
Abstract
A review of the multidisciplinary scientific literature reveals a large variety of amyloid-β (Aβ) oligomeric species, differing in molecular weight, conformation and morphology. These species, which may assemble via either on‐ or off-aggregation pathways, exhibit differences in stability, function and neurotoxicity, according to different experimental settings. The conformations of the different Aβ species are stabilized by intra- and inter-molecular hydrogen bonds and by electrostatic and hydrophobic interactions, all depending on the chemical and physical environment (e.g., solvent, ions, pH) and interactions with other molecules, such as lipids and proteins. This complexity and the lack of a complete understanding of the relationship between the different Aβ species and their toxicity is currently dictating the nature of the inhibitor (or inducer)-based approaches that are under development for interfering with (or inducing) the formation of specific species and Aβ oligomerization, and for interfering with the associated downstream neurotoxic effects. Here, we review the principles that underlie the involvement of different Aβ oligomeric species in neurodegeneration, both in vitro and in preclinical studies. In addition, we provide an overview of the existing inhibitors (or inducers) of Aβ oligomerization that serve as potential therapeutics for neurodegenerative diseases. The review, which covers the exciting studies that have been published in the past few years, comprises three main parts: 1) on- and off‐fibrillar assembly mechanisms and Aβ structural polymorphism; 2) interactions of Aβ with other molecules and cell components that dictate the Aβ aggregation pathway; and 3) targeting the on‐fibrillar Aβ assembly pathway as a therapeutic approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Today, 2021, the scientific community is still seeking an understanding of the progressive loss of memory and cognitive skills that characterize the irreversible brain disorder known as Alzheimer's disease (AD) [1]. The etiology of the disease remains a central question in AD research, but until recently, current thinking was largely dominated by the 'amyloid aggregation' concept that the oligomerization and accumulation of aggregates or fibrils of the amyloid-β (Aβ) peptide in the brain ultimately lead to neuronal injury (reviewed in [2]). However, the 'amyloid aggregation' hypothesis has been—and continues to be—challenged by multiple reports demonstrating that the neurotoxic species are soluble Aβ oligomers, and not mature Aβ aggregates or cross-β-structured fibrils [3]. These studies demonstrate that the prevention of Aβ aggregation or fibrillization does not in stave off AD [4], and that upon disease onset, AD progression is continuously fueled by a supply of Aβ [5]. Moreover, neuronal dysfunction does not necessarily occur in areas in the brain where mature Aβ fibers (i.e., plaques) accumulate [6]. Support for this last finding may be drawn from studies in AD mouse models showing that neuronal death is induced by non-fibrillar Aβ oligomeric species (prior to the development of AD plaques) [7].
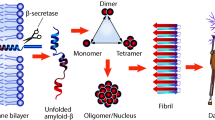
The notion that Aβ oligomers, and not fibrils, are the toxic species that lead to AD has gained growing support in recent years [8]. The Aβ oligomerization chain starts with the reversible equilibrium between Aβ monomers in an α-helical conformation and those in a β-sheet conformation (where the forces driving the process to the β-sheet conformation remain unknown, despite intensive research on this issue). The β-sheet conformation facilitates the initial, i.e., primary, nucleation process of aggregation of Aβ monomers into intermediate low-molecular weight (LMW) Aβ oligomeric species (monomer through octamer, at 4–10 kDa), also termed on-pathway oligomers, which have a high β‐sheet content [9] and are soluble and highly toxic [10]. The on-pathway oligomers aggregate, elongate, and bind with other Aβ monomers to form highly ordered high-molecular weight (HMW) oligomers (multiples of pentamers and hexamers, at 30–60 kDa), protofibrils and mature, non-toxic Aβ fibrils (Fig. 1). In parallel, in an alternative pathway, a variety of factors (e.g., metals, cell components, environmental factors, and inhibitors) promote the formation of disordered, unstructured, non-β-sheet off-pathway Aβ oligomers (Fig. 1) [11]. In addition to the primary nucleation, a secondary nucleation mechanism, in which new fibrils are generated on the surfaces of pre-existing fragments of full-length fibrils, is thought to lead to a considerable increase in the levels of toxic oligomers [12]. These fragments derive from the dissociation of mature fibrils into oligomers under certain unnatural conditions (not spontaneously). Although not completely understood at the molecular level [13], the combination of primary (monomer fibrillization and elongation) and secondary (fibril fragmentation and elongation) nucleation processes contributes to the formation of intermediate Aβ oligomeric species—both toxic and non-toxic [12].

Schematic illustration of the Aβ on- and off-aggregation pathways. The β-sheet conformation adopted by Aβ monomers enables them to aggregate into LMW oligomers with a high β-sheet content. These oligomers, in turn, further aggregate to highly ordered HMW oligomers, protofibrils and fibrils in a process designated on-pathway aggregation. Different factors, e.g., metals, cell components, environmental factors, and inhibitors, may affect this process, causing a shift of the Aβ monomers and LMW oligomers to an alternative oligomerization process, designated the off-pathway. The oligomeric species and Aβ aggregates so produced differ from those generated in the on-aggregation pathway in terms of structure, characteristics and, importantly, toxicity
Intensive experimental and computational efforts have thus been invested in comprehensively elucidating, at the molecular level, the mechanisms underlying the assembly of the different Aβ oligomeric species during the primary and secondary nucleation steps, namely, in understanding how certain cellular factors modulate Aβ assembly and trigger the formation of different oligomeric entities with different degrees of cellular toxicity [14,15,16]. These studies have focused on revealing the structural changes that Aβ species undergo in each nucleation step and the interactions of the different Aβ species with other cellular biomolecules, such as small molecules and membranes. It is thus known that Aβ oligomers—being generated via different oligomerization pathways, i.e., on- and off‐fibrillar assembly mechanisms—constitute a heterogeneous population of polymorphic transient intermediates, which are difficult to distinguish one from the other. The challenge of quantifying and determining the specific structures, characteristics, and toxicity of these Aβ oligomers [17] is exacerbated by the low concentrations of the Aβ oligomeric intermediates, relative to those of the monomeric and the fibrillar Aβ species, throughout the on- and off‐fibrillar oligomerization pathways. Despite this challenge, recent progress in biophysical (e.g., single-molecule fluorescence) and biochemical approaches has opened the way to overcoming the intrinsic difficulties in investigating Aβ oligomeric species [18, 19]. As reviewed here, these tools are now allowing the direct observation and characterization of individual molecular species of Aβ oligomers generated during the on- and off-pathway aggregation processes. They have, for example, facilitated the recognition that similar specific on-pathway oligomeric species may be generated in both cellular compartments and extracellular brain tissue, which would suggest common mechanisms of toxicity for these Aβ oligomers [9, 16]. With the aid of these tools, conformational conversions from Aβ monomers to disordered vs. β-sheet-rich oligomers were identified as key determinants in the off- vs. on-pathway oligomerization mechanisms, respectively. Both on-pathway and off-pathway oligomerization lead to the formation of toxic oligomers, with the conformational change from monomers to fibrillar structures taking place very slowly (of the order of days) and allowing a substantial period of time for cellular protein quality control to sequester potentially harmful misfolded oligomeric species. Importantly, only the disordered oligomers produced in the off-pathway are cleared by the cell degradation machinery, resulting in reduced toxicity for the off-pathway mechanism of aggregation [11]. In the on-pathway generation of β-sheet-rich oligomeric intermediates, there appears to be resistance to clearance of the oligomers by selective autophagy and lysosomal degradation, with the resultant accumulation of on-pathway oligomeric intermediates leading eventually to cell toxicity [20].
The above-mentioned technological advances notwithstanding, it remains difficult, if not impossible, to study the formation and effects of individual Aβ oligomeric species, and studies aimed at elucidating their formation and roles in Aβ-related diseases (and hence at the development of therapeutics) are, therefore, usually performed on bulk Aβ oligomeric species [21]. In this review, we briefly summarize recent studies and theories designed to elucidate the structural and assembly principles of Aβ oligomers and the pathways by which they induce cell toxicity. We follow by exploring natural compounds and drugs in use or under development (including peptide or protein domains and artificial and biological membranes) with the potential to control the Aβ oligomerization pathway (i.e., change the pathway from on- to off-oligomerization, or vice versa) by inducing or disrupting the formation of different types of oligomer.
Aβ42 and Aβ40 isoforms
Among the diverse structural polymorphs (including intermediate oligomeric species and fibrils) that aggregate via the on- and off-fibrillar assembly pathways (Fig. 1), of particular interest are the distinct structural species generated via the on-pathway, which comprises nucleation (either primary or secondary) of Aβ oligomers, followed by the formation of cross-β-structured fibrils—the feature in the brain that characterizes AD [1, 22]: the intra- and inter-neuronal accumulation of Aβ fibrils—preceding plaque formation—is commonly considered to be the causative mechanism in the development of AD [1].
The on- and off-fibrillar assembly pathways are initiated by the cleavage of the hydrophobic transmembrane (plasma and mitochondrial membranes) portion of the amyloid precursor protein (APP) by β- and γ-secretases. This enzymatic cleavage gives rise to the production of 37–49 amino acid peptides, including the two main Aβ peptide isoforms, Aβ40 and Aβ42, where Aβ42 has two additional hydrophobic residues (Ile and Ala) at the C‐terminus [21]. Of these two Aβ isoforms, each with its unique characteristics, Aβ42 plays a more pathogenic role in AD etiology and progression and consequently has received more attention in the literature [23, 24]. The results of studies conducted some years ago indicate that the molar ratio between the two isoforms does not remain constant and that this fluidity in the ratio dictates the distribution and the forms of the different intermediate Aβ oligomeric species generated in both the on- and the off- Aβ oligomerization pathways. Of note, two such studies showed that the molar ratio of Aβ42–Aβ40 in the brains of patients with familial (also known as early onset) and sporadic (also known as late onset) AD was elevated, possibly providing a clue to the pathology of AD. Upon their co-incubation, and depending on the experimental conditions, Aβ42 and Aβ40 isoforms may affect the aggregation of one another, e.g., it was shown that at a high Aβ40/Aβ42 ratio, Aβ40 acted as an inhibitor of Aβ42, and vice versa [25], and cross-seeding of Aβ40 with pre-formed Aβ42 seeds accelerated the aggregation of Aβ40 [26].
Structural landscapes in Aβ
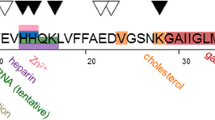
When initially generated, Aβ monomers are intrinsically disordered, lacking a fixed or ordered three-dimensional structure. The conformational changes that these monomers undergo prior to aggregation produce an ensemble comprising fully unfolded, short-lived (partially folded), and completely folded Aβ species in different proportions [1, 15], with the abundance of the fractions of the different conformational species being influenced by multiple Aβ intramolecular and intermolecular interactions [27]. It, therefore, appears that Aβ has a non-smooth structural stability landscape, which is able to tolerate a diverse population of partially folded intermediate species that are the building blocks of the on- and off-oligomerization pathways [28]. The flexible structures of the partially folded Aβ monomers make these species susceptible to self-association and interaction with one another via their self-recognition sites i.e., hydrophobic cores, mostly residues 17–21 and 30–36, thereby promoting oligomerization (via either the on- or the off-oligomerization pathway) and aggregation [1, 15]. The intermolecular interactions (by hydrogen bonding) between the flexible Aβ species are strong enough to overcome unfavorable entropy effects (i.e., decreasing randomness associated with transition from monomers to an oligomer), indicating that Aβ oligomerization (via either on- or off-oligomerization pathway) is a spontaneous (and dominant) process. In light of the propensity of the different partially folded species to simultaneously and rapidly interconvert [29], it is difficult to characterize them definitively [30]. In contrast, mature Aβ fibrils are less flexible than the partially folded monomeric and oligomeric Aβ species and exhibit high structural stability, which has enabled their structural characterization.
Three-phase sequence of Aβ oligomerization
Despite the above-mentioned characterization difficulties, studies to date have provided a fairly detailed picture of the entire three-phase sequence of Aβ oligomerization, starting from an early oligomerization phase (switch from monomer to dimer to trimer) and continuing through a phase in which intermediate oligomeric species are formed, to a final growth phase [31]. Below we examine in some detail the intermediate stage, in which the oligomers cluster and reorganize in a process that is slow relative to the other two phases, and the final growth stage, in which fibrils are formed.
The polymorphic oligomeric assemblies generated in the intermediate phase form a nucleus, which, in turn, promotes the formation of larger—often transient—aggregates. These aggregates, designated on-pathway oligomers, then convert into β-strand protofibrils and fibrils in the growth phase [1, 15]. The parallel pathway to this principal aggregation/fibrillation on-oligomerization pathway leads to the formation of non-toxic unstructured off-pathway oligomers that may be spherical, globular, or amorphous in shape and have different (or zero) β-sheet contents [32]. These unstructured off-pathway oligomers cannot be recognized by the antibodies that are used to detect monomeric species, on-pathway oligomers, or fibrillar species [33]. Nevertheless, despite not having a specific structure, off-pathway oligomers are stable, thus constituting the final products of the off-pathway process [32] (Fig. 1).
To recap, primary nucleation of a pool of Aβ monomers initiates the separation into on- and off-oligomerization pathways [12]. The on- and off-oligomerization pathways, which proceed in concert and are regulated by cross-talk, are driven by various aggregation and fragmentation interactions between monomers and fibrils, with these interactions being referred to as 'secondary nucleation' [12]. In the on-oligomerization pathway, the first nucleation process is rate limiting for Aβ fibrillation [1], since the secondary nucleation is significantly faster than the primary nucleation, with the interactions of monomers with existing fibrils enhancing the formation of new fibrils [12]. Off-pathway oligomers are generated from either mature fibrils (via fragmentation in secondary nucleation) or from monomers (via elongation in primary nucleation). These off-pathway, unstructured Aβ42 oligomers may switch to on-pathway oligomers and then to parallel cross-β structures (i.e., fibrils) [34,35,36] but only after they convert—for reasons as yet unknown—back to monomers.
On-pathway oligomers are formed mainly via secondary nucleation processes that enhance the rate of fibril formation in a feedback loop [12], in which the quantity of oligomers is increased by the fragmentation of fibrils, with the fragments seeding further aggregation upon interaction with monomers. Moreover, at a critical Aβ fibril concentration, secondary nucleation exceeds primary nucleation as a source of new Aβ oligomers with highly ordered structures [12, 37]. Thus, the secondary nucleation of the on-pathway oligomerization contributes significantly (and more than the primary nucleation) to an increase in the concentration of toxic oligomeric species derived from non-toxic monomers [12, 37]. The increasing concentration of toxic, highly structured Aβ oligomers, in turn, contributes to the onset of AD and enhances its progression.
Interactions of Aβ with other molecules and with cell components dictate the Aβ aggregation pathway
The structures of the different Aβ oligomeric species and, particularly, the changes in these structures as a result of interactions of Aβ species with other molecules play major roles in the pathogenesis of AD. Numerous recent studies have, therefore, focused the interactions of different Aβ monomers and oligomers with the other molecules and cell components that dictate Aβ aggregation pathway, i.e., the on vs. the off-pathway, and the consequent cell toxicity.
Both intra- and extracellular stimuli influence the aggregation rate and the particular pathway of Aβ aggregation and hence the structures and sizes of the different types of monomeric, oligomeric and fibrillar species. These stimuli may include changes in pH and temperature and interactions with bi- and/or multivalent metals (and other molecules). We begin this section by discussing recent studies that demonstrate the effects of metal ions on the Aβ aggregation pathway and the formation of different Aβ molecular species that affect cellular toxicity. We then go on to review the work on cell components, with emphasis on lipid membranes.
Interactions of Aβ with metal ions
Several metal ions, such as Zn2+, Cu2+, Fe2+ and Fe3+, that are known to be key factors in the Aβ aggregation process are abundant in the Aβ senile plaques (i.e., protein deposits) found in the brains of AD patients, and the dysregulation of these ions is thus another hallmark of AD [38,39,40,41]. It has been shown that the effect of these ions is concentration dependent. The Zn2+ ion, for example, exhibits a neuroprotective effect towards Aβ-induced toxicity at sub-stoichiometric (below 1:1 Zn/Aβ molar ratio) concentrations [42, 43], whereas stoichiometric concentrations cause an enhancement of Aβ toxicity [43, 44]. An explanation for this dose-dependent effect is that at a 1:1 Zn2+:Aβ molar ratio, Zn2+ reduces Aβ fibrillation levels and promotes the off-pathway aggregation mechanism for both Aβ40 and Aβ42, leading to the formation spherical Zn2+-Aβ oligomers exhibiting a reduced β-sheet content, a highly exposed hydrophobic region, and increased stability [45]. However, unlike other non-toxic off-pathway Aβ oligomers, these unique Zn2+-Aβ off-pathway oligomers are more toxic than the (metal-deficient) wild-type Aβ oligomers: Zn2+-Aβ off-pathway oligomers impair the viability of human neuroblastoma BE(2)-C cells, and in wild-type mice, they inhibit hippocampal long-term potentiation (LTP) and enhance hippocampal microglia activation, indicating neuro-inflammation, as is typical of AD [45]. Even though the structure of Zn2+-Aβ oligomers is clearly different from that of wild-type Aβ oligomers, it is still unclear why the Zn2+-Aβ oligomers exhibit increased cell toxicity vs. the metal-deficient Aβ oligomers. Importantly, the effects Zn2+ on the Aβ aggregation pathway are reversible, as shown in laboratory experiments in which Aβ oligomers were treated with the Zn2+ chelator EDTA; the treatment led to the on-pathway formation of β-sheet-rich Aβ oligomers, which are less toxic than Zn2+-Aβ oligomers [45]. The distinct structure and enhanced toxicity of Zn2+-Aβ oligomers vs. wild-type Aβ oligomers clearly make them a potential target for AD therapeutics. These off-pathway Zn2+-Aβ oligomers were also investigated in a more wide-ranging study, in which the effects of zeolite Y, a crystalline microporous aluminosilicate containing different metal cations (Na+, Mg2+, Fe3+, Zn2+ or Cu2+) and/or their combinations, on Aβ40 aggregation was tested [46]. Here, zeolites containing different metals or combinations of metals showed different effects on Aβ40 aggregation, depending on the identity of the metal ion in the zeolite assembly. All the tested zeolites accelerated Aβ40 nucleation, with NaY, MgY and FeY inducing Aβ40 on-pathway aggregation and fibrillation, and ZnY and CuY inhibiting the formation Aβ40 fibrils and arresting the production of Aβ40 in a high-oligomeric state. The oligomers formed in the presence of either ZnY or CuY were structurally different from the untreated control Aβ40 oligomers, with CuY-treated Aβ40 forming a mixture of different oligomeric species that underwent interconversion either to fibrillar on-pathway oligomers or exclusively to off-pathway oligomers, lacking the ability for seeding aggregation of Aβ40. A recent review of Cu2+-mediated Aβ aggregation [47] indicated that—as opposed to the dose-dependent effect of different Zn2+ concentrations on Aβ aggregation—sub‐stoichiometric Cu2+ levels (vs. the Aβ molar concentration) accelerated the formation of fibrillar Aβ aggregates, while excess Cu2+ led to the production of amorphous toxic fibrillar aggregates but in much lower concentrations than the Aβ fibrillar aggregates induced by sub‐stoichiometric (or zero) concentrations of Cu2+.
In agreement with the above experimental studies, recent molecular dynamics (MD) simulations revealed that interactions between either Zn2+ or Cu2+ with Aβ42 monomers reduced the β-sheet content of the resulting oligomers, while Fe2+-Aβ oligomers exhibited an enhancement in β-sheet content of Aβ42, significantly contributing to Aβ42 fibril formation [48]. Fe3+ also promotes the β-sheet conformation and subsequent fibrillization, but to an even greater extent than that observed for Fe2+ [49]. Importantly, complementary experimental and computational (MD) studies have revealed different mechanisms of action for the different metals and hence different effects on Aβ42 aggregation. For example, Zn2+ binding to Aβ42 has a significant effect on the N-terminal domain of Aβ42, causing a loss of the helical structure in this region and subsequently contributing to the formation of an S‐shaped conformation. The overall non-fibrillar structure of this conformation derives from three separate, but connected (via coil and turn regions) β-sheet strands, i.e., N-terminal, central, and C-terminal strands (comprising residues V12–V18, V24–G33, and V36–V40, respectively) [12, 37, 50]. In the same way, Cu2+, although inducing the formation of a β-sheet conformation locally in the N-terminal domain of Aβ42, confers an overall effect that enhances the content of random coils in the overall Aβ42 peptide structure, consequently reducing Aβ42 fibrillation. In contrast, Fe2+ binding to Aβ42 leads to the production of a Glu22‐Lys28 salt bridge in Aβ42, which stabilizes and promotes β-sheet formation, thereby suggesting that Fe2+ promotes Aβ42 oligomerization and fibrillation by enhancing Aβ42 self-association [48]. It is clear that the presence of different metals differently affects the outcome of Aβ peptide aggregation by altering the pathway (on vs. off) and modulating the biological toxicity of Aβ. Thus, there is a crucial link between the dysregulation of these metals in the brain and AD pathogenesis that can be exploited both for purposes of diagnosis and to delineate therapeutic targets for AD.
Interactions of Aβ with membranes
Other key determinants redirecting the Aβ aggregation pathway and influencing the pathway kinetics are interactions of Aβ with the plasma cell membrane and the inner cell (e.g., mitochondrial) membranes. The interaction with these membranes modulates the Aβ fibrillization process; for example, it was previously shown that in the presence of membranes containing clusters of monosialotetrahexosylganglioside (GM1) (a phospholipid that is abundant in neurons and plays an important physiological role in brain neuroplasticity and in the function and growth of neurons) Aβ monomers exhibited accelerated aggregation and formation of fibrils rich in an anti-parallel β-sheet structure, with increased toxicity towards PC12 neuronal cells. These fibrillary structures were different from the structures that Aβ fibrils form in aqueous solution, with the latter being less toxic and containing mostly parallel β-sheets [51]. In an effort to understand this effect, a recent study examined the interactions of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) or 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG) with two different Aβ42 mutants: (i) a non-toxic L34T variant having a mutation that weakens the hydrophobic interaction in the I32-L34 region of Aβ and thus disrupts formation of the β-barrel conformation in Aβ [52], and (ii) a toxic G37C Aβ42 variant that has a predominant anti-parallel β-sheet structure, even though it was generated from both on- and off-self-assembly processes. The interaction of the artificial membranes with the G37C Aβ42 variant, but not with the L34T mutant, reduced Aβ fibrillation and promoted the formation of smaller aggregates, which disrupted the membranes, suggesting that the interaction between the G37C Aβ42 variant and DOPC and DOPG phospholipids favored an aggregation pathway that differed from that in solution and that promoted membranal damage and hence cell toxicity [53]. A recent study that utilized different phospholipid compositions that mimic the outer leaflet of the synaptic plasma membrane showed that phospholipids, such as cholesterol, sphingomyelin and gangliosides, induced Aβ40 to form a large portion of toxic on-pathway α-helical structures at the early stages of oligomerization and mature Aβ40 fibers at a later stage, which is indicative of on-pathway oligomerization [54]. Thus, although it is commonly believed that the different Aβ oligomeric species formed in aqueous solution may be more toxic than the mature fibrils, in membrane-mediated aggregation, it is the Aβ amyloid fibrils that are more toxic than the oligomers, as they direct the cells to apoptosis [55]. Similarly, as discussed above, the Aβ42 fibrils that are formed in vitro in the presence of GM1-containing biological membranes are more toxic than Aβ42 oligomers, since they activate caspase 3 via binding to toll-like receptor s (TLRs) and inflammasomes, leading to apoptosis [56]. Figure 2 shows a schematic representation of the two parallel aggregation pathways of Aβ peptides and their intermediate structural species—one in solution and the other in membranes—in the brain, both within the cells and in the extracellular matrix (ECM).

On and off Aβ aggregation pathways in solution and in membranes. In the presence of membranes (ii), endogenous Aβ forms a large portion of on-pathway molecules with an α-helical structure in the early aggregation stages [57], followed by the formation of mature anti-parallel β-sheet fibrils, which exhibit higher toxicity than the parallel β-sheet Aβ fibrils formed in aqueous solution (i) [58]. Binding to membrane components (e.g., GM1) allows the anti-parallel β-sheet Aβ fibrils to bind to molecules, such as toll-like receptors (TLRs), and thereby to initiate cell death by apoptosis [55]. Similarly, in the presence of DOPC and DOPG, Aβ forms globular aggregates that are toxic to cells [59]. Red circles indicate the toxic species in each pathway; large arrows indicate accelerated formation of the species
Interactions of Aβ with small molecules
The presence of small molecules in the ECM may also affect the Aβ aggregation pathway and the formation of different Aβ structural species on membrane surfaces. For example, sucrose and glucose, at a concentration of 10 mM, were shown to accelerate the formation of disordered, unstructured, non-β-sheet off-pathway Aβ42 oligomers, which promote mitochondrial dysfunction by causing membrane damage, as was shown by their ability to penetrate both DOPC giant unilamellar vesicles (GUVs) membrane mimetics and intact human neuroblastoma cells (SH-EP) [60]. In that study, it was also demonstrated that glucose favors binding to monomeric and oligomeric Aβ42 species that are in their early oligomeric state, and not to mature Aβ42 fibers. These observations indicate that there may be an Aβ42 monomer ↔ oligomer equilibrium that can be shifted towards distinct oligomers in the case of hyperglycemia. In other words, excess glucose may shift this equilibrium towards distinct unstructured oligomers (which are neurotoxic, even though they are off-pathway oligomers) that are active against the cell membranes of neurons, providing support for a link between AD and other amyloid diseases, including type 2 diabetes [60].
As indicated in the studies discussed above, Aβ–membrane interactions play a major role in Aβ aggregation pathways and cell toxicity, and these interactions are, therefore, considered a potential target for AD treatment. Several Aβ peptide aggregation inhibitors, such as Aβ39-42, EGCG, CLR01 and bacoside, have indeed shown a protective effect against Aβ-induced membranal damage [61, 62]. Notably, our own recent work demonstrated that an Aβ42 variant, carrying the F19S and L34P surface mutations, which we designated Aβ42DM (see also, next section) serves as a potent inhibitor of Aβ42 aggregation and toxicity (Fig. 3). At a low Aβ42DM:Aβ42 molar ratio this variant significantly inhibited the interactions of wild-type Aβ42 with artificial and neuronal cell membranes, thereby abrogating the membrane damage induced by Aβ42. Of note, at micromolar concentrations, Aβ42DM self-assembles to form fibrils (like wild-type Aβ42). We demonstrated that at low nanomolar concentrations, Aβ42DM binds to wild-type Aβ42 and modifies its aggregation pathway to form distinct off-pathway oligomers [63].

Aggregation kinetics and morphology of Aβ42 aggregates in the on and off pathways of oligomerization. The orange dashed graphs show thioflavin T (ThT) fluorescence of Aβ42 (4 μM) as a function of time in the absence (on-pathway) or presence (off-pathway) of the Aβ42DM double mutant (62 nM) at 37 °C with continuous shaking. Below the graphs are circular dichroism (CD) spectra of Aβ42 in the absence or presence of Aβ42DM. CD spectra of Aβ42 (40 µM) or a mixture of Aβ42 (40 µM) and Aβ42DM (62 nM) were obtained at a wavelength of 185–260 nm using a quartz cuvette with a path length of 1 mm. Samples were analyzed at different times of incubation (t = 0, 6 and 24 h) in 10 mM sodium phosphate buffer. Each inset shows (left) TEM images of Aβ42 aggregates in the absence or presence of Aβ42DM after 24 h of incubation at 37 °C with continuous shaking. Each inset also includes (right images) dot blot analyses of Aβ42 (4 μM) incubated for 24 h in the presence or absence of Aβ42DM (62 nM), as assessed using anti-A11 antibodies (upper images). The total amount of Aβ42 (lower images) was detected by blotting with anti-Aβ42 antibodies. Adapted from references [63] and [80]
Targeting the on and off pathways of Aβ assembly and aggregation as a therapeutic approach
In this section, we start by discussing the well-established inhibitors that modulate the Aβ aggregation pathway and their mechanisms of inhibition of Aβ aggregation and toxicity. We follow with a brief review of the work on novel inhibitors. Over the years, numerous inhibitors—namely, small molecules, short peptides, and antibodies—that target Aβ40 and/or Aβ42 production (outside the scope of this review) or aggregation (within the scope of this review) have been developed and tested in vitro, in cell-based assays, and in vivo, as recently reviewed [64]. In parallel, a great deal of effort has been invested in elucidating the modes of action of these inhibitors and their consequent effects on Aβ-mediated cell toxicity. It has been observed that while some inhibitors bind monomeric Aβ peptides, thereby preventing their oligomerization, other inhibitors facilitate Aβ oligomerization, but modify the aggregation pathway in a way that non-toxic off-pathway aggregates are dominantly formed. These off-pathway aggregates are characterized by Aβ structures that differ from those of Aβ oligomers in the absence of inhibitor (i.e., on-pathway Aβ oligomers), with the former mostly lacking the ability to form mature fibrils.
Natural small molecule Aβ inhibitors
Some of the best known Aβ inhibitors that exert a neuroprotective effect (both in cells and in vivo) are plant-based polyphenols, with curcumin being the best studied polyphenol for AD therapeutics. Among the many in vitro and in vivo studies showing that curcumin exerts the protective effect [65, 66], a recent study demonstrated that this biochemical does not prevent the formation of Aβ40 β-sheet enriched fibrillar aggregates, but, rather, it induces the formation of two distinct non-toxic off-pathway oligomeric species with different solubilities [67]. A similar effect was observed for the polyphenol, ( −)-epigallocatechin-3-gallate (EGCG) [32], that is present in green tea. A recent very detailed study of EGCG revealed the mechanism by which EGCG remodeled Aβ40 assemblies to form off-pathway non-toxic oligomers with reduced seeding competency. It was suggested that Aβ40 self-assembles to form low-ordered oligomers with many "independent binding sites for EGCG" [68]. The EGCG-Aβ40 complex exhibits an affinity constant (Kd) that is about ~ tenfold lower than that of an Aβ40 oligomer-Aβ40 monomer complex, allowing EGCG to bind Aβ40 oligomers in preference to Aβ40 monomers, thereby attenuating the oligomer aggregation pathway to form Aβ40 oligomers with an overall decreased solvent exposure [68].
Other small molecules, some natural and others synthetic, have also been shown to modulate the Aβ aggregation to the off-pathway, thus preventing Aβ fibrillation. One such molecule is ATP [69]. It has been suggested that the hydrophobic adenosine in ATP makes strong contacts with the backbone atoms of Aβ42, thereby preventing the initial binding and rebinding of Aβ42 monomers to Aβ42 oligomers [70]. Another natural small molecule that was recently shown to interrupt the Aβ aggregation process is sclerotiorin (SCL), which consists of an aromatic ring system with an aliphatic diene side chain, where the aromatic ring system is highly oxygenated. It has been suggested that the hydrophobic diene side chain interacts with the C-terminal hydrophobic regions in Aβ42 peptides, thereby interfering with β-sheet formation in Aβ42 and hence with Aβ42 oligomerization. In addition, the oxygenated aromatic ring system may bond covalently to polar amino acids (such as Lys16 or Lys28) in Aβ42, thus destabilizing the β-sheet conformation in the Aβ42 oligomer and, instead, stabilizing small Aβ42 spherical oligomers (with a reduced β-sheet content) that do not have Aβ42 seeding capability. Consequently, these small Aβ42 spherical oligomers are characterized by reduced cell uptake and toxicity in human neuroblastoma cells (PC12) due to their reduced β-sheet content, which probably leads to nondestructive interactions with the cell membrane [71].
Synthetic small molecule inhibitors of Aβ
A different approach to reduce the toxicity of Aβ peptides rests on the use of N, N-dimethyl-p-phenylenediamine (DMPD) to redirect the aggregation of Aβ40, either in the absence or presence of a metal (Zn2+ or Cu2+), into off-pathway non-toxic species [72]. DMPD is a redox-active small molecule that structurally modulates Aβ40 assembly through intramolecular cross-linking with critical residues within the self-recognition K16-A21 region of the Aβ40 peptide to form cross-β-sheet structures. An in vivo evaluation of DMPD inhibition of Aβ40 aggregation and its toxicity potency showed a significant reduction of both intracellular soluble Aβ40 and Aβ42 and amyloid plaques in 5XFAD mice, which exhibited restored memory and learning abilities relative to non-treated mice [72]. Similar to DMPD, another small molecule, L2B, was designed to specifically bind metal/Aβ40 and metal/Aβ42 complexes (i.e., Zn/Aβ and Cu/Aβ), which are known to function as toxic assemblies, better than to metal-free Aβ, thus redirecting the aggregation of the metal/Aβ40 and metal/Aβ42 complexes into off-pathway non-toxic oligomers, as was shown both in vitro and in vivo in 5XFAD mice. In the L2B-treated mice, the cognitive deficits associated with the AD model were significantly improved, and the metal/Aβ in their brains exhibited reduced amyloid pathology [73].
Another promising inhibitor—currently in clinical development for the topical treatment of dry age-related macular degeneration (AMD) and glaucoma [74], in which Aβ peptides play a crucial role—is (R)-(2-[2-amino-3-(1H-indol-3-yl)-propionylamino]-2-methyl-propionic acid (MRZ-99030). This small (289 Daltons) dipeptide, which was designed to modulate Aβ42 aggregation into off-pathway oligomers, has been shown to reverse the synapto-toxic effect of Aβ42 and improve the Aβ42-mediated cognitive deficits in vivo in rats and mice [74]. MRZ-99030—chosen from a screen of 40 rationally designed small molecules and peptides—consists of two elements that break β-sheets, an aromatic moiety and α-aminoisobutyric acid. The molecule was thus designed to interrupt the aromatic stacking of Aβ peptides and hence amyloid aggregation [75]. MRZ-99030 prevents the formation of toxic Aβ42 oligomers by promoting the formation of large off-pathway amorphous non-amyloidogenic oligomers and by reducing the proportion of toxic soluble oligomeric Aβ species in the Aβ population [76].
Another group of potent Aβ inhibitors with the potential to attenuate the Aβ aggregation pathway are foldamers, which are artificial molecular architectures consisting of monomers or oligomers that exhibit an ordered structure in solution and function as molecular chaperones [77, 78]. A recent study demonstrated that screening of an oligoquinolines-based foldamer library for binding to Aβ42 yielded a unique dianionic tetraquinoline that was able to induce a strong α-helical Aβ40 or Aβ42 structure by binding to the central α-helical domain of the Aβ monomer [78]. It is assumed that the carboxylate groups in this dianionic tetraquinoline foldamer form a salt bridge with the positively charged Lys16 in the Aβ peptide and also interact with and stabilize the Leu17-Phe20 hydrophobic region of the Aβ peptide, thus inhibiting intramolecular interactions within the Aβ peptide that facilitate its oligomerization. This dianionic tetraquinoline foldamer was thus shown to reduce Aβ self-association and thereby to modulate Aβ aggregation into non-toxic oligomers and to disrupt pre-formed neurotoxic Aβ oligomers, as demonstrated in both in vitro and in cell-based assays in mouse N2a neuroblastoma cells.
Protein-based inhibitors of Aβ
In addition to the small molecules described above, larger peptides and small proteins also exhibit the potential to serve as Aβ inhibitors by inducing off-pathway Aβ aggregation processes. Our previous work (described in the previous section) demonstrated that Aβ42DM—a non-self-aggregating Aβ42 variant carrying the surface mutations, F19S and L34P—binds to both intra- and extracellular Aβ42 and directs its aggregation into non-toxic Aβ42DM/Aβ42 assemblies, in which Aβ42 exhibits a different secondary structure with a reduced β-sheet content vs. untreated Aβ42 oligomers [63, 79]. In addition, the results showed no cross-seeding between the Aβ42DM/Aβ42 assemblies and Aβ42, a reduction in neuronal cell Aβ42 uptake, reduced damage to the plasma and mitochondrial membranes, and significantly reduced toxicity and Aβ42-mediated apoptosis in neuronal cells. We assume that the inhibitory capacity of Aβ42DM derives from its two mutations: the F19S mutation in the Leu17-Phe20 hydrophobic pocket, which inhibits Aβ42 self-assembly, and the L34P mutation in the Ile23-Val36 hydrophobic region, in which the proline residue sterically disrupts Aβ42 self-assembly. We hypothesize that Aβ42DM, which does not have the capacity to disrupt pre-formed Aβ42 aggregates, exerts its inhibitory action on the aggregation of Aβ42 by binding to low-oligomeric weight Aβ42 species, thereby modulating the Aβ42 aggregation pathway.
In seeking for Aβ42-based inhibitors that may modulate the Aβ42 aggregation pathway, we recently developed an efficient strategy for mapping oligomerization landscapes encompassing single and double mutations (including hot spots, cold spots, solubility switches, and correlated mutations that impact aggregation) by combining experimental Aβ42 library screening in neuronal cells with NGS analysis (unpublished). By applying this strategy, we identified Aβ42 mutants with improved solubility. In related work (unpublished), we developed an approach using fractional solubility selections, NGS analysis, and data normalization to accurately quantify aggregation over a broad dynamic range for thousands of Aβ42 mutants in a single experiment; this strategy enabled us to map in reasonable detail the single-mutant oligomerization landscape of Aβ42. Currently, the remaining barrier to generating truly comprehensive oligomerization landscapes, encompassing all possible combinations of multiple mutations, is the inadequate coverage of sequence space in experimentally tractable library screening methodologies. To overcome this barrier, we have conducted preliminary studies to integrate state-of-the-art ML methods with our unpublished approaches, enabling us to achieve a more extensive coverage and more accurate predictions of mutant peptide aggregation ability. We are currently in the process of combining Aβ42 library solubility screening, NGS analysis, ML-derived predictive models, and normalization/validation using aggregation measurements on purified Aβ42 peptides. Our novel, single-step approach for comprehensively mapping oligomerization landscapes and predicting aggregation of Aβ42 peptides with multiple mutations will facilitate the discovery of the most soluble (namely, having the lowest propensity for aggregation) peptide that can be produced from the Aβ42 scaffolds. As these Aβ42 peptide mutants can also be engineered as polyvalent constructs with potentially improved affinity for wild-type Aβ42, the impact of polyvalency on the putative monomeric Aβ42-based inhibitor should also be defined. It will then be necessary for us and others to elucidate the mechanisms responsible for enhanced inhibition of aggregation and to exploit polyvalency to engineer highly effective inhibitors of Aβ42 aggregation using our new strategy.
A different study from our group showed that an engineered variant of the B1 domain of protein G (designated HTB1M2), which was identified by Aβ42 affinity screens performed in a yeast surface display format, modulates Aβ42 aggregation to form an Aβ42 structure with reduced cell permeation capabilities and toxicity (80). HTB1M2 was evolved from a focused library in which the HTB1 was randomly mutated in positions identified as ‘stability patches,’ namely, surface areas with an increased potential to evolve into efficient protein − protein interfaces. Indeed, HTB1M2 was shown to bind to and modulate the aggregation of Aβ42 to produce non-toxic off-pathway aggregates, having a different structural morphology (smaller number of fibrils and a narrower width than untreated Aβ42 fibrils) but with a mechanism of inhibition that still remains elusive.
Conclusions and future directions
To obtain a comprehensive view of the dynamics of the critical Aβ structural species that are generated in the multi-step process of Aβ oligomerization and that play significant roles in AD pathology, it is necessary to combine computational and experimental approaches. The complementing of conventional biophysical methods (i.e., X-ray crystallography and NMR spectroscopy) and numerous conventional and novel spectroscopy approaches with computational methods, i.e., MD simulations and machine learning (ML) analysis, will ultimately provide the breadth and depth of knowledge required to reveal the multiple diverse components of the aggregation pathway and to unravel its mode of action.
Overall, modulating the on and off Aβ aggregation pathways and manipulating the formation of the various components in these pathways appears to be the way forward to establishing an effective strategy to inhibit Aβ peptide neurotoxicity, as is indicated by the in vitro and in vivo studies detailed above. As revealed by our group and by others, a key determinant in the reduced toxicity of the various off-pathway (vs. on-pathway) oligomeric species formed upon treatment with different inhibitors may derive from the modulation of the interactions between off-pathway Aβ oligomeric species and cell components, particularly cell membranes. The outcome of these interactions may be reduced membranal damage (namely, changes in the membrane rigidity and the formation of pores) and decreased uptake of Aβ into cells, and hence less intracellular damage, such as the dysregulation of intracellular calcium levels and mitochondrial activity that eventually leads to apoptosis, as has been demonstrated by us and by others [63, 79, 80]. From these studies, we understand the critical need to evaluate the interactions of both the plasma membrane and the intracellular (in particular mitochondrial) membranes with (and their disruption by) the different Aβ structural species produced in both the on- and off-oligomerization pathways. With an eye to the development of efficient inhibitors against Aβ aggregation and toxicity, it is necessary to test potential inhibitors with respect to their effects on the Aβ oligomerization pathway, on the different oligomeric species, and on the interaction of these species with membranes. We believe that such a strategy would reveal a reliable way to block Aβ neurotoxicity as a means to treating AD.
The substantial corpus covering the role of Aβ aggregation in the etiology of AD does not yet provide a comprehensive picture of the structural determinants of the Aβ aggregation pathway, particularly since current knowledge derives mainly from computational tools and in vitro studies (experiments in cells and preclinical animal models are lacking, which is important for drug development). Moreover, methods for elucidating the structural determinants of the oligomerization pathway typically include the use of inhibitors (disrupting different stages in the oligomerization process) that are, in most cases, non-specific for particular structural species of Aβ and, therefore, have limited utility in elucidating the different steps/stages of aggregation and the different oligomeric species and their specific contribution to neurotoxicity and cell death. Table 1 summaries the status of each inhibitor in terms of clinical trials. An alternative (and efficient) strategy for overcoming the above drawbacks and hence for elucidating the structural determinants of Aβ aggregation and the specific role played by different oligomeric species in neurotoxicity may lie in mutating candidate residues in the Aβ sequence and testing the consequent effects on Aβ aggregation. The idea here is that when a mutation is generated, there is a change in structure that affects the oligomeric state of the peptide. If we could mutate all the residues, one at a time, and then determine the structural change that each mutation causes and the resulting oligomeric state of the peptide, then we could start to fully understand the molecular determinants (i.e., the effects of single mutations) that dictate whether the peptide will oligomerize via the on- or the off-pathway. Furthermore, if we could determine this factor for each and every position and mutation in the Aβ peptide, then we would be able to open the way to controlling these pathways and hence Aβ toxicity. In a first attempt to apply this idea, the group of Fowler recently combined a yeast growth-based aggregation assay with deep mutational scanning to evaluate the effects of 791 of the possible 798 single amino acid substitutions on the aggregation propensity of Aβ42 [81]. The output of their study was a solubility score for each Aβ42 variant generated by the selection, which they derived by following the frequency of each variant by high-throughput DNA sequencing, i.e., next-generation sequencing (NGS). This study, designed to comprehensively evaluate the aggregation landscape of Aβ42, constitutes the first large-scale, high-throughput, cell-based mutational analysis of Aβ, and comprehensively indicates the physicochemical properties of the amino acids that will reduce, enhance, or have no affect Aβ solubility and/or aggregation. We note here that this new research direction could also be applied to other amyloids.
The oligomerization/aggregation landscapes of Aβ, like the one mentioned above, link the amino acid sequence to the oligomerization properties of the mutated peptide and shed light on the consequences for Aβ peptide oligomerization of any possible mutation or combination of mutations in the peptide. Accurate and comprehensive mapping of such landscapes are key to understanding the mechanisms of Aβ oligomerization pathways, the evolutionary origins of the different Aβ oligomeric species, and their roles in biological processes, and hence to engineering binders and inhibitors to alter Aβ function. Studies of oligomerization landscapes may identify several common features, such as solubility-switch and correlated solubility residues, and hot and cold spots, where: solubility-switch residues are those in which a mutation can reverse the solubility of Aβ; correlated solubility residues are those in which co-occurring mutations work in concert to alter solubility/aggregation; hot-spot residues are those that can be optimized to enhance the solubility of an oligomer vs. a monomer or an insoluble aggregate; and cold-spot residues are those that are suboptimal for solubility enhancement, meaning that mutations in these residues are crucial to improve solubility, i.e., to reduce aggregation (the terms hot and cold spots are taken from the field of protein–protein interactions). Oligomerization landscapes can be mapped via mutagenesis and experimental aggregation measurements both in vitro and in cells, but despite recent advances [81,82,83], such methods demonstrate limitations of scale, being restricted to the analysis of single-mutant libraries. In the future, exploitation of NGS, typically analyzing only single-mutant libraries and then reconstructing multi-mutant libraries, may improve coverage and provide a powerful means to improve solubility and generate comprehensive oligomerization landscapes.
Availability of data and materials
Not applicable.
References
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10:698–712
Klein WL, Krafft GA, Finch CE (2001) Targeting small Amyloid beta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci 24:219–224
Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, and Borchelt DR (2005) Persistent amyloidosis following suppression of Amyloid beta production in a transgenic model of Alzheimer disease. PLoS Med 2, e355
Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM (2009) Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci 29:10706–10714
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 96:3228–3233
Hayden EY, Teplow DB (2013) Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther 5:60
Ono K, Condron MM, Teplow DB (2009) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A 106:14745–14750
Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB (2003) Amyloid beta -protein (Amyloid beta) assembly: Amyloid beta 40 and Amyloid beta 42 oligomerize through distinct pathways. Proc Natl Acad Sci U S A 100:330–335
Arosio P, Michaels TC, Linse S, Mansson C, Emanuelsson C, Presto J, Johansson J, Vendruscolo M, Dobson CM, Knowles TP (2016) Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat Commun 7:10948
Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP (2013) Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110:9758–9763
Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440:352–357
Miller Y, Ma B, Nussinov R (2010) Polymorphism in Alzheimer Amyloid beta amyloid organization reflects conformational selection in a rugged energy landscape. Chem Rev 110:4820–4838
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14:837–842
Misra P, Kodali R, Chemuru S, Kar K, Wetzel R (2016) Rapid alpha-oligomer formation mediated by the Amyloid beta C terminus initiates an amyloid assembly pathway. Nat Commun 7:12419
Orte A, Birkett NR, Clarke RW, Devlin GL, Dobson CM, Klenerman D (2008) Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc Natl Acad Sci U S A 105:14424–14429
Pitschke M, Prior R, Haupt M, Riesner D (1998) Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat Med 4:832–834
Klaips CL, Jayaraj GG, Hartl FU (2018) Pathways of cellular proteostasis in aging and disease. J Cell Biol 217:51–63
Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, Xu HE (2017) Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38:1205–1235
Tran L, Ha-Duong T (2015) Exploring the Alzheimer amyloid-beta peptide conformational ensemble: A review of molecular dynamics approaches. Peptides 69:86–91
Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT (2009) Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem 1:326–331
Economou NJ, Giammona MJ, Do TD, Zheng X, Teplow DB, Buratto SK, Bowers MT (2016) Amyloid beta-Protein Assembly and Alzheimer’s Disease: Dodecamers of Amyloid beta42, but Not of Amyloid beta40, Seed Fibril Formation. J Am Chem Soc 138:1772–1775
Jan A, Gokce O, Luthi-Carter R, Lashuel HA (2008) The ratio of monomeric to aggregated forms of Amyloid beta40 and Amyloid beta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem 283:28176–28189
Tran J, Chang D, Hsu F, Wang H, Guo Z (2017) Cross-seeding between Amyloid beta40 and Amyloid beta42 in Alzheimer’s disease. FEBS Lett 591:177–185
Chang YJ, Chen YR (2014) The coexistence of an equal amount of Alzheimer’s amyloid-beta 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. FEBS J 281:2674–2687
Vendruscolo M, Paci E, Karplus M, Dobson CM (2003) Structures and relative free energies of partially folded states of proteins. Proc Natl Acad Sci U S A 100:14817–14821
Kad NM, Myers SL, Smith DP, Smith DA, Radford SE, Thomson NH (2003) Hierarchical assembly of beta2-microglobulin amyloid in vitro revealed by atomic force microscopy. J Mol Biol 330:785–797
Wetzel R (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 39:671–679
Garai K, Frieden C (2013) Quantitative analysis of the time course of Amyloid beta oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Amyloid beta. Proc Natl Acad Sci U S A 110:3321–3326
Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 15:558–566
Cremades N, Dobson CM (2018) The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol Dis 109:178–190
Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE (2010) EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A 107:7710–7715
Karamanos, T. K., Jackson, M. P., Calabrese, A. N., Goodchild, S. C., Cawood, E. E., Thompson, G. S., Kalverda, A. P., Hewitt, E. W., and Radford, S. E. (2019) Structural mapping of oligomeric intermediates in an amyloid assembly pathway. Elife 8
Wu JW, Breydo L, Isas JM, Lee J, Kuznetsov YG, Langen R, Glabe C (2010) Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. J Biol Chem 285:6071–6079
Liang C, Ni R, Smith JE, Childers WS, Mehta AK, Lynn DG (2014) Kinetic intermediates in amyloid assembly. J Am Chem Soc 136:15146–15149
De Benedictis, C. A., Vilella, A., and Grabrucker, A. M. (2019) The Role of Trace Metals in Alzheimer’s Disease. Exon Publications, 85–106.
Lovell M, Robertson J, Teesdale W, Campbell J, Markesbery W (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158:47–52
Frederickson CJ, Koh JY, Bush AI (2005) The neurobiology of zinc in health and disease. Nat Rev Neurosci 6:449–462
Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci 26:207–214
Garai K, Sahoo B, Kaushalya S, Desai R, Maiti S (2007) Zinc lowers amyloid-β toxicity by selectively precipitating aggregation intermediates. Biochemistry 46:10655–10663
Lovell MA, Xie C, Markesbery WR (1999) Protection against amyloid beta peptide toxicity by zinc. Brain Res 823:88–95
Bishop GM, Robinson SR (2004) The Amyloid Paradox: Amyloid-β-Metal Complexes can be Neurotoxic and Neuroprotective. Brain Pathol 14:448–452
Lee MC, Yu WC, Shih YH, Chen CY, Guo ZH, Huang SJ, Chan JC, Chen YR (2018) Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci Rep 8:1–16
Lucas MJ, Keitz BK (2018) Influence of zeolites on amyloid-β aggregation. Langmuir 34:9789–9797
Borghesani V, Alies B, Hureau C (2018) Cu(II) binding to various forms of amyloid-β peptides: are they friends or foes? Eur J Inorg Chem 2018:7–15
Boopathi S, Kolandaivel P (2016) Fe2+ binding on amyloid β-peptide promotes aggregation. Proteins: Structure. Function, and Bioinformatics 84:1257–1274
Vahed, M., Sweeney, A., Shirasawa, H., and Vahed, M. (2019) The initial stage of structural transformation of Aβ42 peptides from the human and mole rat in the presence of Fe2+ and Fe3+: Related to Alzheimer's disease. Computational biology and chemistry 83, 107128
Grasso G, Rebella M, Muscat S, Morbiducci U, Tuszynski J, Danani A, and Deriu MA (2018) Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid beta Assemblies. Int J Mol Sci 19
Matsuzaki K (2014) How do membranes initiate Alzheimer’s disease? Formation of toxic amyloid fibrils by the amyloid β-protein on ganglioside clusters. Acc Chem Res 47:2397–2404
Qian Z, Zhang Q, Liu Y, Chen P (2017) Assemblies of amyloid-β30–36 hexamer and its G33V/L34T mutants by replica-exchange molecular dynamics simulation. PLoS ONE 12:e0188794
Henry S, Vignaud H, l. n., Bobo, C., Decossas, M., Lambert, O., Harte, E., Alves, I. D., Cullin, C., and Lecomte, S. (2015) Interaction of Aβ1–42 amyloids with lipids promotes “off-pathway” oligomerization and membrane damage. Biomacromol 16:944–950
Cheng, Q., Hu, Z.-W., Doherty, K. E., Tobin-Miyaji, Y. J., and Qiang, W. (2018) The on-fibrillation-pathway membrane content leakage and off-fibrillation-pathway lipid mixing induced by 40-residue β-amyloid peptides in biologically relevant model liposomes. Biochim Biophy Acta (BBA)-Biomembr. 1860, 1670–1680
Itoh N, Takada E, Okubo K, Yano Y, Hoshino M, Sasaki A, Kinjo M, Matsuzaki K (2018) Not Oligomers but Amyloids are Cytotoxic in the Membrane-Mediated Amyloidogenesis of Amyloid-β Peptides. ChemBioChem 19:430–433
Takada E, Okubo K, Yano Y, Iida K, Someda M, Hirasawa A, Yonehara S, Matsuzaki K (2020) Molecular Mechanism of Apoptosis by Amyloid β-Protein Fibrils Formed on Neuronal Cells. ACS Chem Neurosci 11:796–805
Cheng Q, Hu ZW, Doherty KE, Tobin-Miyaji YJ, Qiang W (2018) The on-fibrillation-pathway membrane content leakage and off-fibrillation-pathway lipid mixing induced by 40-residue beta-amyloid peptides in biologically relevant model liposomes. Biochim Biophys Acta Biomembranes 1860:1670–1680
Matsuzaki K (2014) How do membranes initiate Alzheimer’s Disease? Formation of toxic amyloid fibrils by the amyloid beta-protein on ganglioside clusters. Acc Chem Res 47:2397–2404
Henry S, Vignaud H, Bobo C, Decossas M, Lambert O, Harte E, Alves ID, Cullin C, Lecomte S (2015) Interaction of Amyloid beta(1–42) amyloids with lipids promotes “off-pathway” oligomerization and membrane damage. Biomacromol 16:944–950
Kedia N, Almisry M, Bieschke J (2017) Glucose directs amyloid-beta into membrane-active oligomers. Phys Chem Chem Phys 19:18036–18046
Malishev R, Shaham-Niv S, Nandi S, Kolusheva S, Gazit E, Jelinek R (2017) Bacoside-A, an Indian traditional-medicine substance, inhibits β-amyloid cytotoxicity, fibrillation, and membrane interactions. ACS Chem Neurosci 8:884–891
Malishev R, Nandi S, Kolusheva S, Levi-Kalisman Y, Klärner F-G, Schrader T, Bitan G, Jelinek R (2015) Toxicity inhibitors protect lipid membranes from disruption by Aβ42. ACS Chem Neurosci 6:1860–1869
Oren O, Ben Zichri S, Taube R, Jelinek R, Papo N (2020) An Aβ42 double mutant inhibits Aβ42-induced plasma and mitochondrial membrane disruption in artificial membranes, isolated organs and intact cells. ACS Chem Neurosci 11(7):1027
Lee SJC, Nam E, Lee HJ, Savelieff MG, Lim MH (2017) Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chem Soc Rev 46:310–323
Chainoglou E, Hadjipavlou-Litina D (2020) Curcumin in Health and Diseases: Alzheimer’s Disease and Curcumin Analogues, Derivatives, and Hybrids. Int J Mol Sci 21:1975
Reddy PH, Manczak M, Yin X, Grady MC, Mitchell A, Tonk S, Kuruva CS, Bhatti JS, Kandimalla R, Vijayan M (2018) Protective effects of Indian spice curcumin against amyloid-β in Alzheimer’s disease. J Alzheimers Dis 61:843–866
Thapa A, Jett SD, Chi EY (2016) Curcumin attenuates amyloid-β aggregate toxicity and modulates amyloid-β aggregation pathway. ACS Chem Neurosci 7:56–68
Ahmed R, VanSchouwen B, Jafari N, Ni X, Ortega J, Melacini G (2017) Molecular mechanism for the (−)-epigallocatechin gallate-induced toxic to nontoxic remodeling of Aβ oligomers. J Am Chem Soc 139:13720–13734
Kurisaki I, Tanaka S (2019) ATP converts Aβ42 oligomer into off-pathway species by making contact with its backbone atoms using hydrophobic adenosine. J Phys Chem B 123:9922–9933
Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, Hyman AA (2017) ATP as a biological hydrotrope. Science 356:753–756
Wiglenda T, Groenke N, Hoffmann W, Manz C, Diez L, Buntru A, Brusendorf L, Neuendorf N, Schnoegl S, Haenig C (2020) Sclerotiorin stabilizes the assembly of nonfibrillar amyloid beta42 oligomers with low toxicity, seeding activity, and beta-sheet content. J Mol Biol 432:2080–2098
Derrick JS, Kerr RA, Nam Y, Oh SB, Lee HJ, Earnest KG, Suh N, Peck KL, Ozbil M, Korshavn KJ (2015) A redox-active, compact molecule for cross-linking amyloidogenic peptides into nontoxic, off-pathway aggregates: in vitro and in vivo efficacy and molecular mechanisms. J Am Chem Soc 137:14785–14797
Beck MW, Oh SB, Kerr RA, Lee HJ, Kim SH, Kim S, Jang M, Ruotolo BT, Lee J-Y, Lim MH (2015) A rationally designed small molecule for identifying an in vivo link between metal–amyloid-β complexes and the pathogenesis of Alzheimer’s disease. Chem Sci 6:1879–1886
Rammes G, Gravius A, Ruitenberg M, Wegener N, Chambon C, Sroka-Saidi K, Jeggo R, Staniaszek L, Spanswick D, O’Hare E (2015) MRZ-99030–A novel modulator of Aβ aggregation: II–Reversal of Aβ oligomer-induced deficits in long-term potentiation (LTP) and cognitive performance in rats and mice. Neuropharmacology 92:170–182
Frydman-Marom A, Rechter M, Shefler I, Bram Y, Shalev DE, Gazit E (2009) Cognitive-performance recovery of Alzheimer’s disease model mice by modulation of early soluble amyloidal assemblies. Angew Chem Int Ed 48:1981–1986
Parsons CG, Ruitenberg M, Freitag CE, Sroka-Saidi K, Russ H, Rammes G (2015) MRZ-99030–A novel modulator of Aβ aggregation: I-Mechanism of action (MoA) underlying the potential neuroprotective treatment of Alzheimer’s disease, glaucoma and age-related macular degeneration (AMD). Neuropharmacology 92:158–169
Guichard G, Huc I (2011) Synthetic foldamers. Chem Commun 47:5933–5941
Kumar S, Henning-Knechtel A, Chehade I, Magzoub M, Hamilton AD (2017) Foldamer-mediated structural rearrangement attenuates Aβ oligomerization and cytotoxicity. J Am Chem Soc 139:17098–17108
Oren O, Banerjee V, Taube R, Papo N (2018) An Aβ42 variant that inhibits intra-and extracellular amyloid aggregation and enhances cell viability. Biochemical Journal 475:3087–3103
Banerjee V, Oren O, Dagan B, Taube R, Engel S, Papo N (2018) An engineered variant of the B1 domain of protein G suppresses the aggregation and toxicity of intra-and extracellular Aβ42. ACS Chem Neurosci 10:1488–1496
Gray VE, Sitko K, Kameni FZN, Williamson M, Stephany JJ, Hasle N, and Fowler DM (2019) Elucidating the Molecular Determinants of Amyloid beta Aggregation with Deep Mutational Scanning. G3 (Bethesda). 9(11):3683–3689
Fowler DM, Fields S (2014) Deep mutational scanning: a new style of protein science. Nat Methods 11:801–807
Fowler DM, Stephany JJ, Fields S (2014) Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat Protoc 9:2267–2284
Radbakhsh S, Barreto GE, Bland AR, Sahebkar A (2021) Curcumin: A small molecule with big functionality against amyloid aggregation in neurodegenerative diseases and type 2 diabetes. BioFactors 47:570–586
Ide K, Yamada H, Takuma N, Harada S, Nakase J, Ukawa Y, Sagesaka YM (2015) Effects of green tea consumption on cognitive dysfunction in an elderly population: a randomized placebo-controlled study. Nutr J 15:49–58
Funding
This work was supported by the European Research Council “Ideas program” ERC-2013-StG (contract grant number: 336041) and the Israel Science Foundation (grant number 615/14) to Niv Papo.
Author information
Authors and Affiliations
Contributions
O.O. and N.P. wrote the paper. All authors edited the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest with respect to publication of this paper.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Oren, O., Taube, R. & Papo, N. Amyloid β structural polymorphism, associated toxicity and therapeutic strategies. Cell. Mol. Life Sci. 78, 7185–7198 (2021). https://doi.org/10.1007/s00018-021-03954-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03954-z