
Abstract
In a pursuit of combating acquired immunodeficiency syndrome, the targets human immunodeficiency virus (HIV) protease, reverse transcriptase and HIV specific proteins were exhaustively explored. Resistance through mutations in these targets has impeded the further developments in HIV research. HIV-1 integrase (HIV-1 IN) has emerged as a newer target and 3′-processing, and strand transfer inhibition has proved potential intervention in HIV replication. In present study, in line with our interests in coumarin derivatives as potential bioactive molecules, 4D-quantitative structure–activity relationship (4D-QSAR) is proposed. Fifty-seven structurally diverse coumarin derivatives were subjected to 4D-QSAR studies. Quantum mechanics-based geometry optimization and molecular dynamics simulation were carried out on individual compound. The conformational ensemble generated for each compound was aligned with most active compound, and Coulombic and Lennard-Jones interaction energy descriptors were computed. After selecting the best variables in MATLAB, partial least square regression (PLS) analysis was carried out on 44 training set and 13 test set compounds. The model with ten latent variables was found best with R 2 calculated = 0.903015, R 2 cross-validated = 0.599553, R 2 predicted = 0.688525, root-mean-square error (RMSE) calculated = 0.21276, RMSE predicted = 0.371579 and prediction bias = −0.15362. Docking studies were carried out on AutoDock Vina, which were in good agreement with the PLS model, suggesting the importance of few descriptors of Coulombic interaction energy and VWD interactions with VAL79, VAL77, ARG199 and GLU157. These results may be useful in designing more potent HIV-1 IN inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acquired immunodeficiency syndrome (AIDS) is a disease caused by human immunodeficiency virus (HIV). Since its first report in 1981, it has caused more than 30 million deaths worldwide (Curran, 1983). Most of the current approved drugs are developed targeting the two essential enzymes, viral reverse transcriptase and protease (Johnson et al., 2010). HIV-1 integrase (HIV-1 IN), the enzyme essential in viral replication, has emerged as most promising target as it does not have human homologue, and former two targets are eloped with emergence of highly resistant viral strains (Jaskolski et al., 2011). HIV-1 IN comprises of single polypeptide chain with 288 amino acid residues and three domains, N-terminal domain (residues 1–54), catalytic domain (residues 55–209) and C-terminal domain (residues 210–288). HIV-1 IN catalyzes the insertion of viral DNA into the host genome in two biochemical steps (a) 3′-processing which removes two nucleotides from each 3′-ends of the viral cDNA to produce reactive 3′-hydroxyl ends and (b) strand transfer which joins 3′-viral DNA ends into the host DNA through a nucleophilic transesterification reaction (Schroder et al., 2002). Several HIV-1 IN inhibitors have been reported, but only two compounds, raltegravir and elvitegravir, strand transfer inhibitors, are approved for clinical use (US Food and Drug Administration, 2014). Major impediments in the development of HIV-1 IN strand transfer inhibitors are cross-resistance due to the similar mechanism of action of developed agent, HIV-1 IN polymorphism which is less studied and the fact that the agent has to act on HIV-1 IN—viral DNA complex. The inhibitors of 3′-processing step are thought to suppress both 3′-processing and strand transfer steps (Korolev et al., 2011). Zhao’s (1997) report on coumarin derivatives opened the avenue for coumarin-based HIV-1 IN inhibitors. Many coumarin derivatives were reported subsequently as HIV-1 IN inhibitors as 3′-processing and strand transfer inhibitors (Mao et al., 2002; Chiang et al., 2007; Tewtrakul et al., 2007; Liu et al., 2009; Kostova et al., 2006; Al-Mawsawi et al., 2006; Bailly et al., 2005; Olmedo et al., 2012; Mahajan et al., 2009; Spino et al., 1998). Hansch (Hansch and Fujita, 1964) and Wilson (Free and Wilson, 1964) established QSAR, a tool which establishes the quantitative relationship between structural, physicochemical and conformational properties with biological activity. Since its inception, QSAR approaches have evolved from 2D-QSAR to more complex 4D quantitative structure–activity relationship (4D-QSAR). QSAR approaches are further classified as receptor-dependent and receptor-independent QSAR methods based on the descriptors calculated with or without receptor. Comparative molecular field analysis (CoMFA; Cramer et al., 1988) and comparative molecular similarity indices analysis (CoMSIA; Klebe et al., 1994) are most acclaimed 3D-QSAR approaches exploited in successful design of many therapeutic agents. Hopfinger et al. (1997) proposed a 4D-QSAR where conformational flexibility of ligand and freedom of alignment in three dimensional spaces as required in 3D-QSAR is utilized. In this approach, grid cell occupancy descriptors, GCODs, are calculated by aligning the set of molecules in defined grid box which resembles active site of receptor. GOCDs are the impetus of interaction pharmacophore elements, IPEs for atom sets like polar positive, polar negative, aromatic, hydrogen bond acceptor, and hydrogen bond donor. Partial least squares (PLS; Dijkstra, 2010) are one of the chemometric methods commonly envisaged in many QSAR models. In present work, 4D-QSAR approach called LQTAgrid-QSAR (LQTA, Laboratório de Quimiometria Teórica e Aplicada) introduced by Martins et al. (2009) has been used to build a 4D-QSAR model. Such 4D-QSAR model was developed in a search of important structural features required in the coumarin derivatives and subsequent design of more active molecules. In this approach, conformational ensemble was generated for individual compound called conformational ensemble profile (CEP) by molecular dynamics (MD) simulations using GROMACS 4.6.3 (Berendsen et al., 1995; Hess et al., 2008; Pronk et al., 2013).
Experimental
Methods
Dataset
Fifty-seven coumarin derivatives presented in Table 1 have been taken from different literatures (Zhao et al., 1997; Nunthaboot et al., 2006; Mao et al., 2002, Chiang et al., 2007, Su et al., 2006; Al-Mawsawi et al., 2006). These coumarin derivatives have been reported in these literatures as 3′-processing inhibitors. All the derivatives selected have biscoumarin component common in their structure. Based on the substitution pattern in these derivatives, 13 compounds were selected as test set and remaining 44 compounds as training set. From the literature, the reported 3′-processing IC50 in µM was transformed to pIC50 by taking negative logarithm of IC50 values in moles.
Computer hardware and software
Computational work was done on Ubuntu Linux 12.0 and windows XP operating system. Various software used include AutoDock 4.2 with MGLTools 1.5.6 (Morris et al., 2009), AutoDock Vina (Trott and Olson, 2010), Marvin Sketch, UCSF Chimera 1.8rc (Pettersen et al., 2004), Discovery Studio 3.5 (Accelrys Inc.), ArgusLab 4.0.1, Firefly 801 Quantum Chemistry Package (Granovsky, Firefly version 8.0; Schmidt et al., 1993), MATLAB 7.7.0 (R2008b) (MathWorks, Inc.), PLS_Toolbox version 7.51 (Eigenvector Research, Inc.), Pymol version 1.3 (from Schrodinger, LLC), LQTAgrid (Martins et al., 2009), ERM algorithm, a MATLAB .m script (Ballabio et al., 2014) and GROMACS 4.6.3.
Geometry optimization
General 4D-QSAR methodology is depicted in Fig. 1. Structures of all compounds were drawn using Marvin Sketch. Geometry optimization was carried out in ArgusLab 4.0.1 on semi empirical quantum mechanical basis with parameterized model number 3 (PM3) hamiltonian, until restricted closed-shell Hartree–Fock self-consistent field formalism converses to 10−10 kcal/mol and steepest descent geometry search criteria until gradient converses to 10−6 kcal/mol. Gasteiger partial atomic charges of optimized molecules were computed in UCSF Chimera.

Schematic representation of 4D-QSAR methodology
GROMACS coordinates and topology files
PRODRG2 server (Schuttelkopf et al., 2004) was used to generate coordinate and topology files of all the compounds from dataset. As the structures were optimized, energy minimization option in PRODRG was not chosen. Appropriate add hydrogen or hybridization patch was used during the generation of coordinate and topology files. The Gasteiger charges computed in UCSF Chimera were manually loaded in PRODRG-generated topology files.
Molecular dynamics simulation
Molecular dynamics simulation was carried out in GROMACS 4.6.3 in order to obtain CEP of each compound. During MD simulation, GROMOS96 ffG43a1 force field was used in an explicit water model in a cubic box of 1 Å volume. MD simulation included heating the system at 50, 100, 200 and 350 K for 20 picoseconds (ps) simulation time with 1-femtosecond (fs) step size. Particle mesh Ewald (PME) method was used to compute long-range electrostatics, and the Van der Waal (VWD) interaction energies were calculated with a cutoff radius of 1 Å. Compound and solvent water were separately coupled during the simulation. The pressure and temperature of the system were controlled by Parrinello–Rahman coupling and velocity rescaling thermostat (V-rescale), respectively. The system was then cooled down to 300 K. The trajectory generated was recorded every 2000 simulation steps, which is 2 ps simulation time.
Alignment
Conformations of all the compounds generated at 300 K simulation were subjected to alignment. Compound 16, the most active among all compounds, was chosen as reference compound. The atoms chosen for alignment included the common structural component (biscoumarin part) as shown in Fig. 2. The CEP of all other conformations of the rest of the compounds was aligned against the reference compound. In the alignment step, trajectories generated at 20–100 ps time with 2-ps increment were aligned. The alignments of conformers of the few most active (36, 29, 35 and 34) and least active compounds (3, 33, 7, 4 and 17) from dataset aligned with conformers of most active compound 18 are shown in Fig. 3.

Structural component (biscoumarin part) used in alignment

Alignment of conformers generated during MD simulation (CEP) a aligned CEP of most active (reference) compound 16; b CEP of active compound 36 aligned with compound 16 (compound 16 shown blue color); c CEP of active compound 29; d CEP of active compound 35; e CEP of active compound 34; f CEP of least active compound 3; g CEP of less active compound 33; h CEP of less active compound 7; i CEP of less active compound 4; j CEP of less active compound 17 (Color figure online)
Descriptors of interaction energy
The grid box of size 18 × 18 × 18, large enough to accommodate the CEPs, was chosen. LQTAgrid module was used with a hypothetical N-terminal of protein as –NH3+ probe to generate matrix of interaction energy descriptors. The electrostatic property in terms of Coulombic (C) potential function and steric 3D property in terms of Lennard-Jones (LJ) potential function was generated as a descriptor matrix of 11,664 descriptors containing 5832 LJ and 5832 Coulombic potential-based descriptors.
Variable selection and model development
The dimension of the descriptor matrix generated by LQTAgrid was 57 × 11,664. Descriptor matrix was refined by eliminating descriptors having correlation lower than 0.3 leaving 4248 descriptors. V-WSP variable reduction MATLAB routine which is an unsupervised variable reduction based on V-WSP algorithm (Ballabio et al., 2014) was subsequently applied on 4248 descriptors with 0.85 absolute correlations which gave 357 most suitable descriptors. The dataset was split into training set of 44 compounds and test set of 13 compounds. Test set compounds were selected so as to include highest and lowest activities as well as structural diversity. The PLS model was built using PLS_Toolbox (Eigenvector Research, Inc) in MATLAB workspace. The model built by using absolute values, ten latent variables and Venetian blinds with six splits which includes one sample per split was found to be the best model.
Docking studies
Till date, neither full-length structure of integrase from HIV-1 nor integrase complex with its DNA counterpart is available. Due to emergence of resistance strains against HIV-1 IN catalytic site inhibitors like raltegravir, development of allosterical site inhibitors can be extremely advantageous approach (Al-Mawsawi et al., 2006). Through structure-based design, a new allosteric region in HIV-1 IN has been identified and is available at RCSB protein data bank (PDB code, 3NF7). The QSAR methodology proposed in this work is receptor independent, but to understand the structural features important for HIV-1 IN inhibition, docking studies were carried out using AutoDock Vina from molecular graphics laboratory, Scripps Research Institute. The accuracy of docking was validated by docking the co-crystallized ligand, 5-[(5-chloro-2-oxo-2, 3-dihydro-1H-indil-1-yl) methyl]-1, 3-benzodioxole-4-carboxylic acid, into the active site. The root-mean-square deviation (RMSD) between docked pose and original co-crystallized pose was 0.104 Å (Fig. 4).

Docked conformer of ligand (red) and original pose of co-crystallized ligand (green) (Color figure online)
Results and discussion
4D-QSAR model
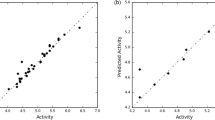
The objective of current 4D-QSAR studies was to build the best 4D-QSAR model with good predictive abilities. The strategy of MD simulation and subsequent generation of interaction energy contributions was thought to emulate interaction of important residues at the binding site of HIV-1 IN with compounds under investigation. The number of refined descriptors was 357, and multiple linear regression (MLR), a multivariate chemometric tool, cannot be a good model. This is because the number of independent variable matrix exceeds dependent variable vector, and generated model could have the over-fitting. PLS regression is best in this situation, and in current investigation, PLS regression was carried out. The data were split into 44 training set compounds and 13 test set compounds with the help of random_select.m script for MATLAB. The PLS model development was carried out by using PLS_Toolbox from Eigenvector Research, Inc. Data were preprocessed by autoscaling the raw data. The leave-one-out cross-validation method was found inappropriate for current data as the data constitute more than 20 samples. Thus, Venetian blind cross-validation method with six sample splits with one sample per split was adopted. The model with 10 latent variables was found best among 20 models. This PLS model showed R 2 calculated = 0.903015, R 2 cross-validated = 0.599553 and R 2 predicted = 0.688525. All these regression coefficients are within acceptable limits. The full model including test set and training set showed R 2 = 0.853. The other statistical findings include root-mean-square error (RMSE) calculated = 0.21276, RMSE predicted = 0.371579 and prediction bias = −0.15362. The pIC50 values predicted with the residuals are shown in Table 2. The plots of predicted activity against measured activity, scores on latent variables and measured activity against residuals are shown in Figs. 5, 6 and 7, respectively.

Predicted pIC50 values against measured pIC50 values for training set and test set compounds

Scores on latent variables

Predicted pIC50 residuals against measured pIC50 values
Docking studies
During development of current 4D-QSAR model, NH3 + probe was exploited while generating interaction energy contributions. The interaction energy contribution between NH3 + field point and CEP at each grid point of 18 × 18 × 18 grid box spaced at 1 Å grid was generated. VAL150, LYS188, ARG199, HIS183, ILE151, MET154 and LEU158 are important residues in the binding site of HIV-1 IN. First three residues are important for hydrogen bonding, and other residues are important for hydrophobic interactions. Docking studies were carried out using AutoDock Vina on heteroatom stripped and optimized model protein of HIV-1 IN catalytic domain (PDB, 3NF7). During docking, the grid box of size 11 × 11 × 11 was used spaced at 1 Å and with x, y, z center 9.918, −26.384, −10.818, respectively. The VWD interaction constitutes Coulombic contributions, and electrostatic, hydrophobic interaction constitutes steric LJ potential contributions in 4D-QSAR descriptors. The interactions of docked conformers of few most active and least active compounds from dataset with important residues are presented in Table 3 and shown in Fig. 8.

Interaction of active and less active compounds with important residues
Docking studies revealed that for HIV-1 IN inhibitory activity electrostatic, hydrophobic interactions are necessary between ligand and the important residues, LYS188, HIS183 and ARG199. The most active compounds were found to make important VWD interactions with VAL79, VAL77, ARG199 and GLU157, whereas least active compounds could not make such interactions. The water molecule, HOH272, at binding site was found to make interaction with most of the HIV-1 IN inhibitors. The 4D-QSAR studies resulted in a model where the descriptors 17_17_9_NH3 +_Coulombic (C), 17_17_8_NH3 +_C, 18_15_9_NH3 +_C, 18_16_18_NH3 +_LJ, 8_13_11_NH3 +_LJ, 18_15_10_NH3 +_C, 17_15_12_NH3 +_LJ, 18_17_8_NH3 +_C, 18_17_6_NH3 +_C, 17_16_10_NH3 +_C were found most contributing to the final model. Out of these, seven descriptors are Coulombic contribution descriptors and three are LJ steric interaction energy contributions. These descriptors as field points on the structures of most active compound 16 and least active compound 3 and the VWD and electrostatic, hydrophobic surfaces around these molecules are shown in Fig. 9.

Interpretation of interaction energy descriptors. a Interaction energy descriptors around most active compound 16; b Interaction energy descriptors around least active compound 3; c VW surface; d Hydrophobic surface around most active compound 16; e VW surface; f hydrophobic surface around least active compound 3
Conclusion
4D-QSAR model was built using GROMACS-based MD simulation on coumarin derivatives as HIV-1 IN inhibitors. In the model, interaction energy descriptors were constructed on CEP of each compound. PLS regression was carried out on selected descriptor matrix of 357 descriptors. The selected model with ten latent variables showed R 2 calculated 0.903015, R 2 cross-validated 0.599553 and R 2 predicted 0.688525. The results of 4D-QSAR are in good agreement with the docking studies, suggesting that the VW interactions are important for higher activity of these compounds. The 4D-QSAR model generated can be used for development of HIV-1 IN 3′-processing inhibitors. As a part of future work, we are designing coumarin-based molecules with substituents which will mainly contribute to VW interactions.
References
Al-Mawsawi LQ, Fikkert V, Dayam R, Witvrouw M, Burke TR Jr, Borchers CH, Neamati N (2006) Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc Natl Acad Sci USA 103:10080–10085. doi:10.1073/pnas.0511254103
Bailly F, Queffelec C, Mbemba G, Mouscadet JF, Cotelle P (2005) Synthesis and HIV-1 integrase inhibitory activities of caffeic acid dimers derived from Salvia officinalis. Bioorg Med Chem Lett 15:5053–5056. doi:10.1016/j.bmcl.2005.07.091
Ballabio D, Consonni V, Mauri A, Claeys-Bruno M, Sergent M, Todeschini R (2014) A novel variable reduction method adapted from space-filling designs. Chemom Intell Lab Syst 136:147–154
Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91:43–56
Chiang CC, Mouscadet JF, Tsai HJ, Liu CT, Hsu LY (2007) Synthesis and HIV-1 integrase inhibition of novel bis- or tetra-coumarin analogues. Chem Pharm Bull 55:1740–1743
Cramer RD III, Patterson DE, Bunce JD (1988) Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 110:5959–5967
Curran JW (1983) AIDS—two years later. N Engl J Med 309:609–611
Dijkstra TK (2010) Latent variables and indices: herman wold’s basic design and partial least squares. In: Vinzi VE, Chin WW, Henseler J, Wang H (eds) Handbook of partial least squares, 1st edn. Springer, NewYork, pp 23–46
Free SM, Wilson JWA (1964) Mathematical contribution to structure-activity studies. J Med Chem 7:395–399
Granovsky AA, Firefly version 8.0, www http://classic.chem.msu.su/gran/firefly/index.html
Hansch C, Fujita TA (1964) Method for the correlation of biological activity and chemical structure. J Am Chem Soc 86:1616
Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447
Hopfinger A, Wang S, Tokarski J, Jin B, Albuquerque M, Madhav P, Duraiswami C (1997) Construction of 3D-QSAR models using the 4D-QSAR analysis formalism. J Am Chem Soc 119:10509–10524
Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A (2011) Structural studies of retroviral integrase. In: Neamati N (ed) HIV-1 integrase: mechanism and inhibitor design, 1st edn. Wiley, New York, pp 35–49
Johnson VA, Brun-Vezinet F, Clotet B, Gunthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD (2010) Update of the drug resistance mutations in HIV-1. Top HIV Med 18:156–163
Klebe G, Abraham U, Mietzner T (1994) Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem 37:4130–4146
Korolev SP, Agapkina YY, Gottikh MB (2011) Clinical use of inhibitors of HIV-1 integration: problems and prospects. Acta Naturae 3:12–28
Kostova I, Raleva S, Genova P, Argirova R (2006) Structure-activity relationships of synthetic coumarins as HIV-1 inhibitors. Bioinorg Chem Appl 2006:68274. doi:10.1155/BCA/2006/68274
Liu M, Cong XJ, Li P, Tan JJ, Chen WZ, Wang CX (2009) Study on the inhibitory mechanism and binding mode of the hydroxycoumarin compound NSC158393 to HIV-1 integrase by molecular modeling. Biopolymers 91:700–709. doi:10.1002/bip.21211
Mahajan DH, Pannecouque C, De Clercq E, Chikhalia KH (2009) Synthesis and studies of new 2-(coumarin-4-yloxy)-4,6-(substituted)-S-triazine derivatives as potential anti-HIV agents. Arch Pharm 342:281–290. doi:10.1002/ardp.200800149
Mao PCM, Mouscadet JF, Leh H, Auclair C, Hsu LY (2002) Chemical modification of coumarin dimer and HIV-1 integrase inhibitory activity. Chem Pharm Bull 50:1634–1637. doi:10.1248/cpb.50.1634
Martins JPA, Barbosa EG, Pasqualoto KFM, Ferreira MMC (2009) LQTA-QSAR: a new 4D-QSAR methodology. J Chem Inf Model 49:1428–1436
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 16:2785–2791
Nunthaboot N, Tonmunphean S, Parasuk V, Wolschann P, Kokpol S (2006) Three-dimensional quantitative structureeactivity relationship studies on diverse structural classes of HIV-1 integrase inhibitors using CoMFA and CoMSIA. Eur J Med Chem 41:1359–1372
Olmedo D, Sancho R, Bedoya LM, Lopez-Perez JL, Del Olmo E, Munoz E, Alcami J, Gupta MP, San Feliciano A (2012) Molecules 17:9245–9257. doi:10.3390/molecules17089245
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Pronk S, Paill S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F (2002) HIV-1 integration in the human genome favors active genes and local hotspots. Cell 10:521–529
Schuttelkopf AW, van Aalten DMF (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D 60:1355–1363
Spino C, Dodier M, Sotheeswaran S (1998) Anti-HIV coumarins from Calophyllum seed oil. Bioorg Med Chem Lett 8:3475–3478
Su C, Mouscadet J, Chiang C, Tsai H, Hsu L (2006) HIV-1 integrase inhibition of biscoumarin analogues. Chem Pharm Bull 54:682–686
Tewtrakul S, Subhadhirasakul S, Cheenpracha S, Karalai C (2007) HIV-1 protease and HIV-1 integrase inhibitory substances from Eclipta prostrata. Phytother Res 21:1092–1095
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461. doi:10.1002/jcc.21334
U.S. Food and Drug Administration. http://www.fda.gov/downloads/drugs/developmentapprovalprocess/developmentresources/ucm385696.pdf Accessed 15 July 2014
Zhao H, Neamati N, Hong H, Mazumder A, Wang S, Sunder S, Milne GWA, Pommier Y, Burke TR Jr (1997) Coumarin-based inhibitors of HIV integrase-1. J Med Chem 40:242–249
Acknowledgments
We acknowledge constant encouragement of Prof. M. N. Navale, President, Sinhgad Technical Education Society and Matteo Cassotti, Milano Chemometrics and QSAR Research Group, Department of Earth and Environmental Sciences, University of Milano-Bicocca for helpful suggestions in MATLAB usage.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Patil, R.B., Sawant, S.D. 4D-QSAR studies of coumarin derivatives as HIV-1 integrase 3′-processing inhibitors. Med Chem Res 24, 3062–3076 (2015). https://doi.org/10.1007/s00044-015-1359-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-015-1359-z