
Abstract
The synthesis of some new (E)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-ones directly linked to either pyrazole, pyrazoline, pyrazolidine counterparts, or to substituted thio and hydrazono functionalities is described. Six of the newly synthesized compounds were selected by the National Cancer Institute (NCI) to be evaluated for their in vitro antitumor activity according to the protocol of the NCI in vitro disease-oriented human cells screening panel assay. The results revealed that the pyrazole derivative 5c was found to be the most active member in this screen as evidenced by its ability to exert potential growth inhibitory activity against most of the tested subpanel tumor cell lines with selective influence on leukemia subpanel tumor cell lines (GI50 values 2.01–3.03 μM). Moreover, a comparative study for log GI50 values of both compound 5c and 5-fluorouracil (5-FU) revealed that compound 5c showed higher potency than 5-FU against most of the tested subpanel tumor cell lines. Thus compound 5c could be considered as a suitable lead towards the design of broad spectrum antitumor active agents targeting various human tumor cell lines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer poses a serious human health problem despite much progress in understanding its biology and pharmacology. Although cancer research has led to a number of new and effective solutions, the medicines used as treatments have clear limitations due to lack of selectivity leading to toxicity, metastatic spreading and the intrinsic or acquired resistance developed after few therapeutic cycles (Braña and Ramos, 2001; Cozzi, 2003). At the same time, random screening remains one of the main routes to discover new leads with antineoplastic activity and the National Cancer Institute (NCI), Bethesda, USA, is still playing an articular role in this field, with special emphasis on novel chemical structures that have not had extensive clinical evaluation (Cocco et al., 2000).
Among the wide variety of heterocycles that have been explored for developing pharmaceutically important molecules, the 1,2,4-triazines have received great attention as chemotherapeutic agents. Azanucleosides (6-azacytosine and 6-azauracil), structurally based on the 1,2,4-triazine scaffold were proved to display antitumor (Creasey et al., 1963), antiviral (Sidwell et al., 1968), and antifungal activities (Sangshetti and Shinde, 2010). In addition, some 1,2,4-triazin-6(1H)-ones were reported to display significant broad spectrum antitumor activity against lymphoblastic leukemia CEM, myeloid leukemia K562, and lung adenocarcinoma A549 cancer cell lines (Gucky et al., 2009) while, some 1,2,4-triazine 5-one derivatives were found to exhibit strong antiproliferative effect on human leukemia K-562 cell line (Krauth et al., 2010). Moreover, particular interest has been focused on 6-azaisocytosine (3-amino-1,2,4-triazin-5(2H)-one), an isosteric isomer of 6-azacytosine and 6-azauracil as potential transcription inhibitor (Pal chykovska et al., 2004). On the other hand, a literature survey revealed that some pyrazoles and pyrazole containing compounds have been implemented as antileukemic (Daidone et al., 2004a; Chou et al., 2007; Manetti et al., 2008), antitumor (Li et al., 2006; Xia et al., 2007; Xia et al., 2008; Farag et al., 2008), and antiproliferative agents (Schenone et al., 2004; Daidone et al., 2004b), beside their capability to exert remarkable anticancer effects through inhibiting different types of enzymes that play important roles in cell division (Warshakoon et al., 2006; Huang et al., 2007; Zhu et al., 2007). Furthermore, some thioethers were found to show enhanced antimicrobial and antitumor activities (Gulerman et al., 2001; Khalil et al., 2003) beside being a common structural subunit in SDABOs (dihydro-alkylthio-benzyl-oxopyrimidines) which possess antiproliferative as well as antiviral activities (Manetti et al., 2005). Addditionally, hydrazono derivatives with their effective contribution as antineoplastic agents (Remers, 2004) were not far of our attention.
Inspired by the above-mentioned facts and as a continuation of an ongoing research program aimed at the discovery of novel chemotherapeutic agents (Rostom et al., 2009; Ashour and Abdel Wahab, 2009; Rostom et al., 2011), it seemed worthwhile to synthesize new structure hybrids (A–C) incorporating the 1,2,4-triazin-5-one scaffold linked to a pyrazole, pyrazoline, pyrazolidine ring or substituted thioether and hydrazono moieties (Fig. 1) which are believed to be responsible for the biological significance of some relevant natural and synthetic chemotherapeutic agents. This combination was suggested in an attempt to investigate the possible synergistic influence of such structure hybridization on the anticipated biological activity hoping to discover a new lead structure that would have a significant antitumor potential. In addition, variation in the nature and size of substituents was also attempted, as it would offer variable electronic, lipophilic, and steric environment that would influence the targeted biological activity. The present work reports the synthesis and the results of preliminary antitumor screening of compounds selected by the NCI.

The structure of some reported antitumor 1,2,4-triazines and the newly synthesized compounds
Chemistry
Synthesis of the intermediate and target compounds was accomplished according to the steps depicted in Schemes 1 and 2. In Scheme 1, the starting thione 1 and the hydrazine intermediate 3 were prepared according to previously reported reaction conditions (Slouka, 1962; Osman et al., 2007). Heating 1 with phenacyl or 4-substituted phenacyl bromide in ethanol gave rise to the substituted 2-oxoethylsulfanyl analogs 2a–e. 1H-NMR spectra of compounds 2a–e revealed singlets at 4.45 and 4.38 ppm for compounds 2a, b and two doublets at 3.67–3.87 ppm for compounds 2c–e attributed to SCH2 protons. In addition, investigation of 1H-NMR spectra for compounds 2a–e revealed presence of two doublets for ethenyl C1 and C2 protons at 6.65–6.81 and 7.56–7.75 ppm with coupling constant 16.05–16.7 Hz indicating their existence as E-isomers (Williams and Fleming, 1980). 13C-NMR spectrum of compound 2e as an example showed a signal at 50.55 ppm corresponding to SCH2 carbon and other signals were observed at their expected chemical shifts. Condensation of the hydrazine derivative 3 with 4-substituted benzaldehydes, furfural, 5-nitro-2-furfural, isatin, and N-methyl isatin in boiling ethanol resulted in the formation of the corresponding hydrazones 4a–g. 1H-NMR spectra of compounds 4a–e showed singlets for the N=CH protons in the range 7.82–8.08 ppm indicating existence of these compounds in the E configuration around the N=C (Pretsch et al., 2000, pp 211–212). 13C-NMR spectrum of compound 4a provided further confirmation of the chemical structure. On the other hand, reaction of 3 with phenacyl or 4-substituted phenacyl cyanides in ethanol/acetic acid mixture furnished the target 5-aminopyrazoles 5a–d. Inspection of 1H-NMR spectra of compounds 5a–d indicated presence of a singlet at 5.88–5.92 ppm corresponding to pyrazole C4-proton in addition to D2O exchangeable singlets at 7.06–7.10 ppm due to NH2 protons. Additionally, condensation of 3 with an equimolar amount of acetyl acetone in ethanol gave rise to the requisite 3,5-dimethyl pyrazole 6. 1H-NMR spectrum revealed presence of three singlets at 2.22, 2.46, and 6.24 ppm interpreted for two CH3 groups and pyrazole C4-proton. Heating the same intermediate 3 with diethyl malonate in a mixture of ethanol/glacial acetic acid afforded the pyrazolidine-3,5-dione 7. 1H-NMR spectrum for this compound showed a singlet for the pyrazolidine C4-protons at 2.45 ppm in addition to a D2O exchangeable singlet at 8.70 ppm due to pyrazolidine NH proton. Reaction of 3 with an equimolar amount of ethyl acetoacetate in boiling ethanol yielded the ethyl butanoate ester 8. 1H-NMR spectrum of compound 8 revealed a singlet for the C-3 methyl group at a high chemical shift (1.94 ppm) confirming that the configuration at C-3 is E (Pretsch et al., 2000, pp 211–212). Attempts to cyclize the ethyl butanoate ester to the corresponding pyrazolinone 9 using high boiling point solvents were unsuccessful; however fusion of the ethyl butanoate ester in an oil bath at 160 °C afforded the respective pyrazolinone 9 in a good yield. 1H-NMR spectrum of 9 showed a singlet at 2.47 ppm assigned for CH3 protons and a singlet at 3.44 ppm attributed to pyrazoline C4 proton, while its MS spectrum revealed a molecular ion peak at m/z 285 (21 %) which is in accordance with its molecular formula. It should be noted that the pyrazolinone derivative 9 could be directly prepared from the hydrazine intermediate 3 by fusing the latter compound with ethyl acetoacetate in an oil bath at 160 °C.

Reagents and reaction conditions: i R1COCH2Br, absolute ethanol, reflux, ii hydrazine 98 %, absolute ethanol, reflux, iii aldehydes or ketones, absolute ethanol, glacial acetic acid, reflux, iv R1COCH2CN, ethanol, glacial acetic acid, reflux, v acetylacetone, absolute ethanol, reflux, vi diethyl malonate, glacial acetic acid, reflux, vii ethyl acetoacetate, absolute ethanol, reflux, viii ethyl acetoacetate, fusion, oil bath, 160 °C, ix fusion, oil bath, 160 °C

Reagents and reaction conditions: i thiocarbohydrazide, ethanol, glacial acetic acid, stir, r.t., ii 1 N sodium hydroxide, iii methyl iodide or ethyl iodide or benzyl chloride, dry dimethyl formamide, anhydrous potassium carbonate, stir, r.t., iv 4-nitrobenzaldeyhde, absolute ethanol, glacial acetic acid, reflux, v ClCH2CH2R1.HCl, potassium hydroxide, stir, r.t.
Referring to Scheme 2, stirring 10 (Rohmer, 1898) with thiocarbohydrazide in ethanol containing a catalytic amount of glacial acetic acid resulted in the formation of the respective thiocarbohydrazone 11. IR spectra of the latter compound displayed absorption band characteristic for C=O group at low frequency; 1661 cm−1, while its 1H-NMR spectrum showed a D2O exchangeable singlet for the NH proton at a high chemical shift; 10.3 ppm confirming presence of a hydrogen bond between the C=O and NH groups (Pretsch et al., 2000, pp. 290–291). This led to the suggestion that the configuration around the C=N is Z. Heating 11 in 1 N sodium hydroxide gave rise to the corresponding 4-amino-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one 12. IR spectrum for compound 12 lacked absorption bands for OH group and showed absorption bands characteristic for NH2 and N-CS moieties whereas, its 1H-NMR spectrum verified its structure. Stirring the aminothione 12 with an equivalent amount of methyl iodide, ethyl iodide or benzyl chloride in dry DMF containing anhydrous potassium carbonate afforded the S-alkyl derivatives 13a-c in good yields. IR, 1H-NMR, and 13C-NMR spectral data confirmed the chemical structure of these derivatives. Additionally, synthesis of the azomethine derivative 14 was achieved by condensing the thione 12 with 4-nitrobenzaldehyde in boiling ethanol containing few drops of glacial acetic acid. IR spectrum of 14 lacked absorption bands characteristic for NH2 group, while its 1H-NMR spectrum revealed singlets attributed to the nitrophenyl group in addition to a singlet for the N=CH proton at 8.89 ppm indicating that the configuration around the C=N is E (Pretsch et al., 2000, pp 211–212). Moreover, the MS spectrum of 14 showed a molecular ion peak at m/z 369 (20.5 %) which matches with its molecular formula. Finally, alkylation of the azomethine 14 with hydrochloride salts of 2-substituted ethyl chloride derivatives in aqueous KOH furnished the corresponding 2-substituted ethylsulfanyl derivatives 15a–c. Inspection of 1H-NMR spectra of the latter compounds indicates presence of signals for SCH2 and NCH2 protons at their expected chemical shifts. Furthermore, 13C-NMR spectrum of compound 15b displayed signals at 28 and 58.20 ppm corresponding to SCH2 and NCH2 in addition to signals of piperidine carbons which appeared at their expected values.
In vitro antitumor screening
Primary in vitro one-dose assay
Out of the newly synthesized compounds, six derivatives namely: 2c, 5c, 7, 12, 14, and 15a were selected by the National Cancer Institute (NCI) in vitro disease-oriented human cells screening panel assay to be evaluated for their in vitro antitumor activity.
An effective one-dose assay has been added to the NCI-60 cell screen in order to increase compound throughput and reduce data-turnaround time to suppliers while maintaining efficient identification of active compounds (Weislow et al., 1989; Monks et al., 1991; Boyd and Paull, 1995). All compounds submitted to the NCI-60 cell screen are tested initially at a single high dose (10 μM) in the full NCI-60 cell panel including leukemia, non-small cell lung, colon, CNS melanoma, ovarian, renal, prostate, and breast cancer cell lines. Only compounds which satisfy pre-determined threshold inhibition criteria would proceed to the five-dose screen. The threshold inhibition criteria for proceeding to the five-dose screen was designed to efficiently capture compounds with anti-proliferative activity, and it is based on careful analysis of historical Development Therapeutic Program (DTP) screening data. Data are reported as a mean graph of the percent growth of treated cells, and presented as percentage growth inhibition (GI%) caused by the test compounds (Table 1).
The obtained results revealed that compound 2c was able to exhibit promising broad spectrum anticancer activity particularly against breast cancer MCF7, MDA-MB-48, Ovarian cancer IGROV 1, and renal cancer RXF 393 cell lines (GI% values 80.83, 57.34, 80.44, and 62.53, respectively). However, its overall antitumor profile did not meet the pre-determined threshold inhibition criteria and therefore was not sufficient to proceed to the five-dose screen. Compounds 12 and 14 displayed remarkable growth inhibitory activity towards breast cancer MCF7, ovarian cancer IGROV 1, and melanoma SK-MEL-2 cell lines (GI% range 70.32–77.03). Moreover, compound 15a exhibited significant activity against breast cancer MCF7 and SR cell lines (GI% values 69.03, 120.44, and 66.41, respectively). It should be noted here that compound 15a showed lethal effect towards the CCRF-CEM cell line with 20.44 %. On the other hand, compound 7 was proved to be the weakest anticancer member in this screen owing to its low potency and narrow margin of activity, which was against only leukemia HL-60(TB) and with GI% value of 54.26. Whereas, compound 5c was found to be the most active member in this preliminary screen as evidenced by its ability to exert potential growth inhibitory activity against most of the tested subpanel tumor cell lines. Consequently, it passed successfully this assay and was carried over to the five-dose screen against a panel of about 60 different tumor cell lines.
In vitro full panel (five-dose) 60-cell line assay for compound 5c
About 60 cell lines of nine tumor subpanels, including leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancer cell lines, were incubated with five concentrations (0.01–100 μM) for each compound and were used to create log concentration % growth inhibition curves. Three response parameters (GI50, TGI, and LC50) were calculated for each cell line. The GI50 value (growth inhibitory activity) corresponds to the concentration of the compounds causing 50 % decrease in net cell growth, the TGI value (cytostatic activity) is the concentration of the compounds resulting in total growth inhibition and the LC50 value (cytotoxic activity) is the concentration of the compounds causing net 50 % loss of initial cells at the end of the incubation period (48 h). Subpanel and full panel mean-graph midpoint values (MG-MID) for certain agents are the average of individual real and default GI50, TGI, or LC50 values of all cell lines in the subpanel or the full panel, respectively.
In the present study, compound 5c exhibited potential antitumor activities against most of the tested subpanel tumor cell lines (GI50 and TGI values <100 μM). This compound showed a distinctive pattern of sensitivity against some individual cell lines (Table 1), as well as a broad spectrum (MG-MID) of antitumor activity (Table 2).
A deep insight into the obtained results (Table 2) revealed that compound 5c exhibited remarkably high activity against renal cancer CAKI-1 cell line with GI50 value of 0.37 μM. It also displayed a distinguished sensitivity profile towards all leukemia cell lines with GI50 range of 2.01–3.03 μM beside a potential activity against seven cell lines with GI50 range of 1.23–1.63 μM. In addition, compound 5c showed appreciable growth inhibitory potential against 58 cell lines with GI50 range of 1.88–18.7 μM. Further interpretation of the obtained data revealed that compound 5c was able to totally inhibit the growth of 50 cell lines at 4.6–84.8 μM. Moreover, 5c was cytotoxic against three cell lines; ovarian cancer OVCAR-3, NCI/ADR-RES, and prostate cancer DU-145 (LC50 values 31.8, 96.4, and 55.4 μM, respectively).
The results also revealed that compound 5c displayed high growth inhibitory potential (GI50 MG-MID 3.98 μM) (Table 3), together with reasonable cytostatic (TGI MG-MID 35.48 μM) and mild cytotoxic (LC50 MG-MID 97.72 μM) activities (Table 4).
The ratio obtained by dividing the compound full panel MG-MID (μM) by its individual subpanel MG-MID (μM) is considered as a measure of compound selectivity. Ratios between 3 and 6 refer to moderate selectivity, ratios >6 indicate high selectivity towards the corresponding cell line, while compounds meeting neither of these criteria are rated non-selective (Acton et al., 1994). In this context, compound 5c was found to be non-selective with broad spectrum antitumor activity against the nine tumor subpanels tested with selectivity ratios ranging between 0.59 and 1.47 at the GI50 MG-MID level. Moreover, log GI50 values for compound 5c was illustrated with respect to 5-fluorouracil as a comparative study for anticancer potency (Table 5), where values of −0.4 and less are considered to be of high anticancer activity. The comparative study indicated that compound 5c displayed anti-tumor activity against all human tumor cell lines higher than 5-fluorouracil.
The above results revealed that this compound could be an appropriate candidate for further derivatization in order to explore the scope and limitations of its potential hoping to find more selective and active anticancer agents.
Experimental
All reagents and solvents were purchased from commercial suppliers and were dried and purified when necessary by standard techniques. Melting points were determined in open glass capillaries using Stuart capillary melting point apparatus (Stuart Scientific Stone, Staffordshire, UK) and are uncorrected. Infrared (IR) spectra were recorded on Perkin-Elmer 1430 infrared spectrophotometer (Perkin Elmer, Beaconsfield, UK) and measured by ύ cm−1 scale using KBr cell. 1H-NMR spectra were scanned on Jeol-500 MHz spectrometer (Jeol, Tokyo, Japan) and Varian Mercury VX-300 using tetramethylsilane (TMS) as internal standard and DMSO-d 6 as the solvent (chemical shifts are given in δ ppm). Splitting patterns were designated as follows: s: singlet; brs: broad singlet; d: doublet; dd: doublet of doublet; t: triplet; m: multiplet. 13C-NMR proton decoupled spectra were recorded on a Varian Mercury VX-300 spectrometer in DMSO-d 6 and measured in δ scale. Mass spectra were run on a Finnigan mass spectrometer model SSQ/7000 (70 eV) or on a gas chromatograph/mass spectrometer Schimadzu GCMS-QP 2010 Plus (70 eV). Elemental analyses were performed on Elementar Vario E1 and were found within ±0.4 % of the theoretical values. Follow up of the reactions and checking the purity of the compounds was made by thin layer chromatography (TLC) on silica gel-precoated aluminum sheets (Type 60 GF254; Merck; Germany) and the spots were detected by exposure to UV lamp at λ 254 nm for few seconds. Compounds 1 (Slouka, 1962), 3 (Osman et al., 2007), and 10 (Rohmer, 1898) were prepared according to previously reported reaction conditions.
3-[(2-Aryl-2-oxoethyl)sulfanyl]-6-[(E)-2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(2H)-ones (2a–e)
A mixture of the thione 1 (2.2 g, 10 mmol) and the appropriate phenacyl bromide (10 mmol) in absolute ethanol (10 ml) was heated under reflux for 2 h. The reaction mixture was cooled to room temperature and the separated product was filtered, washed with ethanol, dried and crystallized from dioxane/water.
(E)-6-[2-(Furan-2-yl)ethenyl]-3-[(2-phenyl-2-oxoethyl)sulfanyl]-1,2,4-triazin-5(2H)-one (2a)
Brown solid (76 %); m.p.: 192–194 °C; IR (KBr, cm−1): 3146 (NH), 3095 (CH furan), 2964, 2916 (CH2), 1692, 1659(C=O), 1625 (C=N), 1552, 1526, 1482 (C=C, δ NH), 1290, 1076 (C–S–C), 1232, 1014 (C–O–C), 744 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 4.45 (s, 2H, SCH2), 6.54–6.59 (m, 1H, furan C4-H), 6.77 (d, J = 3.9 Hz, 1H, furan C3-H), 6.81 (d, J = 16.7 Hz, 1H, ethenyl C1-H), 7.23–7.34 (m, 5H, phenyl-H), 7.56 (d, J = 16.7 Hz, 1H, ethenyl C2-H), 7.78 (s, 1H, furan C5-H), 12.40 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C17H13N3O3S (339.37): C, 60.17; H, 3.86; N, 12.38; found: C, 59.78; H, 3.47; N, 12.73.
(E)-6-[2-(Furan-2-yl)ethenyl]-3-[{2-(4-methylphenyl)-2-oxoethyl}sulfanyl]-1,2,4-triazin-5(2H)-one (2b)
Yellow solid (63 %); m.p.: 218–220 °C; IR (KBr, cm−1): 3275 (NH), 3075 (CH furan), 2921, 2850 (CH2, CH3), 1705, 1653 (C=O), 1628 (C=N), 1529, 1473 (C=C, δ NH), 1281, 1087 (C–S–C), 1194, 1017 (C–O–C), 750 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 2.34 (s, 3H, CH3), 4.38 (s, 2H, SCH2), 6.57–6.62 (m, 1H, furan C4-H); 6.75 (d, J = 3.9 Hz, 1H, furan C3-H), 6.79 (d, J = 16.7 Hz, 1H, ethenyl C1-H), 7.33 (d, J = 8.4 Hz, 2H, methylphenyl C3,5-H), 7.56 (d, J = 16.7 Hz, 1H, ethenyl C2-H), 7.75 (s, 1H, furan C5-H), 7.85 (d, J = 8.4 Hz, 2H, methylphenyl C2,6-H), 12.39 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C18H15N3O3S (353.39): C, 61.18; H, 4.28; N, 11.89; found: C, 61.53; H, 3.88; N, 11.53.
(E)-3-[{2-(4-Chlorophenyl)-2-oxoethyl}sulfanyl]-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(2H)-one (2c)
Yellow solid (78 %); m.p.: 228–230 °C; IR (KBr, cm−1): 3171 (NH), 3097 (CH furan), 2962, 2915 (CH2), 1672 (C=O), 1615 (C=N), 1588, 1533, 1483 (C=C, δ NH), 1286, 1091 (C–S–C), 1257, 1013 (C–O–C), 748 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 3.67, 3.78 (2 × d, J = 12.2 Hz, each 1H, S-CH2), 6.51–6.54 (m, 1H, furan C4-H), 6.65–6.80 (m, 2H, ethenyl C1-H and furan C3-H), 7.48, 7.57 (2 × d, J = 8.4 Hz, each 2H, chlorophenyl C3,5-H and chlorophenyl C2,6-H), 7.69–7.74 (m, 2H, ethenyl C2-H and furan C5-H), 8.32 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C17H12ClN3O3S (373.81): C, 54.62; H, 3.24; N, 11.24; S, 8.58; found: C, 54.62; H, 3.13; N, 11.06; S, 8.59.
(E)-3-[{2-(4-Bromophenyl)-2-oxoethyl}sulfanyl]-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(2H)-one (2d)
Yellow solid (80 %); m.p.: 218–220 °C; IR (KBr, cm−1): 3164 (NH), 3018 (CH furan), 2964, 2901 (CH2), 1673 (C=O), 1608 (C=N), 1582, 1506, 1478 (C=C, δ NH), 1287, 1069 (C–S–C), 1255, 1011 (C–O–C), 732 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 3.67, 3.77 (2 × d, J = 12.2 Hz, each 1H, S-CH2), 6.51–6.56 (m, 1H, furan C4-H), 6.67 (d, J = 16.05 Hz, 1H, ethenyl C1-H), 6.79 (d, J = 3.1 Hz, 1H, furan C3-H), 7.50, 7.61 (2 × d, J = 8.0 Hz, each 2H, bromophenyl C3,5-H and bromophenyl C2,6-H), 7.69–7.75 (m, 2H, ethenyl C2-H and furan C5-H), 8.32 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C17H12BrN3O3S (418.26): C, 48.82; H, 2.89; N, 10.05; S, 7.67; found: C, 49.09; H, 2.52; N, 9.87; S, 7.67.
(E)-6-[2-(Furan-2-yl)ethenyl]-3-[{2-(4-nitrophenyl-2-oxoethyl}sulfanyl]-1,2,4-triazin-5(2H)-one (2e)
Orange solid (60 %); m.p.: >300 °C; IR (KBr, cm−1): 3113 (NH), 3025 (CH furan), 2900, 2856 (CH2), 1675 (C=O), 1621 (C=N), 1575, 1472 (C=C, δ NH), 1519, 1347 (NO2), 1281, 1077 (C–S–C), 1219, 1019 (C–O–C), 754 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 3.74, 3.87 (2 × d, J = 12.6 Hz, each 1H, S-CH2), 6.51–6.57 (m, 1H, furan C4-H), 6.68 (d, J = 16.2 Hz, 1H, ethenyl C1-H), 6.81 (d, J = 3.3 Hz, 1H, furan C3-H), 7.72 (d, J = 16.2 Hz, 1H, ethenyl C2-H), 7.75 (s, 1H, furan C5-H), 7.89, 8.29 (2 × d, J = 8.7 Hz, each 2H, nitrophenyl C2,6-H and C3,5-H), 8.59 (s, 1H, NH, D2O exchangeable); 13C-NMR (300 MHz, DMSO-d 6) δ 50.55 (CH2), 122.11 (furan C4), 122.99 (furan C3), 127.26 (ethenyl C1), 132.90 (nitrophenyl C3,5), 134.49 (ethenyl C2), 137.64 (nitrophenyl C2,6), 152.0 (nitrophenyl C1), 154.54 (furan C5), 156.33 (furan C2), 157.21 (nitrophenyl C4), 158.5 (triazine C6), 161.01 (triazine C3), 175.18 (triazine C5), 175.98 (C=O); Anal. Calcd for C17H12N4O5S (384.37): C, 53.12; H, 3.15; N, 14.58; S, 8.34; found: C, 52.79; H, 2.89; N, 14.84; S, 8.22.
6-[(E)-2-(Furan-2-yl)ethenyl]-3-(2-substituted hydrazono)-1,2,4-triazin-5(2H)-ones (4a–g)
A mixture of the hydrazine 3 (0.48 g, 2.2 mmol) and the proper aldehyde or ketone (2.2 mmol), in absolute ethanol (10 ml) containing a catalytic amount of glacial acetic acid, was heated under reflux for 1 h. The reaction mixture was cooled and the precipitated product was filtered, washed with ethanol, dried and crystallized from the proper solvent.
6-[(E)-2-(Furan-2-yl)ethenyl]-3-[(E)-2-(4-methoxybenzylidene)hydrazono]-1,2,4-triazin-5(2H)-one (4a)
Yellow solid (98 %); m.p.: 282–284 °C (ethanol); IR (KBr, cm−1): 3350, 3250 (NH), 3050 (CH furan), 2912, 2838 (CH3), 1663 (C=O), 1620 (C=N), 1558, 1505, 1464 (C=C, δ NH), 1243, 1025 (C–O–C), 738 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 3.82 (s, 3H, OCH3), 6.54–6.58 (m, 1H, furan C4-H), 6.73 (d, J = 3.3 Hz, 1H, furan C3-H), 6.89 (d, J = 16.2 Hz, 1H, ethenyl C1-H), 6.99 (d, J = 8.7 Hz, 2H, methoxyphenyl C3,5-H), 7.74 (s, 1H, furan C5-H), 7.75–7.85 (m, 3H, ethenyl C2-H and methoxyphenyl C2,6-H), 8.05 (s, 1H, N = CH), 11.6, 12.9 (2 × s, each 1H, 2 NH, D2O exchangeable). 13C-NMR (300 MHz, DMSO-d 6) δ 64.76 (OCH3), 121.38 (furan C4), 121.85 (furan C3), 123.55 (methoxyphenyl C3,5), 128.95 (ethenyl C1), 132.12 (ethenyl C2), 136.01 (methoxyphenyl C1), 138.58 (methoxyphenyl C2,6), 152.80 (N = CH), 153.63 (furan C5), 154.51 (furan C2), 160.87 (triazine C6), 161.56 (methoxyphenyl C4), 170.21 (triazine C3), 174.0 (C=O); Anal. Calcd for C17H15N5O3 (337.33): C, 60.53; H, 4.48; N, 20.76. Found: C, 60.19; H, 4.17; N, 20.52.
3-[(E)-2-(4-Chlorobenzylidene)hydrazono]-6-[(E)-2-(furan-2-yl)ethenyl]1,2,4-triazin-5(2H)-one (4b)
Yellow solid (97 %); m.p.: 276–278 °C (ethanol); IR (KBr, cm−1): 3393, 3300 (NH), 3050 (CH furan), 1663 (C=O), 1636 (C=N), 1553, 1505 (C=C, δ NH), 1273, 1017 (C–O–C), 741 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 6.57–6.62 (m, 1H, furan C4-H), 6.76 (d, J = 3.6 Hz, 1H furan C3-H), 6.88 (d, J = 15.9 Hz, 1H, ethenyl C1-H), 7.51 (d, J = 8.4 Hz, 2H, chlorophenyl C3,5-H), 7.76 (s, 1H, furan C5-H), 7.84 (d, J = 15.9 Hz, 1H ethenyl C2-H), 7.98 (d, J = 8.4 Hz, 2H, chlorophenyl C2,6-H), 8.08 (s, 1H, N=CH), 11.9, 13.1 (2 × s, each 1H, 2 NH, D2O exchangeable); Anal. Calcd for C16H12ClN5O2 (341.75): C, 56.23; H, 3.54; N, 20.49; found: C, 56.08; H, 3.54; N, 20.35.
6-[(E)-2-(Furan-2-yl)ethenyl]-3-[(E)-2-(4-nitrobenzylidene)hydrazono]-1,2,4-triazin-5(2H)-one (4c)
Yellow solid (95 %); m.p.: >300 °C (dimethyl formamide); IR (KBr, cm−1): 3361 (NH), 3045 (CH furan), 1675 (C=O), 1621 (C=N), 1587, 1503 (C=C, δ NH), 1558, 1338 (NO2), 1250, 1016 (C–O–C), 738 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.52–6.56 (m, 1H, furan C4-H), 6.69 (d, J = 3.05 Hz, 1H, furan C3-H), 6.84 (d, J = 20 Hz, 1H, ethenyl C1-H), 7.82 (s, 1H, CH = N), 7.84 (d, J = 20 Hz, 1H, ethenyl C2-H), 8.11 (d, J = 8.4 Hz, 2H, nitrophenyl C2,6-H), 8.14 (s, 1H, furan C5-H), 8.22 (d, J = 8.4 Hz, 2H, nitrophenyl C3,5-H), 12.24, 13.40 (2 × s, each 1H, 2 NH, D2O exchangeable); Anal. Calcd for C16H12N6O4.1/2 H2O (361.31): C, 53.19; H, 3.63; N, 23.26; found: C, 53.44; H, 3.50; N, 23.16.
6-[(E)-2-(Furan-2-yl)ethenyl]-3-[(E)-2-{(furan-2-yl)methylene}hydrazono]-1,2,4-triazin-5(2H)-one (4d)
Yellow orange solid (90 %); m.p.: 264–266 °C (dimethyl formamide); IR (KBr, cm−1): 3325, 3182 (NH), 3025 (CH furan), 1663 (C=O), 1615 (C=N), 1563, 1509 (C=C, δ NH), 1275, 1016 (C–O–C), 745 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.55-6.56 (m, 1H, furan C4-H), 6.62–6.63 (m, 1H, furan C4-H), 6.72 (d, J = 3.1 Hz, 1H, furan C3-H), 6.83 (d, J = 16.1 Hz, 1H, ethenyl C1-H), 7.03 (d, J = 3.05 Hz, 1H, furan C3-H), 7.73 (s, 1H, furan C5-H), 7.78 (d, J = 16.05 Hz, 1H, ethenyl C2-H), 7.82 (d, J = 1.6 Hz, 1H, furan C5-H), 7.97 (s, 1H, CH = N), 11.71, 12.88 (2 × s, each 1H, 2 NH, D2O exchangeable); Anal. Calcd for C14H11N5O3. 2H2O (333.30): C, 50.45; H, 4.54; N, 21.01; found: C, 50.06; H, 4.32; N, 21.13.
6-[(E)-2-(Furan-2-yl)ethenyl]-3-[(E)-2-{(5-nitrofuran-2-yl)methylene}hydrazono]-1,2,4-triazin-5(2H)-one (4e)
Orange solid (96 %); m.p.: >300 °C (dioxane); IR (KBr, cm−1): 3400, 3150 (NH), 3025 (CH furan), 1663 (C=O), 1628 (C=N), 1595, 1497 (C=C, δ NH), 1567, 1349 (NO2), 1249, 1018 (C–O-C), 738 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.56–6.57 (m, 1H, furan C4-H), 6.74 (d, J = 3.8 Hz, 1H, furan C3-H), 6.84 (d, J = 16.1 Hz, 1H, ethenyl C1-H), 7.42 (d, J = 3.8 Hz, 1H, nitrofuran C3-H), 7.73 (s, 1H, furan C5-H), 7.78 (d, J = 16.1 Hz,1H, ethenyl C2-H), 7.80 (d, J = 3.8 Hz, 1H, nitrofuran C4-H), 8.01 (s, 1H, CH = N), 12.28, 13.14 (2 × s, each 1H, 2 NH, D2O exchangeable); Anal. Calcd for C14H10N6O5.1H2O: C, 46.67; H, 3.36; N, 23.33; found: C, 46.29; H, 3.07; N, 23.43.
3-[2-{6-((E)-2-(Furan-2-yl)ethenyl)-5-oxo-2,5-dihydro-1,2,4-triazin-3-yl}hydrazono]indolin-2-one (4f)
Orange solid (80 %); m.p.: >300 °C (dimethyl formamide/water); IR (KBr, cm−1): 3357, 3181, 3105 (NH), 3042 (CH furan), 1688 (C=O), 1624 (C=N), 1556, 1491 (C=C, δ NH), 1231, 1046 (C–O–C), 741 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.56–6.59 (m, 1H, furan C4-H), 6.76–6.82 (m, 3H, furan C3-H, isatin C4-H and ethenyl C1-H), 6.94–7.23 (2 × t, J = 7.5 Hz, each 1H, isatin C5,6-H), 7.62 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 7.75 (d, J = 1.6 Hz, 1H, furan C5-H), 8.40–8.45 (m, 1H, isatin C7-H), 10.50, 12.90, 13.9 (3 × s, each 1H, 3 NH, D2O exchangeable); Anal. Calcd for C17H12N6O3·2H2O (384.35): C, 53.12; H, 4.20; N, 21.87; found: C, 52.98; H, 3.87; N, 21.94.
3-[2-{6-((E)-2-(Furan-2-yl)ethenyl)-5-oxo-2,5-dihydro-1,2,4-triazin-3-yl}hydrazono]-1-methylindolin-2-one (4g)
Orange solid (87 %); m.p.: 260–262 °C (dimethyl formamide/water); IR (KBr, cm−1): 3344, 3310(NH), 3024 (CH furan), 2932, 2875 (CH3), 1676 (C=O), 1620 (C=N), 1550, 1520 (C=C, δ NH), 1245, 1042 (C–O–C), 741 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 2.7 (s, 3H, CH3), 6.54–6.60 (m, 1H, furan C4-H), 6.78–6.84 (m, 3H, furan C3-H, isatin C4-H and ethenyl C1-H), 6.93–7.21 (2 × t, J = 7.5 Hz, each 1H, isatin C5,6-H), 7.65 (d, J = 16.2 Hz, 1H, ethenyl C2-H), 7.76 (d, J = 1.6 Hz, 1H, furan C5-H), 8.37–8.42 (m, 1H, isatin C7-H), 10.40, 14.0 (2 × s, each 1H, 2NH, D2O exchangeable); Anal. Calcd for C18H14N6O3 (362.34): C, 59.67; H, 3.89; N, 23.19; found: C, 59.45; H, 3.64; N, 23.04.
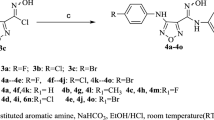
3-(5-Amino-3-aryl-1H-pyrazol-1-yl)-6-[(E)-2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-(2H)-ones (5a–d)
A solution of the hydrazine 3 (0.48 gm, 2.2 mmol) and the selected phenacyl cyanide (2.2 mmol) in ethanol/glacial acetic acid mixture (10 ml) (4:1) was refluxed for 3 h. The reaction mixture was cooled and the precipitated product was filtered, washed with ethanol, dried and crystallized from dioxane.
(E)-3-(5-Amino-3-phenyl-1H-pyrazol-1-yl)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-(2H)-one (5a)
Pale yellow solid (80 %); m.p.: 262–264 °C; IR (KBr, cm−1): 3418, 3344, 3306, 3178 (NH), 3058 (CH furan), 1660 (C=O), 1617(C=N), 1576, 1546 (C=C, δ NH), 1245, 1016 (C–O–C), 752 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 5.92 (s, 1H, pyrazole C4-H), 6.60–6.65 (m, 1H, furan C4-H), 6.86 (d, J = 3.6 Hz, 1H furan C3-H), 6.93 (d, J = 16.2 Hz, 1H, ethenyl C1-H), 7.1 (br.s, 2H, NH2, D2O exchangeable), 7.40–7.50 (m, 3H, phenyl C3,4,5-H), 7.81 (d, J = 1.5 Hz, 1H, furan C5-H), 7.90–7.97 (m, 3H, ethenyl C2-H and phenyl C2,6-H), 13.9 (br.s, 1H, NH, D2O exchangeable); Anal. Calcd for C18H14N6O2 (346.34): C, 62.42; H, 4.07; N, 24.27; found: C, 62.27; H, 3.77; N, 24.0.
(E)-3-[5-Amino-3-(4-methylphenyl)-1H-pyrazol-1-yl]-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-(2H)-one (5b)
Yellow solid (60 %); m.p.: 272–274 °C; IR (KBr, cm−1): 3398, 3347, 3301 (NH), 3023 (CH furan), 2918 (CH3), 1680 (C=O), 1624 (C=N), 1548, 1486 (C=C, δ NH), 1248, 1012 (C–O–C), 743 (oop furan); 1H-NMR (300 MHz, DMSO-d 6) δ: 2.35 (s, 3H, CH3), 5.88 (s, 1H, pyrazole C4-H), 6.56-6.62 (m, 1H, furan C4-H), 6.88 (d, J = 3.6 Hz, 1H furan C3-H), 6.93 (d, J = 16.2 Hz, 1H, ethenyl C1-H), 7.1 (br.s, 2H, NH2, D2O exchangeable), 7.27 (d, J = 8.2 Hz, 2H, methylphenyl C3,5-H), 7.82–7.89 (m, 4H, furan C5-H, ethenyl C2-H and methylphenyl C2,6-H), 13.9 (br.s, 1H, NH, D2O exchangeable); Anal. Calcd for C19H16N6O2 (360.37): C, 63.32; H, 4.48; N, 23.32; found: C, 63.65; H, 4.27; N, 23.13.
(E)-3-[5-Amino-3-(4-chlorophenyl)-1H-pyrazol-1-yl]-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-(2H)-one (5c)
Pale yellow solid (85 %); m.p.: 280–282 °C; IR (KBr, cm−1): 3415, 3300, 3177 (NH), 3025 (CH furan), 1658 (C=O), 1620 (C=N), 1546, 1488 (C=C, δ NH), 1250, 1016 (C–O–C), 762 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 5.89 (s, 1H, pyrazole C4-H), 6.58–6.61 (m, 1H, furan C4-H), 6.81 (d, J = 3.8 Hz, 1H, furan C3-H), 6.89 (d, J = 16.8 Hz, 1H, ethenyl C1-H), 7.07 (br.s, 2H, NH2, D2O exchangeable), 7.48 (d, J = 8.4 Hz, 2H, chlorophenyl C3,5-H), 7.76 (s, 1H, furan C5-H), 7.87 (d, J = 16.8 Hz, 1H, ethenyl C2-H), 7.94 (d, J = 7.3 Hz, 2H, chlorophenyl C2,6-H), 13.98 (s, 1H, NH, D2O exchangeable); 13C-NMR (500 MHz, DMSO-d 6) δ: 85 (pyrazole C4), 112.51 (furan C4), 112.92 (ethenyl C1), 118.41 (furan C3), 124.11(ethenyl C2), 127.83 (chlorophenyl C2,6), 128.56 (chlorophenyl C3,5), 130.70 (chlorophenyl C1), 133.60 (chlorophenyl C4), 140.01 (triazine C6), 144.65 (furan C5), 145.58 (pyrazole C3), 149.93 (furan C2), 151.84 (pyrazole C5), 152.02 (triazine C3), 153.02 (C=O); Anal. Calcd for C18H13ClN6O2 (380.79): C, 56.78; H, 3.44; N, 22.07; found: C, 56.75; H, 3.74; N, 22.02.
(E)-3-[5-Amino-3-(4-bromophenyl)-1H-pyrazol-1-yl]-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-(2H)-one (5d)
Yellowish white solid (78 %); m.p.: 288–290 °C; IR (KBr, cm−1): 3396, 3361, 3294 (NH), 3023 (CH furan), 1662 (C=O), 1624(C=N), 1548, 1488 (C=C, δ NH), 1249, 1012 (C–O–C), 753 (oop furan). 1H-NMR (500 MHz, DMSO-d 6) δ: 5.89 (s, 1H, pyrazole C4-H), 6.58–6.62 (m, 1H, furan C4-H), 6.83 (d, J = 3.8 Hz, 1H, furan C3-H), 6.90 (d, J = 16.4 Hz, 1H, ethenyl C1-H), 7.06 (br.s, 2H, NH2, D2O exchangeable), 7.43 (d, J = 7.3 Hz, 2H, bromophenyl C3,5-H), 7.75 (s, 1H, furan C5-H), 7.89 (d, J = 16.4 Hz, 1H, ethenyl C2-H), 7.90 (d, J = 7.3 Hz, 2H, bromophenyl C2,6-H), 13.95 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C18H13BrN6O2 (425.24): C, 50.84; H, 3.08; N, 19.76; found: C, 50.93; H, 2.82; N, 19.62.
(E)-3-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(2H)-one (6)
A mixture of the hydrazine 3 (0.2 g, 0.9 mmol) and acetylacetone (0.1 g, 0.11 ml, 1 mmol) in absolute ethanol was heated under reflux for 3 h. The reaction mixture was cooled, and the formed precipitate was filtered, washed with ethanol, dried and crystallized from ethanol. Shiny yellow crystals (68 %); M.p. 180–182 °C; IR (KBr, cm−1): 3288, 3116 (NH), 3069 (CH furan), 2988, 2923 (CH3), 1676 (C=O), 1619 (C=N), 1588, 1558, 1540, 1493 (C=C, δ NH), 1239, 1018 (C–O–C), 746 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 2.22, 2.46 (2 × s, each 3H, pyrazole CH3), 6.24 (s, 1H, pyrazole C4-H), 6.54–6.57 (m, 1H, furan C4-H), 6.80 (d, 1H, J = 2.3 Hz, furan C3-H), 6.86 (d, J = 16.1 Hz, 1H, ethenyl C1-H), 7.76 (s, 1H, furan C5-H), 7.86 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 14.16 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C14H13N5O2 (283.29): C, 59.36; H, 4.63; N, 24.72. Found: C, 59.27; H, 4.92; N, 24.42.
(E)-3-(3,5-Dioxopyrazolidin-1-yl)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(2H)-one (7)
A mixture of the hydrazine 3 (0.2 g, 0.9 mmol) and diethyl malonate (0.16 g, 0.15 ml, 1 mmol) in a mixture of ethanol/glacial acetic acid (10 ml) (1:1) was heated under reflux for 8 h and then cooled. The precipitated solid was filtered, washed with ethanol, dried and crystallized from glacial acetic acid. Red crystals (57 %); m.p.: 165–167 °C; IR (KBr, cm−1): 3341, 3101 (NH), 3059 (CH furan), 2903 (CH2), 1707, 1662 (C=O), 1628 (C=N), 1583, 1552, 1500 (C=C, δ NH), 1205, 1029 (C–O–C), 761 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 2.45 (s, 2H, pyrazolidine C4-H), 3.36 (br.s., 1H, pyrazolidine NH), 6.56–6.60 (m, 1H, furan C4-H), 6.87 (d, J = 3.8 Hz, 1H,furan C3-H), 7.0 (d, J = 16.3 Hz, 1H, ethenyl C1-H), 7.79 (s, 1H, furan C5-H), 7.86 (d, J = 16.3 Hz, 1H, ethenyl C2-H), 8.70 (s, 1H, NH, D2O exchangeable), 13.59 (s, 1H, triazine NH, D2O exchangeable); Anal. Calcd for C12H9N5O4 (287.23): C, 50.18; H, 3.16; N, 24.38; found: C, 49.77; H, 3.49; N, 24.56.
(E)-Ethyl 3-[2-{6-(2-(furan-2-yl)ethenyl)-5-oxo-2,5-dihydro-1,2,4-triazin-3-yl} hyrazono]butanoate (8)
A mixture of the hydrazine 3 (0.2 g, 0.9 mmol) and ethyl acetoacetate (0.13 g, 0.12 ml, 1 mmol) in absolute ethanol was heated under reflux for 3 h. The reaction mixture was left to cool to room temperature and the obtained product was filtered, washed with ethanol, dried and crystallized from ethanol. Pale yellow crystals (76 %); m.p.: 156–158 °C; IR (KBr, cm−1): 3296, 3100 (NH), 3038 (CH furan), 2986, 2901 (CH2, CH3), 1732, 1685 (C=O), 1628 (C=N), 1572, 1550, 1500 (C=C, δ NH), 1185, 1016 (C–O–C), 742 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 1.16 (t, J = 6.9 Hz, 3H, CH2-CH 3 ), 1.94 (s, 3H, CH3), 4.07 (q, J = 6.9 Hz, 2H, CH 2 -CH3), 6.50–6.54 (m, 1H, furan C4-H), 6.70 (d, J = 3.1 Hz, 1H, furan C3-H), 6.81 (d, J = 16.1 Hz, 1H, ethenyl C1-H), 7.71 (s, 1H, furan C5-H), 7.77 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 10.72, 12.74 (2 × s, each 1H, 2 NH, D2O exchangeable); Anal. Calcd for C15H17N5O4 (331.33): C, 54.38; H, 5.17; N, 21.14 Found: C, 54.04; H, 5.51; N, 20.78.
(E)-6-[(2-(Furan-2-yl)ethenyl]-3-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-1,2,4-triazin-5(2H)-one (9)
Method A Compound 8 (0.5 g, 1.5 mmol) was heated in an oil bath at 160 °C for 20 min. The reaction mixture was left to cool and the obtained product was triturated with EtOH, filtered, dried, and crystallized from dimethyl formamide/water. Reddish brown solid (72 %).
Method B The hydrazine derivative 3 (0.2 g, 0.9 mmol) was heated with ethyl acetoacetate (0.13 g, 0.12 ml, 1 mmol) in an oil bath at 160 °C for 1 h. The reaction mixture was left to cool and the solidified residue was triturated with ethanol, filtered, dried, and crystallized from dimethyl formamide/water. (65 %); m.p.: 190–192 °C; IR (KBr, cm−1): 3433 (NH), 3066 (CH furan), 2923 (CH3), 1679 (C=O), 1637 (C=N), 1542, 1487 (C=C, δ NH), 1210, 1012 (C–O–C), 743 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 2.47 (s, 3H, pyrazoline C3-CH3), 3.44 (s, 2H, pyrazoline C4-H), 6.58–6.61 (m, 1H, furan C4-H), 6.81 (d, J = 3.1 Hz, 1H, furan C3-H), 6.86 (d, J = 16.1 Hz, 1H, ethenyl C1-H), 7.77 (s, 1H, furan C5-H), 7.89 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 13.38 (s, 1H, NH, D2O exchangeable); MS (m/z, %): 285 (M+, 21.0), 98 (100.0); Anal. Calcd for C13H11N5O3·½H2O (294.27): C, 53.06; H, 4.11; N, 23.80; found: C, 52.91; H, 4.29; N, 23.96.
(2Z, 3E)-4-(Furan-2-yl)-2-[2-(thiocarbohydrazono)]butenoic acid (11)
A mixture of the 2-furylidene pyruvic acid 10 (5 g, 30 mmol) and thiocarbohydrazide (3.2 g, 30 mmol) in a mixture of ethanol (20 ml) and glacial acetic acid (0.5 ml) was stirred at room temperature for 15 min. It was filtered, washed with water, dried and crystallized from dimethyl formamide/water. Orange red solid (79 %); m.p.: 238–240 °C; IR (KBr, cm−1): 3437, 3290, 3200 (OH, NH), 3078 (CH furan), 1661 (C=O), 1626 (C=N), 1568, 1551, 1501 (C=C, δ NH), 1534, 1277, 1119, 965 (N–C=S), 1232, 1016 (C–O–C), 753 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.50 (s, 2H, NH2, D2O exchangeable), 6.55–6.59 (m, 1H, furan C4-H), 6.78–6.83 (m, 2H, furan C3-H and ethenyl C1-H), 7.64 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 7.77 (s, 1H, furan C5-H), 9.68, 10.30 (2s, each 1H, 2NH, D2O exchangeable); Anal. Calcd for C9H10N4O3S (254.27): C, 42.51; H, 3.96; N, 22.03; found: C, 42.74; H, 3.95; N, 22.35.
(E)-4-Amino-6-[2-(furan-2-yl)ethenyl]-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (12)
A solution of the thiocarbohydrazone 11 (2.5 g, 10 mmol) in 1 N NaOH (20 ml) was heated for 20 min. After cooling, the solution was acidified with dil. H2SO4 to pH 6. The formed precipitate was filtered, washed with water, dried, and crystallized from dioxane.
Pale brown crystals (93 %); m.p.: 230–232 °C; IR (KBr, cm−1): 3437, 3254 (NH), 3078 (CH furan), 1660 (C=O), 1625 (C=N), 1593, 1517, 1499 (C=C, δ NH), 1542, 1278, 1104, 961 (N–C=S), 1235, 1013 (C–O–C), 754 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.51 (s, 2H, NH2, D2O exchangeable), 6.55–6.59 (m, 1H, furan C4-H), 6.78–6.83 (m, 2H, furan C3-H and ethenyl C1-H), 7.64 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 7.76 (s, 1H, furan C5-H), 13.90 (s, 1H, NH, D2O exchangeable); Anal. Calcd for C9H8N4O2S (236.25): C, 45.75; H, 3.41; N, 23.72; found: C, 46.02; H, 3.64; N, 23.47.
4-Amino-6-[(E)-2-(furan-2-yl)ethenyl]-3-(substituted sulfanyl)-1,2,4-triazin-5(4H)-ones (13a–c)
A mixture of the thione 12 (0.38 g, 1.6 mmol), anhydrous K2CO3 (0.33 g, 2.4 mmol) and dimethylsulfate, ethyl iodide or benzyl chloride (2.4 mmol) in dry dimethyl formamide (4 ml) was stirred at room temperature for 12 h. The reaction mixture was then diluted with water and the obtained product was filtered, washed with water, dried, and crystallized from ethanol.
(E)-4-Amino-6-[2-(furan-2-yl)ethenyl]-3-(methylsulfanyl)-1,2,4-triazin-5(4H)-one (13a)
Yellow solid (72 %); m.p.: 230–232 °C; IR (KBr, cm−1): 3242, 3180 (NH2), 3075 (CH furan), 2947 (CH3), 1661(C=O), 1621 (C=N), 1544, 1494 (C=C, δ NH), 1286, 1078 (C–S–C), 1231, 1010 (C–O–C), 755 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ 3.32 (s, 3H, CH3, under DMSO), 6.50 (s, 2H, NH2, D2O exchangeable), 6.57 (dd, J = 3.8, 1.55 Hz, 1H, furan C4-H), 6.80 (d, J = 3.8 Hz, 1H, furan C3-H), 6.81, 7.64 (2 × d, J = 16.05 Hz, each 1H, ethenyl C1-H and ethenyl C2-H), 7.77 (s, 1H, furan C5-H); Anal. Calcd for C10H10N4O2S (250.28): C, 47.99; H, 4.03; N, 22.39; S, 12.81. Found: C, 47.65; H, 4.39; N, 22.07; S, 13.12.
(E)-4-Amino-3-(ethylsulfanyl)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(4H)-one (13b)
Brown solid (55 %); m.p.: 118–120 °C; IR (KBr, cm−1): 3293, 3201 (NH2), 3075 (CH furan), 2973, 2926 (CH2, CH3), 1676 (C=O), 1624 (C=N), 1536, 1486 (C=C, δ NH), 1287, 1072 (C–S-C), 1231, 1018 (C–O–C), 737(oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 1.33 (t, J = 7.0 Hz, 3H, CH2–CH 3 ), 4.45 (q, J = 7.0 Hz, 2H, CH 2–CH3), 6.59 (dd, J = 3.1, 1.5 Hz, 1H, furan C4-H), 6.67 (s, 2H, NH2, D2O exchangeable), 6.82–6.85 (m, 2H, ethenyl C1-H and furan C3-H), 7.71 (d, J = 16.8 Hz, 1H, ethenyl C2-H), 7.78 (d, J = 1.6 Hz, 1H, furan C5-H); Anal. Calcd for C11H12N4O2S.1/2 H2O (273.31): C, 48.34; H, 4.79; N, 20.50; S, 11.73; found: C, 48.66; H, 4.39; N, 20.17; S, 11.64.
(E)-4-Amino-3-(benzylsulfanyl)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5(4H)-one (13c)
Pale yellow solid (60 %); m.p.: 142–144 °C; IR (KBr, cm−1): 3290, 3190 (NH2), 3025 (CH furan), 2947(CH2), 1676 (C=O), 1629 (C=N), 1563, 1530, 1484 (C=C, δ NH), 1291, 1081 (C–S–C), 1253, 1015 (C–O–C), 730 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 5.65 (s, 2H, S-CH2), 6.59 (dd, J = 3.4, 1.93 Hz, 1H, furan C4-H), 6.66 (s, 2H, NH2, D2O exchangeable), 6.82–6.86 (m, 2H, ethenyl C1-H and furan C3-H), 7.24–7.37 (m, 5H, phenyl-H), 7.71 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 7.78 (s, 1H, furan C5-H); 13C-NMR (300 MHz, DMSO-d 6) δ: 44.21 (CH2), 123.01 (furan C4), 124.97 (furan C3), 127.0 (ethenyl C1), 128.50 (ethenyl C2), 134.29 (phenyl C4), 137.14 (phenyl C3,5), 139.13 (phenyl C2,6), 143.8 (phenyl C1), 145.71 (furan C5), 152.85 (furan C2), 154.48 (triazine C3), 158.0 (triazine C6), 163.0 (C=O); Anal. Calcd for C16H14N4O2S (326.37): C, 58.88; H, 4.32; N, 17.17; found: C, 58.64; H, 4.07; N, 16.88.
6-[(E)-2-(Furan-2-yl)ethenyl]-4-[(E)(4-nitrobenzylidene)amino]-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (14)
A mixture of the amino thione 12 (0.5 g, 2.1 mmol) and 4-nitrobenzaldehyde (0.32 g, 2.1 mmol) in absolute ethanol (20 ml) containing few drops of glacial acetic acid was heated under reflux for 1 h. The reaction mixture was left to cool to room temperature and the formed precipitate was filtered, washed with ethanol, dried and crystallized from glacial acetic acid. Yellow solid (61 %); m.p.: 256-258 °C. IR (KBr, cm−1): 3207 (NH), 3094 (CH furan), 1701 (C=O), 1620 (C=N), 1595, 1525, 1482 (C=C, δNH), 1520, 1260, 1137, 973 (N–C=S), 1525, 1342 (NO2), 1260, 1015 (C–O–C), 747 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 6.56–6.59 (m, 1H, furan C4-H), 6.81 (d, J = 3.8 Hz, 1H, furan C3-H), 6.84, 7.63 (2 × d, J = 16.1 Hz, each 1H, ethenyl C1-H and ethenyl C2-H), 7.78 (s, 1H, furan C5-H), 8.18, 8.39 (2 × d, J = 8.4 Hz, each 2H, nitrophenyl C2,6-H and nitrophenyl C3,5-H), 8.89 (s, 1H, N = CH), 14.06 (s, 1H, NH, D2O exchangeable); MS (m/z, %): 369 (M+, 20.5), 119 (100.0). Anal. Calcd for C16H11N5O4S (369.35): C, 52.03; H, 3.00; N, 18.96; found: C, 52.35; H, 3.06; N, 18.62.
6-[(E)-2-(Furan-2-yl)ethenyl]-4-[(E)(4-nitrobenzylidene)amino]-3-[(2-substituted ethyl)sulfanyl]-1,2,4-triazin-5(4H)-ones (15a-c)
To a suspension of compound 14 (0.48 g, 1.3 mmol) in aqueous KOH solution (0.22 g, 2 ml, 4 mmol), the appropriate alkylating agent (1.3 mmol) was added. The reaction mixture was stirred at room temperature for 24 h then acidified with glacial acetic acid till pH 6. The separated solid product was filtered, dried, and crystallized from the proper solvent.
6-[(E)-2-(Furan-2-yl)ethenyl]-3-[(2-(morpholin-1-yl)ethyl)sulfanyl]-4-[(E)-(4-nitrobenzylidene)amino]-1,2,4-triazin-5(4H)-one (15a)
Orange solid (52 %); m.p.: 132–134 °C (dimethylformamide); IR (KBr, cm−1): 3068 (CH furan), 2924, 2853, (CH2), 1686 (C=O), 1620 (C=N), 1591, 1557, 1491 (C=C), 1523, 1343 (NO2), 1262, 1069 (C–S–C), 1220, 1012 (morpholine and furan C–O–C), 745 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 2.63 (t, J = 6.9 Hz, 2H, S-CH2), 3.16 (t, J = 6.9 Hz, 2H, N-CH2), 3.54–3.58 (m, 8H, morpholine C2,3,5,6-H2), 6.56–6.59 (m, 1H, furan C4-H), 6.74 (d, J = 3.1 Hz, 1H, furan C3-H), 6.85, 7.54 (2 × d, J = 16.1 Hz, each 1H, ethenyl C1-H and ethenyl C2-H), 7.74 (s, 1H, furan C5-H), 8.12, 8.34 (2 × d, J = 8.4 Hz, each 2H, nitrophenyl C2,6-H and nitrophenyl C3,5-H), 8.99 (s, 1H, N = CH); Anal. Calcd for C22H22N6O5S (482.51): C, 54.76; H, 4.60; N, 17.42; S, 6.65; found: C, 54.39; H, 4.79; N, 17.69; S, 6.75.
6-[(E)-2-(Furan-2-yl)ethenyl]-4-[(E)-(4-nitrobenzylidene)amino]-3-[(2-(piperidin-1-yl)ethyl)sulfanyl]-1,2,4-triazin-5(4H)-one (15b)
Brown solid (60 %); m.p.: 141–143 °C (ethanol/water); IR (KBr, cm−1): 3035 (CH furan), 2933, 2850 (CH2), 1690 (C=O), 1619 (C=N), 1591, 1523, 1497, (C=C), 1523, 1344 (NO2), 1280, 1073 (C–S–C), 1226, 1016 (C–O–C), 757 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 1.39–1.50 (m, 6H, piperidine C3,4,5-H2), 1.73–1.79 (m, 4H, piperidine C2,6-H2), 2.67 (m, 2H, SCH2), 2.89 (m, 2H, NCH2), 6.60–6.66 (m, 1H, furan C4-H), 6.87 (d, J = 3.0 Hz, 1H, furan C3-H), 7.18 (d, J = 16.5 Hz, 1H, ethenyl C1-H), 7.82 (s, 1H, furan C5-H), 7.87 (d, J = 16.5 Hz, 1H, ethenyl C2-H), 8.20, 8.42 (2 × d, J = 8.7 Hz, each 2H, nitrophenyl C2,6-H and C3,5-H), 9.58 (s, 1H, N=CH); 13C-NMR (300 MHz, DMSO-d 6) δ: 28.0 (SCH2), 30.71 (piperidine C4), 31.92 (piperidine C3,5), 58.20 (NCH2), 61.65 (piperidine C2,6), 122.01 (furan C4), 122.80 (furan C3), 133.92 (nitrophenyl C3,5), 135.6 (ethenyl C1), 138.3 (ethenyl C2), 139.57 (nitrophenyl C2,6), 144.30 (nitrophenyl C1), 148.5 (N=CH), 152.0 (furan C5), 154.0 (nitrophenyl C4), 155.0 (furan C2), 158.32 (triazine C3), 161.46 (triazine C6), 165.29 (C=O); Anal. Calcd for C23H24N6O4S (480.54): C, 57.49; H, 5.03; N, 17.49; S, 6.67; found: C, 57.19; H, 4.79; N, 17.46; S, 6.58.
6-[(E)-2-(Furan-2-yl)ethenyl]-4-[(E)-(4-nitrobenzylidene)amino]-3-[(2-(pyrrolidin-1-yl)ethyl)sulfanyl]-1,2,4-triazin-5(4H)-one (15c)
Orange brown solid (40 %); m.p.: 120–122 °C (methanol/water); IR (KBr, cm−1): 3050 (CH furan), 2926, 2854 (CH2), 1687 (C=O), 1620 (C=N), 1591, 1558, 1493 (C=C), 1523, 1343 (NO2), 1260, 1075 (C–S–C), 1224, 1014 (C–O–C), 746 (oop furan); 1H-NMR (500 MHz, DMSO-d 6) δ: 1.87–1.89 (m, 4H, pyrrolidine C3,4-H2), 2.97–3.20 (m, 4H, pyrrolidine C2,5-H2), 3.37 (t, J = 6.9 Hz, 2H, S-CH2), 3.38 (t, J = 6.9 Hz, 2H, N-CH2), 6.60–6.63 (m, 1H, furan C4-H), 6.84 (d, 1H, J = 3.1 Hz, furan C3-H), 7.08 (d, J = 16.1 Hz, 1H, ethenyl C1-H), 7.80 (s, 1H, furan C5-H), 7.83 (d, J = 16.1 Hz, 1H, ethenyl C2-H), 8.18, 8.40 (2 × d, J = 8.4 Hz, each 2H, nitrophenyl C2,6-H and nitrophenyl C3,5-H), 9.50 (s, 1H, N=CH); Anal. Calcd for C22H22N6O4S (466.51): C, 56.64; H, 4.75; N, 18.01; S, 6.87; found: C, 56.29; H, 4.79; N, 17.70; S, 7.12.
In vitro antitumor screening
Preliminary in vitro one-dose antitumor screening
Anti-tumor activity screening for compounds 2c, 5c, 7, 12, 14, and 15a at a dose of 10 μM utilizing 55 different human tumor cell lines, representing leukemia, melanoma and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney was carried out according to standard procedure (Skehan et al., 1990; Rubinstein et al., 1990). The human tumor cell lines of the cancer screening panel are grown in RPMI 1640 medium containing 5 % fetal bovine serum and 2 mmol l-glutamine. For a typical screening experiment, the tumor cells were inoculated into 96-well microtiter plates in 100 μl at plating densities ranging from 5,000 to 40,000 cells/well. Density of the inoculum depends on the type of tumor cell and its growth characteristics. After cell inoculation, the microtiter plates were incubated at 37 °C, 5 % CO2, 95 % air, and 100 % relative humidity for 24 h prior to addition of experimental drugs. After 24 h, two plates of each cell lines were fixed in situ with trichloroacetic acid (TCA), to represent a measurement of the cell population for each cell line at the time of test compound addition (time zero, Tz). Tested compounds were solubilized in dimethyl sulfoxide at 400-fold the desired final maximum test concentration and stored frozen prior to use. At the time of test compound addition, an aliquot of frozen concentrate was thawed and diluted to twice the desired final maximum test concentration with complete medium containing 50 μg/ml gentamicin. The percentage growth of the tumor cells were calculated relative to time zero.
Full in vitro five-dose antitumor assay
For compounds passed on for the five-dose assay, the compounds were tested at five different concentrations (10−4, 10−5, 10−6, 10−7, and 10−8 M). Following drug addition, the plates were incubated for an additional 48 h at 37 °C, 5 % CO2, 95 % air, and 100 % relative humidity. The cells were assayed by using the sulforhodamine B assay. Sulforhodamine B (SRB) solution (100 μl) at 0.4 % (w/v) in 1 % acetic acid was added to each well, and plates were incubated for 10 min at room temperature. After staining, unbound dye was removed by washing five times with 1 % acetic acid and the plates were air dried. Bound stain was subsequently solubilized with 10 mM trizma base, and the absorbance was read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology is the same except that the assay was terminated by fixing settled cells at the bottom of the wells by gently adding 50 μl of 80 % TCA.
References
Acton EM, Narayanan VL, Risbood PA, Shoemaker RH, Vistica DT, Boyd MR (1994) Anticancer specificity of some ellipticinium salts against human brain tumors in vitro. J Med Chem 37:2185–2189
Ashour HMA, Abdel Wahab AE (2009) Synthesis and biological evaluation of novel pyrazoles and pyrazolo[3,4-d]pyrimidines incorporating a benzenesulfonamide moiety. Arch Pharm Chem Life Sci 342:238–252
Boyd MR, Paull KD (1995) Practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Rev Res 34:91–109
Braña MF, Ramos A (2001) Naphthalimides as anticancer agents: synthesis and biological activity. Curr Med Chem Anti Cancer Agents 1:237–255
Chou L-C, Huang L-J, Yang J-S, Lee F-Y, Teng C-M, Kuo S-C (2007) Synthesis of furopyrazole analogs of 1-benzyl-3-(5-hydroxymethyl-2-furyl)indazole (YC-1) as novel antileukemic agents. Bioorg Med Chem 15:1732–1740
Cocco MT, Congiu C, Onnis V (2000) Synthesis and antitumor activity of 4-hydroxy-2-pyridone derivatives. Eur J Med Chem 35:545–552
Cozzi P (2003) The discovery of a new potential anticancer drug: a case history. Il Farmaco 58:213–220
Creasey WA, Fink ME, Handschumacher RE, Calabresi P (1963) Clinical and pharmacological studies with 2′,3′,5′-triacetyl-6-azauridine. Cancer Res 23:444–453
Daidone G, Maggio B, Raffa D, Plescia S, Schillaci D, Raimondi MV (2004a) Synthesis and in vitro antileukemic activity of new 4-triazenopyrazole derivatives. Il Farmaco 59:413–417
Daidone G, Raffa D, Maggio B, Raimondi MV, Plescia F, Schillaci D (2004b) Synthesis and antiproliferative activity of triazenoindoles and triazenopyrazoles: a comparative study. Eur J Med Chem 39:219–224
Farag AM, Mayhoub AS, Barakat SE, Bayomi AH (2008) Regioselective synthesis and antitumor screening of some novel N-phenyl pyrazole derivatives. Bioorg Med Chem 16:881–889
Gucky T, Frysova I, Slouka J, Hajduch M, Dzubak P (2009) Cyclocondensation reaction of heterocyclic carbonyl compounds, Part XIII: synthesis and cytotoxic activity of some 3,7-diaryl-5-(3,4,5-trimethoxyphenyl)pyrazolo[4,3-e][1,2,4]triazines. Eur J Med Chem 44:891–900
Gulerman NN, Dogan HN, Rollas S, Johansson C, Celik C (2001) Synthesis and structure elucidation of some new thioether derivatives of 1,2,4-triazoline-3-thiones and their antimicrobial activities. Il Farmaco 56:953–958
Huang S, Lin R, Yu Y et al (2007) Synthesis of 3-(1H-benzimdazol-2-yl)-5-isoquinolin-4-yl-pyrazolo[1,2-b]pyridine, a potent cyclin dependent kinase 1 (CDK 1) inhibitor. Bioorg Med Chem Lett 17:1243–1245
Khalil AA, Abdel Hamide SG, Al-Obaid AM, El-Subbagh HI (2003) Substituted quinazolines, Part 2. Synthesis and in vitro anticancer evaluation of new 2-substituted mercapto-3H-quinazoline analogs. Arch Pharm Chem Life Sci 336:95–103
Krauth F, Dahse H, Ruttinger H, Frohberg P (2010) Synthesis and characterization of novel 1,2,4-triazine derivatives with antiproliferative activity. Bioorg Med Chem 18:1816–1821
Li J, Zhao YF, Zhao XL, Yuan XY, Gong P (2006) Synthesis and anti-tumor activities of novel pyrazolo[1,5-a] pyrimidines. Arch Pharm Chem Life Sci 339:593–597
Manetti F, Est JA, Clotet-Codina I et al (2005) Parallel solution-phase and microwave-assisted synthesis of new S-DABO derivatives endowed with subnanomolar anti-HIV-1 activity. J Med Chem 48:8000–8008
Manetti F, Brullo C, Magnani M et al (2008) Structure-based optimization of pyrazolo[3,4-d]pyrimidines as Abl inhibitors and antiproliferative agents toward human leukemia cell lines. J Med Chem 51:1252–1259
Monks A, Scudiero DA, Skehan P et al (1991) Feasibility of a high flux anticancer drug screen utilizing a derive panel of human tumor cell lines in culture. J Natl Cancer Inst 83:757–766
Osman SAM, Swellem RH, El-Shehry MF (2007) 6-(2-Furylvinyl)-1,2,4-triazinone derivatives as a source of polyfunctional mono-and biheterocycles. Egypt J Chem Special Issue (M.Sidky):91–101
Pal chykovska LH, Platonov MO, Alexeeva IV, Shved AD (2004) Design of the potential transcription inhibitors based on the 6-azacytosine and 6-aza-iso-cytosine. Nonempirical quantum chemical analysis, synthesis and physico-chemical studies. Biopolimery I Kletka 20:131–142
Pretsch E, Buhlmann P, Affolter C (2000) Structure determination of organic compounds. Springer, Berlin
Remers WA (2004) Antineoplastic agents. In: Block JH, Beale JM (eds) Wilson and Gisvold’s text book of organic medicinal and pharmaceutical chemistry, 11th edn. Lippincott Williams and Wilkins, Philadelphia, pp 390–453
Rohmer H (1898) Ueber Condensationen des Furfurols und Furfuracroleïns. Ber Dtsch Chern Ges 31:281–284
Rostom SAF, Ashour HMA, Abd El Razik HA (2009) Synthesis and biological evaluation of some novel polysubstituted pyrimidine derivatives as potential antimicrobial and anticancer agents. Arch Pharm Chem Life Sci 342:299–310
Rostom SAF, Badr MH, Abd El Razik HA, Ashour HMA, Abdel Wahab AE (2011) Synthesis of some pyrazolines and polymethoxy chalcones as anticancer and antimicrobial agents. Arch Pharm Chem Life Sci 344:572–587
Rubinstein LV, Shoemaker RH, Paull KD et al (1990) Comparison of in vitro anticancer-drug-screening data generated with a tetrazonium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst 82:1113–1117
Sangshetti JN, Shinde DB (2010) One pot synthesis and SAR of some novel 3-substituted-5,6-diaryl-1,2,4-triazines as antifungal agents. Bioorg Med Chem Lett 20:742–745
Schenone S, Bruno O, Ranise A, Bondavalli F et al (2004) New pyrazolo[3,4-d]pyrimidines endowed with A431 antiproliferative activity and inhibitory properties of Src phosphorylation. Bioorg Med Chem Lett 14:2511–2517
Sidwell RW, Dixon GJ, Sellers SM, Schabel FM (1968) In vivo antiviral properties of biologically active compounds: II. Studies with influenza and vaccinia viruses. Appl Microbiol 16:370–392
Skehan P, Storeng R, Scudiero DA et al (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112
Slouka J (1962) Die Synthese einiger ungesättigter Derivate von 6-Azauracil. J Für Prakt Chem 4:220–224
Warshakoon NC, Wu S, Boyer A et al (2006) Design and synthesis of a series of novel pyrazolopyridines as HIF 1-α prolyl hydroxylase inhibitors. Bioorg Med Chem Lett 16:5687–5690
Weislow OW, Kiser R, Fine DL, Bader J, Shoemaker RH, Boyd MR (1989) New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J Natl Cancer Inst 81:577–586
Williams DH, Fleming I (1980) Spectroscopic methods in organic chemistry. McGraw-Hill, London, pp 143–145
Xia Y, Dong Z-W, Zhao B-X et al (2007) Synthesis and structure-activity relationships of novel 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide derivatives as potential agents against A 549 lung cancer cells. Bioorg Med Chem 15:6893–6899
Xia Y, Fan C-D, Zhao B-X, Zhao J, Shin D-S, Miao J-Y (2008) Synthesis and structure-activity relationships of novel 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide hydrazone derivatives as potential agents against A 549 lung cancer cells. Eur J Med Chem 43:2347–2353
Zhu G-D, Gong J, Gandhi VB et al (2007) Design and synthesis of pyridine-pyrazolopyridine-based inhibitors of protein kinase B/Akt. Bioorg Med Chem 15:2441–2452
Acknowledgments
The authors are deeply thankful to the staff members of the Department of Health and Human Services, National Cancer Institute (NCI), Bethesda, Maryland, USA for carrying out the anticancer screening of the newly synthesized compounds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ashour, H.M., El-Wakil, M.H., Khalil, M.A. et al. Synthesis of some (E)-6-[2-(furan-2-yl)ethenyl]-1,2,4-triazin-5-ones and their biological evaluation as antitumor agents. Med Chem Res 22, 1909–1924 (2013). https://doi.org/10.1007/s00044-012-0192-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-012-0192-x