associated with moderate (vomiting, diarrhea, and bone marrow suppression) to severe adverse effects (bloody urine, gonadal toxicity, mouth sores, and joint pain) (Brock 1996; Emadi et al. 2009). Thus, phosphoramide mustard prodrugs activated by PSA (Scheme 1) are anticipated to demonstrate prostate tumor selectivity and improved safety profiles. The activated prodrug effectively moves the site of activation to prostate cancer cells by PSA from the liver where cyclophosphamide is activated by cytochrome P450 enzymes (Jiang et al. 2006). The feasibility of coupling the cyclophosphamide metabolite, 4-aminocyclophosphamide, to various peptides as model prodrugs has been previously explored and demonstrated (Jiang and Hu 2007; Hu and Wu 2007; Wu and Hu 2016). Using a similar strategy, phosphoramide mustard can be readily attached to our optimized PSA substrates (Aloysius and Hu 2015a). Evaluation of various D-retro-inverso-7-AMC-peptide surrogates revealed that the optimal sequences for rapid PSA cleavage were glutaryl-Ser-Ala-Ser-Chg-Gln-AMC and GABA ← mGly-Ala-Ser-Chg-Gln-AMC, whereas GABA ← D-Ser-Ψ[NH-CO-NH]-Ala-Ser-Chg-Gln-AMC and GABA ← mGly-Ala-Ser-Chg-Gln-AMC were more resistant to non-PSA-mediated hydrolysis in mouse and human plasma when compared with the known substrate glutaryl-Hyp-Ala-Ser-Chg-Gln (Aloysius and Hu 2015a).

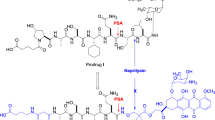
Proposed PSA activation mechanisms of phosphoramide mustard prodrugs
The importance of incorporating a linker between the promoiety and cytotoxic agent has been demonstrated for several prodrugs (Garsky et al. 2001; Hu et al. 2011; Ménez et al. 2008; Poreba 2020; Jiang and Hu 2013). We previously reported a series of self-immolative linkers to enable ready attachment of highly specific PSA substrates to small cytotoxic agents such as phosphoramide mustard (Hu et al. 2011; Wu and Hu 2016). Herein we report the synthesis and evaluation of a series of peptide-linked phosphoramide mustard conjugates incorporating the optimized PSA substrates glutaryl-Ser-Ala-Ser-Chg-Gln and GABA ← mGly-Ala-Ser-Chg-Gln (Aloysius and Hu 2015a) as prodrugs activated by PSA to release phosphoramide mustard in and around prostate cancer cells and tissues.
Results and discussion
Prodrug design principle
To minimize steric hindrance near the scissile bond, attachment of the PSA peptide substrate is typically carried out by inserting a linker between the cytotoxic drug and the PSA-cleavable promoiety (DeFeo-Jones et al. 2000; Garsky et al. 2001; Wong et al. 2001; Jiang and Hu 2007; Ménez et al. 2008). In the present study, peptides were coupled to phosphoramide mustard using a 4-amino-2-fluorobenzyl alcohol linker (NH2-2-F-Bn-OH) as previously reported (Hu and Wu 2007; Hu et al. 2011; Jiang and Hu 2007).
As proof-of-concept, prodrug I was synthesized using 4-amino-2-fluorobenzyl alcohol as the linker and the known PSA substrate, glutaryl-Hyp-Ala-Ser-Chg-Gln (Wu and Hu 2016). Following a single intravenous (IV) administration to Nu/Nu mice at 2 mg/kg, prodrug I was rapidly cleared from blood in vivo (Table 1). The high mouse blood clearance (165 mL/min/kg) of prodrug I suggested that there was significant extrahepatic metabolism given that murine hepatic blood flow is ~90 mL/min/kg (Sohlenius-Sternbeck 2006). These observations led to additional efforts to further improve the PSA specificity of glutaryl-Hyp-Ala-Ser-Chg-Gln in terms of cleavage rate and resistance to non-PSA-mediated hydrolysis in matrices that lack active PSA. Similar to prodrug I, the peptide sequences of glutaryl-Ser-Ala-Ser-Chg-Gln and GABA ← mGly-Ala-Ser-Chg-Gln, which we previously optimized (Aloysius and Hu 2015a), were coupled to phosphoramide mustard using a 4-amino-2-fluorobenzyl alcohol linker to obtain phosphoramide mustard prodrugs II and III, respectively (Scheme 2) in order to assess PSA-mediated tumor killing activity and resistance to non-PSA-mediated metabolism.

Prodrug synthesis. Reagents and conditions: (a) Peptide 1, 2, or 3 (1.3 eq.), DIEA (2.6 eq.), DMF, 0 °C; (b) Compound 4, 0 °C, 30 min, then rt, 30 min; (c) 10% DEA, DMF, rt, 30 min
Synthetic strategy
Peptide conjugates I–III were synthesized by first coupling Gln to the 4-amino-2-fluorobenzyl alcohol linker, which was then conjugated to various peptide sequences (Scheme 2). The synthesis of Gln-linker-phosphoramide mustard 4 has previously been reported by Wu et al. (Wu et al. 2011). A combination of solution and solid-phase chemistry procedures was used to synthesize Fm-GABA ← mGly-Ala-Ser-Chg (3). Briefly, Fm-GABA ← mGly-OSu was prepared and coupled to H-Ala-Ser-Chg in solution or by coupling the Fm-GABA ← mGly-OSu ester to H-Ala-Ser-Chg-O-resin at room temperature in 48 h. Standard solid phase procedures were used to prepare Fm-glutaryl-Hyp-Ala-Ser-Chg (1) and Fm-glutaryl-Ser-Ala-Ser-Chg (2) which, along with 3, were then coupled to the α-amino group of Gln-Linker-phosphoramide mustard 4 to synthesize prodrugs I–III, after Fm deprotection.
Evaluation of peptidamidoarylmethyl phosphoramide mustard prodrugs in PSA- and non-PSA-producing cells
The stability of peptide-linked phosphoramide mustard peptide conjugates in 50 mM Tris buffer, pH 8 was first evaluated over a period of 48 h (Fig. 1). Prodrug disappearance was monitored by LC-MS/MS analysis using multiple reaction monitoring (MRM) as shown in Table 2. There was roughly 80, 70 and 60% of prodrugs I, II, and III remaining over a period of 24 h, respectively (data not shown). All prodrugs slowly converted to their corresponding 4-peptidamido-2-fluorobenzyl alcohol conjugate through chemical hydrolysis (see “Non-PSA-Mediated Metabolism”). Overall, peptide conjugate degradation was insignificant over 8 h and did not affect our enzyme assay result interpretation.

Stability of phosphoramide mustard prodrugs in tris buffer. Each prodrug (100 μM) was incubated in 50 mM Tris/HCl buffer, pH 8.0 containing 10 mM CaCl2 and 0.1% TWEEN-20 at 37 °C
PSA cleavage rates were determined by incubating peptide-linked phosphoramide mustard prodrugs with human PSA over a period of 3 h. Substrate disappearance was monitored by LC-MS/MS analysis using MRM, and the fraction of prodrug remaining at any given time point computed by taking the ratio of its peak area response and that of the 0 h time point. As shown in Table 3 and Fig. 2, prodrug I exhibited the fastest cleavage rate with a half-life (t1/2) of 35 min. Compared with prodrug I, the cleavage rates for prodrugs II and III were slower with t1/2 values of 80 and 107 min, respectively. The rank order of prodrug cleavage rates appears to be reversed when compared with our initial substrate optimization studies using 7‐amino‐4‐methylcoumarin (AMC), which suggested that glutaryl-Ser-Ala-Ser-Chg-Gln-AMC and GABA ← mGly-Ala-Ser-Chg-Gln-AMC were cleaved by PSA 1.3- and twofold faster than glutaryl-Hyp-Ala-Ser-Chg-Gln-AMC, respectively (Aloysius and Hu 2015a). Thus, prodrugs II and III were unexpectedly no better than prodrug I as suggested by their longer half-lives. These results were surprising since we anticipated no effects of peptide conjugation with phosphoramide mustard through the 4-amino-2-fluorobenzyl linker on PSA cleavage due to the small size of 4-amino-2-fluorobenzyl phosphoramide mustard. A possible explanation for these results is that conjugation of glutaryl-Ser-Ala-Ser-Chg-Gln and GABA ← mGly-Ala-Ser-Chg-Gln with 4-amino-2-fluorobenzyl phosphoramide mustard may have afforded poorer substrates for PSA. However, because the half-life appeared to increase for our improved peptide substrate glutaryl-Ser-Ala-Ser-Chg-Gln and GABA ← mGly-Ala-Ser-Chg-Gln, a more plausible explanation is that PSA was irreversibly inhibited by the activated phosphoramide mustard or electrophilic quinonimine methide released upon proteolysis, via mechanism-based inhibition. Thus, the faster the release of the aforementioned PSA inactivators, the slower the PSA cleavage rate due to mechanism-based inhibition of the enzyme. It may, thus, be beneficial to explore combinations of our improved PSA peptide substrates with previously synthesized 4-aminoarylmethyl alcohol linkers (Hu and Wu 2007; Hu et al. 2011) to reach the optimal balance between PSA cleavage rate and its mechanism-based inhibition.

Disappearance of phosphoramide mustard prodrugs during PSA hydrolysis. Each prodrug (1 μM) was incubated with PSA (1 μM) in 50 mM Tris/HCl buffer, pH 8.0 containing 10 mM CaCl2 and 0.1% TWEEN-20 at 37 °C
In order to assess cytotoxicity and tumor selectivity, prodrugs I–III were incubated in cultured PSA-producing (LNCaP) and non-PSA-producing (DU145) cell lines for a period of 72 h. Peptide conjugates were designed to be activated extracellularly by PSA secreted from the LNCaP cells, but not in DU145 cells. 4-Amino-2-fluorobenzyl phosphoramide mustard conjugate or phosphoramide mustard itself is expected to be released into the extracellular space around tumor cells upon PSA activation of prodrugs I–III, producing selective cytotoxicity. Thus, increased hydrolysis of intracellular phosphoramide mustard by phosphoramidases, which are typically induced in tumors, should increase prodrug effectiveness. Consistent with our results in Fig. 2, prodrug I showed cytotoxic response in LNCaP with an IC50 of 7.3 μM and a 14-fold selectivity over DU145 (Table 4, Fig. 3). Prodrug III was equally cytotoxic in both cell lines (DU145 data not shown) whereas prodrug II was inactive. These results can be partially explained by the lack of robust PSA activation of prodrugs II and III (Table 3, Fig. 2). The cytotoxicity observed with prodrug III in both LNCaP and DU145 cell lines is unlikely mediated by PSA and may be due to slow prodrug hydrolysis and concomitant premature phosphoramide mustard release as suggested by its lower stability in buffer as compared with prodrugs I (Fig. 1).
Non-PSA-mediated metabolism
To evaluate non-PSA-mediated metabolism, prodrugs I–III were incubated in blood and cultured hepatocytes, biological matrices lacking active PSA. Prodrugs were incubated in heparinized human or mouse blood at 100 μM over a period of 24 h and the fraction of prodrug remaining at various time points determined by LC-MS/MS using MRM. Experiments in human blood indicated that prodrugs I and III were the most stable in human blood with t1/2 values of ~1 h (Table 5 and Fig. 4). In heparinized mouse blood, prodrugs I and II were the most stable in mouse blood overall, with t1/2 values of 9.4 and 12 min, respectively (Table 5 and Fig. 5). As suggested by our metabolic profiling studies, these results can be explained by chemical instability and/or the involvement of different hydrolases in human and mouse blood. In previous substrate optimization studies, the sequence GABA ← mGly-Ala-Ser-Chg-Gln coupled to 7-AMC was the most stable to non-PSA-mediated metabolism in mouse and human plasma (Aloysius and Hu 2015a).

Cytotoxicity of phosphoramide mustard prodrugs in LNCaP cells

Stability of phosphoramide mustard prodrugs in human blood. Each prodrug (100 μM) was incubated in human blood at 37 °C over a period of 24 h

Stability of phosphoramide mustard prodrugs in mouse blood. Each prodrug (100 μM) was incubated in mouse blood at 37 °C over a period of 24 h
To assess the relative propensity of prodrugs I–III to be cleared by the liver, their metabolic stability in cultured hepatocytes was evaluated. In contrast to blood stability results, prodrugs II and III showed marked improvements in stability in cultured human hepatocytes (Table 6, Fig. 6). As expected from substrate optimization studies (Aloysius and Hu 2015a), prodrug III elicited the highest resistance to non-PSA-mediated metabolism with a t1/2 value >48 h in mouse and human hepatocytes (Table 6, Figs 6 and 7). While the metabolic rates of prodrugs I–III in cultured mouse hepatocytes (Fig. 7) were unexpectedly lower compared with humans (Fig. 6) for unknown reasons, relative comparison of prodrug stability was still possible within each in vitro system. Soft spot metabolite identification conducted in hepatocyte incubations enabled the characterization of early steps in non-PSA-mediated metabolism. Prodrugs were incubated in blood and cultured hepatocytes, and prodrug-related fragments were identified and characterized by LC-MS with accurate mass measurements. In human blood and hepatocyte cultures, the earliest degradants were M1a-c, also observed in Tris buffer incubations (~30% formed, Scheme 3) after 48 h (data not shown). Consequently, M1a-c likely resulted from chemical degradation of phosphoramide mustard prodrugs I–III, respectively. It is possible that the chemical degradation observed in Tris buffer was accelerated in cultured human and mouse hepatocytes thereby generating M1a-c, which could be further converted to metabolites M2a-c (Scheme 3) by enzymatic intervention as indicated by the results in Table 7, Figs 8 and 9. Metabolic profiling of blood samples from initial mouse PK studies with prodrug I also indicated that M1a and M2a were major circulating metabolites in vivo (data not shown). Thus, the instability of phosphoramide mustard prodrugs appeared to result from chemical degradation and/or phosphoramidase-mediated hydrolysis to the corresponding 2-fluoro-4-peptidylaminobenzyl alcohol conjugates which were subsequently hydrolyzed by amidases. Despite the anticipated resistance of their aminobenzyl alcohol linker to endopeptidase action, peptide-linked phosphoramide mustard prodrugs I–III may be susceptible to hydrolysis by phosphoramidases which are typically elevated in tumors. Prodrugs become vulnerable to degradation by aminopeptidases and carboxypeptidases, once hydrolyzed by endopeptidases.

Stability of phosphoramide mustard prodrugs in cultured human hepatocytes. Each prodrug (100 μM) was incubated in plated human hepatocytes at 37 °C over a period of 48 h

Stability of phosphoramide mustard prodrugs in cultured mouse hepatocytes. Each prodrug (100 μM) was incubated in plated mouse hepatocytes at 37 °C over a period of 48 h

Metabolism of prodrugs I–III and the metabolites M1a-c and M2a-c identified

Metabolic profiles of prodrugs I–III in cultured human hepatocytes at 1 h

Metabolic profiles of prodrugs I–III in cultured mouse hepatocytes
Overall, in human cell cultures, phosphoramide mustard prodrugs II and III demonstrated improved stability compared with prodrug I, suggesting that non-PSA-mediated metabolism was mitigated. Further improvement of chemical stability and resistance to phosphoramidases can be achieved by exploring additional linkers reported in structure-activity relationship studies (Hu et al. 2011) and/or coupling the peptide sequences to cytotoxic agents other than phosphoramide mustard (Aloysius and Hu 2020). Results from exploratory PK studies with prodrug I in Nu/Nu mice suggest that additional improvements in chemical stability can be beneficial towards reducing in vivo clearance given that significant resistance to non-PSA-mediated metabolism in human hepatocytes was achieved with prodrugs II and III. Noteworthy, the in vitro blood t1/2 value of prodrug I generated in fresh heparinized blood was 9.4 h (Table 5) which is roughly 40-fold longer than its in vivo t1/2 value of 0.23 h (Table 1). This marked difference in t1/2 values points to a disconnect between in vitro and in vivo elimination rates if hydrolysis in blood were a major clearance route for prodrug I in vivo. Thus, it is possible that the in vivo clearance of prodrug I is driven by metabolism by organs other than the liver (e.g., kidney). Although all prodrugs were slowly metabolized in cultured mouse hepatocytes, enhanced resistance to non-PSA-mediated cleavage was clearly observed with the new prodrugs in human cultures.
In summary, we evaluated novel peptide-linked phosphoramide mustard conjugates designed as PSA-targeted prodrugs for their PSA-mediated tumor killing activity and propensity to undergo non-PSA-mediated metabolism. Nu/Nu mouse PK studies indicated that prodrug I (glutaryl-Hyp-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard), despite its excellent in vitro antiproliferative activity in PSA-producing LNCaP cells (IC50 of 7.3 μM) and adequate selectivity over non-PSA-producing DU145, exhibited high clearance with significant extrahepatic metabolism in vivo. In our continued efforts to design prodrugs with reduced non-PSA-mediated metabolism, improved tumor selectivity and toxicity profiles, the newly optimized PSA substrates GABA ← mGly-Ala-Ser-Chg-Gln and glutaryl-Ser-Ala-Ser-Chg-Gln (Aloysius and Hu 2015a) were coupled to phosphoramide mustard and evaluated. In human PSA experiments, prodrug I was cleaved by PSA with a t1/2 value of 35 min, followed by glutaryl-Ser-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard (II, 80 min) and GABA ← mGly-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard (III, 107 min). Time-dependent and mechanism-based inactivation of PSA by the released phosphoramide mustard and/or electrophilic quinonimine methide may be responsible for the unexpectedly slower cleavage rates of prodrugs II and III. Consistent with their slower cleavage rates compared with prodrug I, prodrugs II and III showed insignificant tumor killing activity. To assess non-PSA-mediated hydrolysis of prodrugs I–III, we conducted stability and metabolism studies in blood and cultured hepatocytes, which contain no active PSA. Hepatocyte stability and metabolism results confirmed that P5 substitutions within the peptide promoiety of prodrug I improved prodrug resistance to non-PSA-mediated metabolism. The improvements were more apparent in human hepatocytes as indicated by significant differences in t1/2 values between prodrug I and the newly synthesized prodrug III. Future studies will explore the profiles of prodrugs derived from the combination of the sequence GABA ← mGly-Ala-Ser-Chg-Gln with previously reported self-immolative linkers (Hu et al. 2011) to achieve an adequate balance between PSA cleavage rate and resistance to non-PSA-mediated hydrolysis.
Experimental section
Materials and methods
Male Nu/Nu mice were ordered from Charles River Laboratories (Wilmington, MA). Prodrug I was previously synthesized in our laboratory (Hu and Wu 2007). Purified PSA (99%, 1.37 mg protein/ml) was purchased from Fitzgerald (North Acton, MA). Cell growth medium was prepared by adding L-glutamine (2 mM), fetal bovine serum (10%), penicillin G (100 units/mL), and streptomycin sulfate (100 units/mL) to phenol red-containing RPMI 1640. Cryopreserved human (lot EBP) and mouse (lot Mc559) hepatocytes were supplied by Celsis (Chicago, IL). HEPES buffer, Hank’s Balanced Salt Solution (HBSS), Cryopreserved Hepatocyte Recovery Media (CHRM), Cryopreserved Hepatocyte Plating Media (CHPM), and all cell medium additives were purchased from Invitrogen (Life Technologies, Grand Island, NY). Cell maintenance buffer was prepared by supplementing 500 mL of William’s E medium with 5 mL Penn-Strep Glutamine (100×), 5 μL Dexamethasone (10 mM in DMSO), 5 mL Insulin-Transferrin-Selenium and 7.5 mL 1 M HEPES pH 7.4. Reactions were monitored by TLC and/or LC-MS. Flash column chromatography (FCC) was conducted on a Teledyne ISCO CombiFlash Companion Automated Flash Chromatographic System (Teledyne Technologies, Thousand Oaks, CA) using pre-packed silica gel columns. All 1H and 13C NMR spectra were recorded on a 400 MHz Bruker spectrometer at ambient temperature and calibrated using residual un-deuterated solvents as the internal reference. Accurate mass measurements and metabolite identification were conducted using an LTQ-Orbitrap mass spectrometer (Thermo Fisher, Waltham, MA) equipped with an electrospray ionization source operated in positive ionization mode interfaced with Shimadzu LC-20ADXR pumps, a SIL-20ACXR autosampler, and SPD-M20A diode array detector (Shimadzu, Columbia, MD). Metabolite separation was achieved with a 2.1 × 100 mm, 1.7 µm BEH C18 column (Waters Corporation, Huntingdon Valley, PA) using a water (A)/acetonitrile (B) mobile phase system containing 0.1% formic acid (v/v) and UV detection at 484 nm. Prodrug quantification was performed on a Transcend LX2 system (Thermo Fisher, Waltham, MA) coupled to an API 4000 (AB Sciex, Framingham, MA) using MRM transitions for both sodium adduct and base peak from each prodrug (Table 2). While results were consistent with either transition, only data generated with the sodium adduct MRM transitions are shown.
Synthesis of Fm-GABA←mGly-Ala-Ser-Chg-OH (3)
4-((tert-Butoxycarbonyl)amino)butanoic acid (Boc-GABA)
GABA (1.0 g, 9.8 mmol) was dissolved in 24 mL of 6% NaHCO3 solution at 0 °C. A pre-cooled solution of Boc-anhydride (15 mmol in 10 mL dioxane) was added slowly and the mixture stirred at 0 °C for 1 h, then overnight at room temperature. After removing dioxane under reduced pressure, the aqueous mixture was acidified to pH 1 with 1 N sodium bisulfate and extracted with 50 mL ethyl acetate three times. The combined organic layer was washed with 10 mL water, brine, and dried over sodium sulfate. Ethyl acetate was removed under reduced pressure to afford the intermediate Boc-GABA collected as a white solid in 84% yield (1.7 g).
4-((tert-Butoxycarbonyl)amino)butanoic acid (Boc-GABA)
1H NMR (CDCl3, 400 MHz): δ 3.21 (m, 2H), 2.42 (t, 2H, J = 7.2 Hz), 1.84 (q, 2H, J1 = 7.0 Hz, J2 = 6.4 Hz), 1.46 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 166.7, 141.3, 125.0, 120.1, 67.8, 46.6, 40.8, MS (ESI+): m/z (intensity), 203.7 ([M + H]+, 45%), 406.8 ([2M + H]+, 74%), 306.8 ([2M + H-Boc]+, 100%).
(9H-Fluoren-9-yl)methyl 4-((tert-butoxycarbonyl)amino)butanoate (Boc-GABA-Fm)
Boc-GABA (1.0 g, 9.8 mmol) was pre-activated with CDI (1.6 g, 9.8 mmol) in 2 mL of anhydrous DCM at room temperature under nitrogen atmosphere for 1 h. Fluorenylmethanol (1.3 g, 6.5 mmol) was added and the reaction carried out at room temperature under nitrogen atmosphere for 4 h. The mixture was diluted to 20 mL with DCM, washed sequentially with 20 mL 5% NaHCO3, brine and dried over sodium sulfate. After removing DCM under reduced pressure, the product was collected as an off-white solid in 95% yield (1.2 g). Removal of the Boc-protecting group was carried out with 50% TFA/DCM at room temperature in 30 min. All solvents were removed under a gentle nitrogen stream to afford the intermediate Boc-GABA-Fm, which was washed repeatedly with hexanes (5 × 20 mL) and used for the next step.
(9H-Fluoren-9-yl)methyl 4-((tert-butoxycarbonyl)amino)butanoate (Boc-GABA-Fm)
1H NMR (CDCl3, 400 MHz): δ 7.80 (d, 2H, J = 8.0 Hz), 7.61 (d, 2H, J = 7.4 Hz), 7.44 (t, 2H, J = 7.3 Hz), 7.35 (t, 2H, J = 7.9 Hz), 4.56 (s, 1H), 4.45 (d, 2H, J = 6.7 Hz), 4.24 (t, 1H, J = 7.3 Hz), 3.13 (m, 2H), 2.43 (t, 2H, J = 7.3 Hz), 1.81 (q, 2H, J = 6.8 Hz), 1.46 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 143.8, 141.3, 127.8, 127.1, 125.0, 120.0, 66.2, 46.9, 38.4, 31.5, 28.4, 25.3, MS (ESI+): m/z (intensity), 381.7 ([M + H]+, 10%), 282.0 ([M + H-Boc], 100%).
t-BuO-mGly
Malonic acid (5.0 g, 48 mmol) and t-BuOH (1.8 mL, 19 mmol) were dissolved in 150 mL of ACN at room temperature under nitrogen. EDC (9.2 g, 48 mmol) was added and the reaction conducted at room temperature under nitrogen for 30 min. ACN was removed under reduced pressure and the residue dissolved in 200 mL of ether. The product was back-extracted with two 50 mL portions of saturated NaHCO3, and the combined aqueous layer acidified to pH 2 with 1 N sodium bisulfate. Finally, the product was extracted with three 200 mL portions of DCM which were combined and washed with water, brine, and dried over sodium sulfate. DCM was removed under reduced pressure to afford the intermediate t-BuO-mGly as a white solid in 76% yield (2.3 g).
t-BuO-mGly
1H NMR (CDCl3, 400 MHz): δ 3.28 (s, 2H), 1.42 (s, 9H); 13C NMR (CDCl3, 100 MHz): δ 168.5, 162.1, 83.5, 39.7, 27.9, 27.8, 25.6.
t-BuO-mGly-OSu
To a solution (7 mL, ACN) of t-BuO-mGly (2.3 g, 15 mmol) and HOSu (1.7 g, 15 mmol) was added EDC (3.1 g, 16 mmol), and the mixture stirred at room temperature under nitrogen atmosphere for 1 h. ACN was removed under reduced pressure and the residue dissolved in 200 mL of DCM. The organic layer was washed with 50 mL water (2×), brine and dried over sodium sulfate. DCM was removed under reduced pressure to afford the desired intermediate t-BuO-mGly-OSu as a white solid in 84% yield (3.1 g).
Fm-GABA←mGly
A solution of t-BuO-mGly-OSu (1.8 g, 6.9 mmol) and GABA-Fm (2.0 g, 6.9 mmol) was prepared in 25 mL of anhydrous DCM in the presence of DIEA (1.2 mL, 6.9 mmol) and stirred at room temperature under nitrogen atmosphere for 1 h. The reaction mixture was diluted to 40 mL with DCM, washed with 10 mL water (2×), brine and dried over sodium sulfate. DCM was removed under reduced pressure and the crude mixture purified by FCC to afford the final product in 64% yield (1.9 g). Removal of the t-butyl-protecting group was achieved with 50%TFA/DCM to afford 1.7 g of the free acid Fm-GABA ← mGly, which was carried forward to the next step without further purification.
Fm-GABA ← mGly-Ala-Ser-Chg-OH (3)
The Fm-GABA ← mGly-OSu ester freshly prepared above was coupled to the amino end of H-Ala-Ser-Chg-OH. The tripeptide was generated using standard automated peptide synthesis procedures. The OSu ester (410 mg, 0.87 mmol) was directly coupled (within 1 min) to H-Ala-Ser-Chg-OH (230 mg, 0.73 mmol) in 1 mL ACN/NMP (1:2) in the presence of 1 eq. DIEA. After FCC purification, the final peptide was obtained in 56% yield (270 mg).
Synthesis of 2-fluoro-4-glutaminylaminobenzyl phosphoramide mustard (4)
All intermediates leading to compound 4 were synthesized as previously described (Wu et al. 2011). TFA-Gln was coupled to 4-azido-2-fluorobenzyl phosphoramide mustard linker using a selenocarboxylate/azide amidation strategy to afford 58% yield (462 mg) of crude 2-fluoro-4-glutaminylaminobenzyl phosphoramide mustard.
Synthesis of prodrug II (Glutaryl-Ser-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard)
Fm-glutaryl-Ser-Ala-Ser-Chg-OH was prepared using standard automated peptide synthesis procedures. Fm-glutaryl-Ser-Ala-Ser-Chg-OH (40 mg, 0.058 mmol) and HBTU (22 mg, 0.058 mmol) were dissolved in the presence of 2.6 eq. DIEA in 250 μL of anhydrous DMF at 0 °C. 2-Fluoro-4-glutaminylaminobenzyl phosphoramide mustard (21 mg, 0.044 mmol) was added and the mixture stirred at 0 °C for 30 min, and allowed to warm-up to room temperature in 30 min. The mixture was cooled to 0 °C and 4 mL of ice-cold water added dropwise. After stirring the resulting suspension at 0 °C for an additional 30 min, the precipitate was collected by centrifugation, washed with 2 mL ice-cold water (2×) and dried under reduced pressure. The crude solid was re-dissolved in 250 μL of anhydrous DMF and de-protected with 10% DEA. The mixture was triturated with ice-cold ether and the final peptide prodrug obtained in 26% yield (11 mg) after HLPC purification. Glutaryl-Ser-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard; HRMS (ESI+): m/z calc’d for C38H60Cl2FN9O13P: [M + H]+ = 970.3409, found: 970.3417.
Synthesis of prodrug III (GABA←mGly-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard)
Fm-GABA ← mGly-Ala-Ser-Chg-OH (3, 40 mg, 0.060 mmol) and HBTU (23 mg, 0.060 mmol) were dissolved in the presence of 2.6 eq. DIEA in 250 μL of anhydrous DMF at 0 °C. 2-Fluoro-4-glutaminylaminobenzyl phosphoramide mustard (16 mg, 0.055 mmol) was added and the mixture stirred at 0 °C for 30 min to warm-up to room temperature in 30 min. The mixture was cooled to 0 °C and 4 mL of ice-cold water added dropwise. After stirring the resulting suspension at 0 °C for an additional 30 min, the precipitate was collected by centrifugation, washed with 2 mL of ice-cold water (2×) and dried under reduced pressure. The crude solid was re-dissolved in 250 μL of anhydrous DMF and de-protected with 10% DEA. The mixture was triturated with ice-cold ether and the final peptide prodrug obtained in 28% yield (10 mg) after HLPC purification. GABA ← mGly-Ala-Ser-Chg-Gln-NH-2-F-Bn-phosphoramide mustard; HRMS (ESI+): m/z calc’d for C37H58Cl2FN9O12P: [M + H]+ = 940.3304, found: 940.3311.
PSA assay
Phosphoramide mustard peptide solution concentrations were determined by measuring the UV absorbance at 254 nm using 50 μM solutions prepared (based on weight) in water from 10 mM DMSO stock solutions. Using the extinction coefficients of 2-fluoro-4-glutaminylaminobenzyl phosphoramide mustard at 254 nm (20,800 M−1 cm−1), peptide concentrations were calculated and adjusted accordingly to prepare 100 μM solutions in 50 mM Tris buffer, pH 8 containing 2 mM CaCl2 and 0.1% Tween 20. The stability of peptide prodrugs in 50 mM Tris buffer, pH 8 was determined from 48 h incubations in buffer (Fig. 1). Peptide solutions were prepared in Tris buffer pH 8 at a concentration of 1.11 μM from 1 mM DMSO stock solutions, and warmed up to 37 °C. Reactions were initiated at 37 °C with the addition of 5 μL aliquots of a 10 μM solution of human PSA to 45 μL aliquots of the 1.11 μM peptide solutions in Eppendorf vials (total volume 50 μL, final PSA concentration 1 μM), and time points recorded over a period of 3 h. Reactions were terminated at 0, 10, 20, 30, 60, 120, 180 and 360 min by quenching 2 μL aliquots of incubates with 20 μL of chilled 50% ACN containing 0.1 μM internal standard (IS) and 0.1% formic acid (FA) pre-added to Eppendorf vials. Samples were centrifuged at 3000 RPM for 15 min and the supernatants analyzed by LC-MS/MS in MRM mode as previously described. The fraction of prodrug remaining at each time point was calculated by dividing the prodrug/IS ratio by the 0–h prodrug/IS area ratio and half-life determined from prodrug disappearance rate.
Carcinoma cell culture
Monolayer cultures of PSA-secreting LNCaP and non-PSA-secreting DU145 human prostate carcinoma cells were conducted in growth medium in a CO2 incubator under a humidified atmosphere at 37 °C for 3 weeks prior to initiating cytotoxicity experiments. During cell growth, media were changed every 3 days and trypsinization carried out at 80% confluence with a 1:4 split for subsequent cultures. Cells were grown on 96-well plates for 48 h at initial concentrations of 5000 cells/well. At the end of the 48 h period, the growth medium was replaced by serum-free medium RPMI 1640 medium containing L-glutamine (2 mM), 2% TCM, penicillin G (100 units/mL) and streptomycin sulfate (100 units/mL), and prodrugs added to wells at concentrations ranging from 0.07 to 100 μM. The plates were incubated at 37 °C for 72 h under a humidified CO2 atmosphere, and cell viability determined using the MTT assay. Briefly, 10 μL aliquots of a 12 mM MTT solution were added to wells and the plates incubated in a CO2 incubator under a humidified atmosphere at 37 °C for 4 h. Well contents were then solubilized with 1% SDS (100 μL/well) for 12 h, and OD570 values measured on a Dynatech MR5000 plate reader. Cell viability was computed as a percentage of control growth, and IC50 values determined as the concentration at which cell growth is inhibited by 50%.
Nu/Nu mouse PK study
Three Nu/Nu male mice (weight 25–28 g) received prodrug I at 2 mg/kg by IV injection in a 0.4 mg/mL solution prepared in DMSO: HBSS (10:90). Blood samples (20 μL) were drawn at 5, 15, 30 min, 1, 2, 4, 6, 8 and 24 h and collected on a 96-well plate containing 60 μL of 0.5 M sodium citrate on ice. Prodrug quantification was carried out as described under “Prodrug Quantification by LC-MS/MS in Mouse and Human Blood”.
Blood sample collection for in vitro studies
Blood samples were collected in sodium-heparin tubes from 15 male adult C57BL/6 mice; the use of EDTA as anticoagulant was avoided due to its anticipated inhibitory effect on metalloproteases through ion metal chelation. Likewise, human blood was sampled from three adult male volunteers in heparinized collection tubes and stored on ice until use.
Hepatocyte cultures
All hepatocyte culture experiments were conducted under sterile conditions. Cryopreserved mouse or human hepatocytes (10–14 million cells) were thawed in 50 mL CHRM pre-warmed to 37 °C in a water bath. The cell suspensions were centrifuged at 800 RPM for 10 min and the pellet re-suspended in 5 mL CHRM by gentle mixing. Cell count and viability were determined using AO/PI (acridine orange/propidium iodide) staining on a Nexcelom Cellometer (Nexcelom Bioscience, Lawrence, MA). Cell suspensions with viability greater than 80% were diluted with CHRM to a concentration of 0.4 and 0.7 × 10 ^ 6 cells/mL, for mouse and human hepatocytes, respectively, and seeded at 2.5 mL per well on pre-coated collagen plates. Plates were incubated at 37 °C in a 95% humidified incubator at 5% CO2 for 2–4 h to allow adequate cell attachment before replacing CHRM with the maintenance media spiked with prodrug.
Metabolite profiling by accurate mass spectrometry
For each prodrug, 200 μL aliquots were drawn at 1, 2, 4, 8, and 24 h from blood and plated hepatocyte incubations, and quenched with 400 μL of chilled acetonitrile containing 0.1% FA. Samples were centrifuged at 3000 RPM for 15 min, the supernatant removed and dried under a gentle stream of nitrogen. Residues were reconstituted in 100 μL of acetonitrile:water (1:1) and analyzed by LC-MS system with accurate mass determination. Accurate mass measurements and metabolite identification were performed using an LTQ-Orbitrap mass spectrometer (Thermo Fisher, Waltham, MA) equipped with an electrospray source operated in positive ionization mode interfaced with Shimadzu LC-20ADXR pumps, a SIL-20ACXR autosampler, and SPD-M20A diode array (Shimadzu, Columbia, MD). Metabolite separation was achieved with a 2.1 × 100 mm, 1.7 µm BEH C18 column (Waters Corporation, Huntingdon Valley, PA) using a water (A)/acetonitrile (B) mobile phase system containing 0.1% FA (v/v) and UV detection at 254 nm. The gradient was performed at a total flow rate of 200 μL/min as follows, 2% B from 0 to 3 min, 2–95% B from 3 to 20 min, 95% B from 20 to 25 min, 95–2% B from 25 to 25.1 min, 2% B from 25.1 to 30 min.
Prodrug quantification by LC-MS/MS in mouse and human blood
Prodrug stability to non-PSA-mediated hydrolysis was evaluated in heparinized mouse or human blood by incubating each prodrug at a concentration of 100 μM for a period of 24 h at 37 °C. Reactions were initiated by adding 10 μL of a 10 mM solution of each prodrug to 1 mL of buffer, mouse or human blood and allowing the mixture to shake at 37 °C in a 95% humidified incubator at 5% CO2. Reactions were terminated at 0, 0.5, 1, 2, 4, 6 and 24 h by quenching 50 μL aliquots of incubates with 50 μL of chilled acetonitrile containing 0.2 μM IS and 0.1% FA pre-added to 96-well plates. Precipitated samples were centrifuged at 3000 RPM for 15 min and the supernatants analyzed by LC-MS/MS in MRM mode (Table 2). Prodrugs were quantified using LC-MS/MS on a Transcend LX2 system (Thermo Fisher, Waltham, MA) coupled to an API 4000 (AB Sciex, Framingham, MA). As shown in Table 2, both MRM transitions for sodium adduct and base peak from each prodrug were used for LC-MS/MS quantification. Results were consistent with either transition although only data generated with the sodium adduct MRM transitions are shown. The fraction of prodrug remaining at each time point was calculated by dividing its prodrug/IS area ratio by the prodrug/IS area ratio at 0 h.
Prodrug quantification by LC-MS/MS in mouse and human plated hepatocytes
As previously described for blood incubations, prodrug incubations in cultured hepatocytes were initiated at a concentration of 100 μM by diluting 25 μL of 10 mM DMSO solutions in 2.5 mL of maintenance media added to 1.75 million cells per well. Reactions were carried out at 37 °C in a 95% humidified incubator at 5% CO2. At 0, 0.5, 1, 2, 4, 8, 24 and 48 h, 50 μL aliquots of incubates were sampled and quenched with 50 μL of chilled acetonitrile containing 0.2 μM IS and 0.1% FA pre-added to 96-well plates. Precipitated samples were centrifuged at 3000 RPM for 15 min and the supernatants analyzed by LC-MS/MS in MRM mode. The fraction of prodrug remaining at each time point was calculated.
Abbreviations
- AMC:
-
7-amino-4-methylcoumarin
- ACN:
-
acetonitrile
- Boc:
-
tert-butyloxycarbonyl
- Bn:
-
benzyl
- CDI:
-
carbonyldiimidazole
- Chg:
-
L-cyclohexylglycine
- DCM:
-
dichloromethane
- DEA:
-
diethylamine
- DIEA:
-
N,N-diisopropylethylamine
- DMF:
-
dimethylformamide
- DMSO:
-
dimethyl sulfoxide
- EDC:
-
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FCC:
-
Flash column chromatography
- Fm:
-
fluorenylmethyl
- Fmoc:
-
fluorenylmethoxycarbonyl
- GABA:
-
γ-aminobutyric acid
- HBTU:
-
O-benzotriazole N,N,N’,N’-tetramethyluronium hexa-fluorophosphate
- HOBt:
-
N-hydroxybenzotriazole
- HOSu:
-
N-hydroxysuccinimide
- HPLC:
-
high performance liquid chromatography
- HRMS:
-
high-resolution mass spectrometry
- IPCF:
-
isopropyl chloroformate
- Hyp:
-
trans-4-hydroxy-L-proline
- LC-MS:
-
liquid chromatography-mass spectrometry
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- IS:
-
internal standard
- NMP:
-
N-methyl-2-pyrrolidone
- NMR:
-
nuclear magnetic resonance
- NTR:
-
nitroreductase
- PSA:
-
prostate-specific antigen
- RPMI:
-
Roswell Park Memorial Institute
- TCM:
-
tissue culture medium
- MRM:
-
multiple reaction monitoring
- THF:
-
tetrathydrofuran
- TFA:
-
trifluoroacetic acid
- TLC:
-
thin-layer chromatography.
References
Aloysius H, Hu L (2015a) Improving the specificity of the PSA substrate Glutaryl-Hyp-Ala-Ser-Chg-Gln as a promoiety. Chem Biol Drug Des 86:837–848
Aloysius H, Hu L (2015b) Targeted prodrug approaches for hormone refractory prostate cancer. Med Res Rev 35:554–585
Aloysius H, Hu L (2020) Synthesis and evaluation of new peptide-linked doxorubicin conjugates as prodrugs activated by prostate specific antigen. Med Chem Res 29, https://doi.org/10.1007/s00044-020-02573-w
Ayyıldız SN, Ayyıldız A (2014) PSA, PSA derivatives, proPSA and prostate health index in the diagnosis of prostate cancer. Turk J Urol 40:82–88
Balk SP, Ko Y-J, Bubley GJ (2003) Biology of prostate-specific antigen. J Clin Oncol 21:383–391
Brock N (1996) The history of the oxazaphosphorine cytostatics. Cancer J 78:542–547
Catalona WJ, Smith DS, Ratliff TL, Dodds KM, Coplen DE, Yuan JJ, Petros JA, Andriole GL (1991) Measurement of prostrate-specific antigen in serum as a screening test for prostrate cancer. N. Engl J Med 324:1156–1161
Choi KY, Swierczewska M, Lee S, Chen X (2012) Protease-Activated Drug Development. Theranostics 2:156–178
De Angelis G, Rittenhouse HG, Mikolajczyk SD, Blair Shamel L, Semjonow A (2007) Twenty Years of PSA: From prostate antigen to tumor marker. Rev Urol 9:113–123
DeFeo-Jones D, Garsky VM, Wong BK, Feng DM, Bolyar T, Haskell K, Kiefer DM, Leander K, McAvoy E, Lumma P, Wai J, Senderak ET, Motzel SL, Keenan K, Van Zwieten M, Lin JH, Freidinger R, Huff J, Oliff A, Jones RE (2000) A peptide-doxorubicin prodrug activated by prostate-specific antigen selectively kills prostate tumor cells positive for prostate-specific antigen in vivo. Nat Med 6:1248–1252
Denmeade SR, Lou W, Lövgren J, Malm J, Lilja H, Isaacs JT (1997) Specific and efficient peptide substrates for assaying the proteolytic activity of prostate-specific antigen. Cancer Res 57:4924–4930
Diamandis E (1995) New diagnostic applications and physiological functions of prostate specific antigen. Scand J Clin Lab Investig 55:105–112
DiPaola RS, Rinehart J, Nemunaitis J, Ebbinghaus S, Rubin E, Capanna T, Ciardella M, Doyle-Lindrud S, Goodwin S, Fontaine M, Adams N, Williams A, Schwartz M, Winchell G, Wickersham K, Deutsch P, Yao SL (2002) Characterization of a novel prostate-specific antigen-activated peptide-doxorubicin conjugate in patients with prostate cancer. J Clin Oncol 20:1874–1879
Doan NTQ, Paulsen ES, Sehgal P, Møller JV, Nissen P, Denmeade SR, Isaacs JT, Dionne CA, Christensen SB (2015) Targeting thapsigargin towards tumors. Steroids 97:2–7
Emadi A, Jones RJ, Brodsky RA (2009) Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol 6:638–647
Ganesan S, Keating AF (2015) Phosphoramide mustard exposure induces DNA adduct formation and the DNA damage repair response in rat ovarian granulosa cells. Toxicol Appl Pharm 282:252–258
Garsky VM, Lumma PK, Feng DM, Wai J, Ramjit HG, Sardana MK, Oliff A, Jones RE, DeFeo-Jones D, Freidinger RM (2001) The synthesis of a prodrug of doxorubicin designed to provide reduced systemic toxicity and greater target efficacy. J Med Chem 44:4216–4224
Giang I, Boland EL, Poon GMK (2014) Prodrug applications for targeted cancer therapy. AAPS J 16:899–913
Herschman JD, Smith DS, Catalona WJ (1997) Effect of ejaculation on serum total and free prostate-specific antigen concentrations. Urology 50:239–243
Hu L, Wu X (2007) Chemotherapeutic conjugates and methods of use. US Patent US20100075927 A1
Hu L, Wu X, Han J, Chen L, Vass SO, Browne P, Hall BS, Bot C, Gobalakrishnapillai V, Searle PF, Knox RJ, Wilkinson SR (2011) Synthesis and structure-activity relationships of nitrobenzyl phosphoramide mustards as nitroreductase-activated prodrugs. Bioorg Med Chem Lett 21:3986–3991
Jiang Y, DiPaola RS, Hu L (2009) Synthesis and stereochemical preference of peptide 4-aminocyclophosphamide conjugates as potential prodrugs of phosphoramide mustard for activation by prostate-specific antigen (PSA). Bioorg Med Chem Lett 19:2587–2590
Jiang Y, Han J, Yu C, Vass SO, Searle PF, Browne P, Knox RJ, Hu L (2006) Design, Synthesis, and Biological Evaluation of Cyclic and Acyclic Nitrobenzylphosphoramide Mustards for E. coli Nitroreductase Activation. J Med Chem 49:4333–4343
Jiang Y, Hu L (2013) Peptide conjugates of 4-aminocyclophosphamide as prodrugs of phosphoramide mustard for selective activation by prostate-specific antigen (PSA). Bioorg Med Chem 21:7507–7514
Jiang Y, Hu L (2007) Phenylalanyl-aminocyclophosphamides as model prodrugs for proteolytic activation: synthesis, stability, and stereochemical requirements for enzymatic cleavage. Bioorg Med Chem Lett 17:517–521
Korkmaz A, Topal T, Oter S (2007) Pathophysiological aspects of cyclophosphamide and ifosfamide induced hemorrhagic cystitis; implication of reactive oxygen and nitrogen species as well as PARP activation. Cell Biol Toxicol 23:303–312
Kres W (1995) Current chemotherapy and future directions in research for the treatment of advanced hormone-refractory prostate cancer. Cancer Investig 13:296–312
Kwiatkowska A, Couture F, Ait-Mohand S, Desjardins R, Dory YL, Guérin B, Day R (2019) Enhanced anti-tumor activity of the Multi-Leu peptide PACE4 inhibitor transformed into an albumin-bound tumor-targeting prodrug. Sci Rep 9:2118
Lee J, Huang W, Broering JM, Barron AE, Seo J (2015) Prostate tumor specific peptide–peptoid hybrid prodrugs. Bioorg Med Chem Lett 25:2849–2852
Leinonen J, Zhang. W, Stenman U (1996) Complex formation between PSA isoenzymesand protease inhibitors. J Urol 155:1099–1103
Levesque M, Yu H, D’Costa M, Diamandis E (1995) Prostate specific antigen expression by various tumors. J Clin Lab Anal 9:123–128
McDonald AC, Vira MA, Vidal AC, Gan W, Freedland SJ, Taioli E (2014) Association between systemic inflammatory markers and serum prostate-specific antigen in men without prostatic disease - the 2001-2008 National Health and Nutrition Examination Survey. Prostate 74:561–567
Ménez R, Michel S, Muller BH, Bossus M, Ducancel F, Jolivet-Reynaud C, Stura EA (2008) Crystal structure of a ternary complex between human prostate-specific antigen, its substrate acyl intermediate and an activating antibody. J Mol Biol 376:1021–1033
Nadler RB, Humphrey PA, Smith DS, Catalona WJ, Ratliff TL (1995) Effect of inflammation and benign prostatic hyperplasia on elevated serum prostate specific antigen levels. J Urol 154:407–413
O’Malley KJ, Eisermann K, Pascal LE, Parwani AV, Majima T, Graham L, Hrebinko K, Acquafondata M, Stewart NA, Nelson JB, Yoshimura N, Wang Z (2014) Proteomic analysis of patient tissue reveals PSA protein in the stroma of benign prostatic hyperplasia. Prostate 74:892–900
Otto A, Bar J, Birkenmeier G (1998) Prostate specific antigen forms complexes with human α2-macroglobulin and binds to the α2-macroglobulin receptor/ldl receptor-related protein. J Urol 159:297–303
Poreba M (2020) Protease-activated prodrugs: strategies, challenges, and future directions. FEBS J 287:1936–1969
Shanafelt TD, Lin T, Geyer SM, Zent CS, Leung N, Kabat B, Bowen D, Grever MR, Byrd JC, Kay NE (2007) Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer J 109:2291–2298
Sohlenius-Sternbeck A-K (2006) Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol Vitr 20:1582–1586
Tabchi S, Nair R, Kunacheewa C, Patel KK, Lee HC, Thomas SK, Amini B, Ahmed S, Mehta RS, Bashir Q, Qazilbash MH, Weber DM, Orlowski RZ, Alexanian R, Feng L, Manasanch EE (2019) Retrospective Review of the Use of High-Dose Cyclophosphamide, Bortezomib, Doxorubicin, and Dexamethasone for the Treatment of Multiple Myeloma and Plasma Cell Leukemia. Clin Lymphoma Myeloma Leuk 19:560–569
Tarvainen I, Zimmermann T, Heinonen P, Jäntti MH, Yli-Kauhaluoma J, Talman V, Franzyk H, Tuominen RK, Christensen SB (2020) Missing Selectivity of Targeted 4β-Phorbol Prodrugs Expected to be Potential Chemotherapeutics. ACS Med Chem Lett 11:671–677
Wong BK, DeFeo-Jones D, Jones RE, Garsky VM, Feng DM, Oliff A, Chiba M, Ellis JD, Lin JH (2001) PSA-specific and non-PSA-specific conversion of a PSA-targeted peptide conjugate of doxorubicin to its active metabolites. Drug Metab Dispos 29:313–318
Wu X, Chen Y, Aloysius H, Hu L (2011) A novel high-yield synthesis of aminoacyl p-nitroanilines and aminoacyl 7-amino-4-methylcoumarins: important synthons for the synthesis of chromogenic/fluorogenic protease substrates. Beilstein J Org Chem 7:1030–1035
Wu X, Hu L (2016) Design and synthesis of peptide conjugates of phosphoramide mustard as prodrugs activated by prostate-specific antigen. Bioorg Med Chem 24:2697–2706
Yang Y, Aloysius H, Inoyama D, Chen Y, Hu L (2011) Enzyme-mediated hydrolytic activation of prodrugs. Acta Pharm Sin B 1:143–159
Acknowledgements
We gratefully acknowledge the financial support of grant SNJ‐CCR 700–009 from the State of New Jersey Commission on Cancer Research, a pilot grant from the Gallo Prostate Cancer Center of the Cancer Institute of New Jersey, and grant RSG‐03–004–01‐CDD from the American Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have filed patents on some of the compounds discussed in this paper.
Additional information
Dedicated to Professor Robert P. Hanzlik on the occasion of his retirement after 49 years of dedicated service on the faculty of the Department of Medicinal Chemistry, University of Kansas School of Pharmacy.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aloysius, H., Hu, L. Synthesis and evaluation of new 4-peptidamido-2-fluorobenzyl phosphoramide mustard conjugates as prodrugs activated by prostate-specific antigen. Med Chem Res 29, 1264–1279 (2020). https://doi.org/10.1007/s00044-020-02572-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02572-x