
Abstract
PDGFRA and PDGFRB are classical proto-oncogenes that encode receptor tyrosine kinases responding to platelet-derived growth factor (PDGF). PDGFRA mutations are found in gastrointestinal stromal tumors (GISTs), inflammatory fibroid polyps and gliomas, and PDGFRB mutations drive myofibroma development. In addition, chromosomal rearrangement of either gene causes myeloid neoplasms associated with hypereosinophilia. Recently, mutations in PDGFRB were linked to several noncancerous diseases. Germline heterozygous variants that reduce receptor activity have been identified in primary familial brain calcification, whereas gain-of-function mutants are present in patients with fusiform aneurysms, Kosaki overgrowth syndrome or Penttinen premature aging syndrome. Functional analysis of these variants has led to the preclinical validation of tyrosine kinase inhibitors targeting PDGF receptors, such as imatinib, as a treatment for some of these conditions. This review summarizes the rapidly expanding knowledge in this field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Platelet-derived growth factor (PDGF) receptors (PDGFRs) belong to the family of receptor tyrosine kinases (RTKs) [1,2,3,4]. There are two isoforms of PDGF receptors, PDGFRα and PDGFRβ, which are encoded by two different genes, PDGFRA and PDGFRB, respectively. Five distinct ligands (PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC and PDGF-DD) can bind to one or both PDGFRs in a dimeric state. PDGF was initially purified from platelets in the seventies and can be produced by numerous other cell types, including epithelial and endothelial cells. PDGF receptors are mainly expressed by cells of mesenchymal origin, such as fibroblasts, pericytes and vascular smooth muscle cells, and stimulate their proliferation and motility [1, 2]. Early studies suggested that PDGF ligands and receptors may play a role in cancer development, and genetic alterations in both receptor genes were subsequently identified in hematopoietic, glial and soft-tissue cancers. Recently, mutations in noncancerous disorders have also been found, including skeletal defects, brain calcification, and vascular anomalies. This review focuses on mutations in PDGFRA and PDGFRB that cause human diseases.
Receptor structure and signal transduction
PDGF receptors are members of the RTK class III family, along with c-KIT, colony stimulating factor 1 receptor (CSF1R) and Fms-like tyrosine kinase 3 receptor (FLT3). These receptors are characterized by a glycosylated extracellular domain composed of five immunoglobulin-like modules that bind the ligand, a helical transmembrane domain and an intracellular region with tyrosine kinase activity. More precisely, the cytosolic region contains (1) the juxtamembrane (JM) domain, (2) the highly conserved tyrosine kinase domain composed of the N-lobe, the insert region and the C-lobe containing the activation loop, and (3) a variable C-terminal tail (Fig. 1) [5,6,7].

PDGFR Structure. The receptor consists of an extracellular part composed of five immunoglobulin-like modules (D1-D5) that bind the ligand, a helical transmembrane domain, and an intracellular part containing the juxtamembrane (JM) regulatory region, the kinase domain made of a N- and C-lobe separated by the kinase insert. Adapted in PyMol from the crystal structure of human PDGFRA (PDB 5K5X) [6] and PDGFRB (PDB 3MJG) [7]
PDGF binding induces receptor dimerization. The dimer is further stabilized by conformational changes in the extracellular region, which favor homotypic interactions between Ig-like domains 4 and 5. The transmembrane helix may also contribute to the formation of receptor dimers [6]. This process disrupts the inhibitory juxtamembrane domain. According to recent cryo-electron microscopy data, the active kinase domain adopts an asymmetric dimer conformation [8, 9]. The tyrosine-kinase domain of PDGF receptors transfers a phosphoryl group (PO32−) from adenosine-5′-triphosphate to a tyrosine residue of the substrate [10]. ATP binds to the cleft between the N- and C-lobes of the kinase domain. The adenine moiety binds in a hydrophobic pocket through hydrogen bonds, while the ribose and triphosphate groups contact a hydrophilic channel that extends to the substrate binding site [11]. This leads to the trans-phosphorylation of receptor tyrosine residues. Phosphorylation of the juxtamembrane domain and the activation loop further stabilizes the active conformation of the kinase domain. In addition, multiple phospho-tyrosine residues act as docking sites for signaling molecules, which can themselves be phosphorylated. Phospholipase Cγ (PLCγ), phosphatidylinositol- 3-kinase (PI3K), SRC family kinases and signal transducers and activators of transcription (STATs) are the main signaling mediators activated by PDGF receptors [5, 12, 13]. Adaptor molecules are also recruited to PDGFR and regulate multiple pathways, including the MAP kinase cascade (Fig. 2).

Overview of signaling pathways downstream of PDGF receptors. PDGF binding induces receptor dimerization and trans-phosphorylation. The recruitment of signaling and adaptor molecules to the phosphorylated receptor triggers several signaling cascades: phospholipase C γ (PLCγ), phosphatidylinositol-3-kinase (PI3K), mitogen-activated protein (MAP) kinases and signal transducers and activators of transcription (STATs). P, phosphorylation site
Physiological roles
Analysis of knockout mice has revealed the crucial role of PDGFRs and their ligands in embryonic development. This has been the subject of several excellent reviews [1,2,3]. PDGFRα has early developmental functions in gastrulation and formation of neural crest. It is also essential for organogenesis, particularly by driving the migration and proliferation of mesenchymal cells. Mice lacking PDGFRα are not viable and present multiple defects in the gastrointestinal tract, central nervous system, lungs, skeleton, testis, and kidneys [2]. This phenotype combines the defects observed in PDGF-A and PDGF-C knockout mice [2, 14, 15].
PDGFRβ is mainly implicated in blood vessels formation and kidney development [2]. Mice lacking PDGFRβ or PDGF-B exhibit defects in the development of both systems and die around birth. This is due to the lack of vascular smooth muscle cells, pericytes, and mesangial cells of kidney glomeruli, causing hemorrhages and altering glomerular filtration [16, 17]. In contrast, mice deficient in PDGF-D only present a mild vascular phenotype, with disorganized pericytes in the cardiac vasculature and a slight increase in blood pressure [18].
In adults, PDGF receptors contribute to wound healing and soft-tissue homeostasis. PDGF is released by platelets at the site of injury and promotes chemotaxis and proliferation of fibroblasts and vascular mural cells, as well as the synthesis of extracellular matrix components, all being key steps to initiate wound repair. Although PDGF effectively stimulates healing of difficult wound in patients, it is mostly redundant with other growth factors [19, 20]. In addition, mouse studies suggested new functions for PDGFRβ in bone regeneration, heart regeneration, and adipose tissue homeostasis [21,22,23]. Finally, PDGFRβ signaling is involved in interstitial fluid pressure homeostasis, by counteracting the decrease of interstitial fluid pressure observed during acute inflammatory events. This mechanism implicates the PI3K pathway as well as the α2β1 integrin system to modulate tension between cells and extracellular matrix structures [24, 25]. The importance of PDGFRβ as a regulator of interstitial fluid pressure is further strengthen by the frequent observation of edema in patients receiving a PDGFR inhibitor, such as imatinib (see below for details). Interestingly, imatinib also induces growth deceleration in children treated for chronic myeloid leukemia, suggesting a role for PDGF receptors in bone formation after birth [26].
Hematopoietic neoplasms
The first PDGF receptor gene alterations were described in hematologic malignancies. In myeloid neoplasms associated with hypereosinophilia, chromosomal rearrangements in hematopoietic progenitors produce fusion genes, such as FIP1L1-PDGFRA and ETV6-PDGFRB, which have been reviewed extensively [27,28,29,30]. These fusion proteins are comparable to BCR-ABL1 and consist of an N-terminal domain that originates from the fusion partner and a C-terminal part that contains the kinase domain of PDGFR [29, 30]. These fusion proteins are activated by either disruption of the autoinhibitory juxta-membrane domain (in the case of FIP1L1-PDGFRA for instance) or by constitutive oligomerization driven by the partner protein. Expression of these constitutively activated PDGFR hybrid proteins is controlled by the promoter sequence of the partner gene, explaining why PDGFR fusion proteins accumulate in hematopoietic cells, which do not express wild-type PDGFR [31]. Similar PDGFRB fusions genes (such as EBF1-PDGFRB) have been reported in B-cell acute lymphoblastic leukemia [32].
Beside fusion genes, gain-of-function point mutations in PDGFRA have been described in myeloid neoplasms associated with hypereosinophilia [33]. These mutations are located in the N-lobe of the kinase domain, the insert or the activation loop (Fig. 3). However, this observation awaits confirmation in another cohort of patients.

Localization of PDGFR mutations associated with human diseases. Gain-of-function mutations are marked in bold, and loss-of-function mutations are marked in italics. Uncharacterized variants are indicated with an asterisk (*). Signal peptide (SP); extracellular domain (ECD); transmembrane (TM); juxtamembrane (JM); kinase domain (KD)
A recurrent somatic p.N666S mutation in PDGFRB has also been found in stromal cells of patients affected with a rare lymphoproliferative disorder called unicentric Castleman disease (UCD). This mutation is located in the N-lobe of the kinase domain and triggers PDGFRβ constitutive activation [34].
Finally, a recent study identified a germline activating PDGFRB p.R853W variant in hereditary progressive mucinous histiocytosis, a rare type of histiocytosis transmitted in an autosomal-dominant mode [35].
Tumors of the gastrointestinal tract
Gastrointestinal stromal tumors (GISTs) are soft-tissue tumors of the gastrointestinal tract. Most cases harbor a mutation in KIT, which belongs to the same RTK class as PDGFR. A low proportion of sporadic cases (5–10%) present somatic mutations in PDGFRA. These mutations are mainly located in the activation loop (exon 18) and, to a lesser extent, in the juxtamembrane domain (exon 12) or in the N-lobe of the kinase domain (exon 14) (Fig. 3) [2, 36, 37].
In contrast to GIST, inflammatory fibroid polyps (IFPs) are benign lesions caused by excessive tissue proliferation and inflammatory cell infiltration into the lumen of the gastrointestinal tract. The PDGFRA regions affected by somatic mutations in IFP are the same as those in GIST (exons 12, 14 and 18) (Fig. 3), but they occur more frequently (at a rate of approximately 55%) [38, 39].
Germline PDGFRA mutations have been described in the so-called “PDGFRA-mutant syndrome” in three families with an autosomal-dominant mode of inheritance and in two unrelated individuals [40,41,42,43]. The presence of at least two IFPs (including fibrous tumors) and/or GISTs in one individual or in a family characterize this syndrome. Some patients also have gastrointestinal lipomas and large hands [41]. Mutations in exon 12 (p.Y555C, p.V561D), exon 14 (p.P653L) and exon 18 (p.D846Y) have been identified. To date, this is the only disease associated with germline PDGFRA mutation.
Brain tumors
Both adult and pediatric forms of glioblastoma are associated with somatic alterations in PDGFRA [44, 45]. These variants include missense mutations, in-frame insertions or deletions, as well as gene amplification. Point mutations are located within the extracellular domain, the transmembrane and the kinase domains of the receptor (Fig. 3) [46, 47]. In children, PDGFRA mutations are found in approximately 37% of diffuse intrinsic pontine glioma cases, constituting a particularly aggressive subtype of glioblastoma [48]. In adults, most PDGFRA-amplified tumors harbor a genomic deletion of exons 8 and 9, the region coding for D4 and D5 in the extracellular part of the receptor (Fig. 3) [45]. One adult case with a gene fusion between PDGFRA and VEGFR2 has also been reported [44]. In vitro and in vivo analyses have demonstrated that these oncogenic mutations constitutively activate PDGFRα [44, 45, 47].
More recently, recurrent PDGFRA mutations have been identified in low-grade neural tumors associated with intractable seizures, named septal dysembryoplastic neuroepithelial tumors or myxoid glioneuronal neoplasms [49,50,51]. These PDGFRA variants mostly consist of dinucleotide substitutions at codon K385 in the immunoglobulin-like module D4 of PDGFRα, which is responsible for receptor-receptor interaction and dimerization after ligand binding.
Finally, a subset of gliomas, called IDH-mutant gliomas, are associated with strong PDGFRα expression, in the absence of PDGFRA locus alteration. IDH mutations are responsible for epigenetic alterations and DNA hypermethylation, impairing proper gene regulation by insulators and leading to aberrant PDGFRA activation [52]. This may drive glioma tumorigenesis by the activation of oncogenic signaling pathways in the absence of canonical kinase mutations.
Myofibromas and infantile myofibromatosis
Myofibromas are the most common fibrous tumors in children. They consist of firm, nontender, flesh-colored nodules that may develop in any organ of the body but without metastasis or invasion of adjacent anatomical structures. The presence of multiple myofibromas defines infantile myofibromatosis (IM). Generalized IM refers to visceral involvement and has a poor outcome [53]. Most IM cases are sporadic, but several familial cases have been reported with an autosomal-dominant mode of inheritance (MIM #228550).
Whole-exome sequencing has identified germline heterozygous mutations in PDGFRB cosegregating with the disease in 18 out of 19 unrelated families [54,55,56,57,58,59]. The p.R561C mutation, reported in 14 of the 19 families, is the most commonly reported variant. To date, all identified germline mutations in IM affect the JM domain (Fig. 3) [54]. Functional assays confirmed that these variants confer constitutive activation of PDGFRβ [54, 60]. Furthermore, the ability of several of these variants to transform cells has been demonstrated both in vitro and in vivo [60, 61].
The p.R561C variant seems to be a weak activator of PDGFRβ in vitro compared to other described mutants [60]. Furthermore, a second somatic mutation (p.N666K), identified in a few patient lesions, shows that the presence of two variants in the same allele considerably increases receptor activity [54, 61, 62]. A “two-hit model” for familial IM was proposed, where a weak germline gain-of-function PDGFRB mutation affecting the JM domain needs a somatic second hit to initiate myofibroma development [61]. Furthermore, this two-hit model may explain the suspected incomplete penetrance of familial IM, as reported in three studies [57,58,59], in which the transmission of p.R561C, or any other weakly activated variant, would be necessary but not sufficient for disease development.
Sporadic cases of IM are more frequent. Recently, tumor sequencing analyses identified a broad catalog of gain-of-function mutations in PDGFRB [61,62,63,64,65]. Most of these variants are located in the JM and kinase domains, the classical hotspots for oncogenic mutations in RTK [61,62,63]. These mutations include single-nucleotide variants (SNV) as well as more complex in-frame indels and duplications in the JM domain, which are highly reminiscent of the internal tandem duplications in FLT3 described in acute myeloid leukemia [30, 62]. Interestingly, oncogenic variants in the D5 and transmembrane domains have also been reported [62].
Kosaki overgrowth syndrome
Kosaki syndrome (MIM #616737) is a rare overgrowth syndrome described in 2015 by Takenouchi and Kosaki in two young female patients [66]. To date, a total of nine cases have been reported in the literature [66,67,68,69,70]. The spectrum of symptoms commonly found in patients includes postnatal and somatic overgrowth, distinctive facial features, hyperelastic and fragile skin, white matter lesions, neurologic deterioration, and myofibroma. Vascular lesions, specifically aneurysms, have also been reported in four individuals [67, 69, 70].
Sequencing analyses led to the identification of three de novo germline heterozygous mutations in PDGFRB. The p.P584R and p.W566R variants, reported in five and three individuals, respectively, affect the juxtamembrane part of the receptor [66, 67, 69, 70]. In vitro experiments demonstrate that both mutations confer constitutive signaling as well as transforming capacity to PDGFRβ.
Foster and colleagues [67] identified a third variant, namely, p.S493C, in one patient. Interestingly, this mutation is located in the extracellular part of the receptor within the Ig-like domain 5. Further functional investigations are necessary to confirm the impact of this mutant on receptor activity.
Penttinen premature aging syndrome
Penttinen and colleagues reported for the first time a distinctive progeroid disorder (MIM #601812) in a 10-year-old Finnish boy. The disorder is characterized by a prematurely aged appearance with lipoatrophy, epidermal and dermal atrophy along with hypertrophic lesions that include scars, thin hair, proptosis, underdeveloped cheekbones, and marked acro-osteolysis [71]. Johnston and colleagues linked Penttinen syndrome to the heterozygous PDGFRB p.V665A de novo mutation [72]. Functional explorations of the variant have revealed that p.V665A induces STAT3 and PLCγ phosphorylation, resulting in constitutive activation of PDGFRβ. Nevertheless, the signaling of this mutant appears to be slightly perturbed, as no phosphorylation of other classical PDGFR signaling mediator, such as SRC, AKT, and ERK, was detected. Additionally, He and colleagues observed that this mutant is a particularly strong activator of STAT1 signaling, leading to an interferon-like inflammatory response [73].
Another gain-of-function variant, p.N666S, was found in two patients affected with a severe form of Penttinen syndrome [74]. Similar to the p.V665A mutant, PDGFRB p.N666S seems to be a particularly potent activator of STAT1 phosphorylation.
Aneurysms
Aneurysms are vascular lesions that have been reported in several patients affected with PDGFRB-related disorders.
The first reported case was a young girl initially diagnosed with generalized IM who had aneurysmal dilatations of the renal and iliac arteries reminiscent of fibromuscular dysplasia (FMD) [61, 75]. The tumors and the vascular wall were later shown to harbor a p.D850V somatic mutation in PDGFRB [61, 62]. One additional similar case was reported by Wright et al. [76].
In 2019, Karasozen and colleagues identified different somatic and activating mutations in PDGFRB in 4 of 6 analyzed fusiform cerebral aneurysms [77]. They suggested that the presence of these somatic variants in the cerebral vasculature disrupts proper formation of the media layer surrounding arteries and acts as a driver in the development of aneurysms. Most reported mutations are identical to those found in IM. These findings are consistent with the recent report of a fatal stroke in a patient with Kosaki syndrome as a consequence of a ruptured fusiform cerebral aneurysm [67].
Finally, aneurysms and cardiovascular lesions have been reported in a 13-year-old individual previously diagnosed with Kosaki overgrowth syndrome. In this case, echocardiogram and head and neck angiography revealed an abnormal coronary artery system with saccular aneurysms, as well as tortuosity of the cervical vertebral arteries. The patient suddenly died 6 years later, but no autopsy was performed to confirm aneurysm rupture as the cause of death [70].
In conclusion, these findings clearly establish a link between PDGFRB variants and aneurysms. However, further investigation is needed to determine the exact prevalence of PDGFRB mutations in vascular anomalies. These observations are consistent with the expression and physiological role of PDGFRβ in vascular smooth muscle cells, which migrate and proliferate upon activation of the receptor.
Definition of a mixed PDGFRB syndrome?
To date, distinct entities have been used to classify diseases related to germline PDGFRB gain-of-function mutations. However, many patients present symptoms overlapping between the different syndromes described above. For instance, some patients initially diagnosed with Kosaki syndrome also develop myofibroma, and a few individuals with Penttinen syndrome present overgrowth similar to Kosaki syndrome [66, 69, 78]. Among other examples, one patient harboring the variant p.N666H presented features overlapping between Kosaki and Penttinen syndromes, and the p.Arg561_Tyr562delinsH indel reported in one case of sporadic IM was also found in another individual presenting severe clinical features of IM, Kosaki, and Penttinen syndromes [62, 79, 80].
These observations suggest that a set of features might be shared by patients harboring germline PDGFRB gain-of-function mutations, which could be grouped in a single entity, the PDGFRB mutant syndrome (by analogy with the PDGFRA-mutant syndrome defined above). A recent study [81] has proposed redefining the spectrum of diseases associated with activating PDGFRB variants into two distinct but still overlapping groups: PDGFRB-activating variant spectrum disorder 1 and 2 (PAVS1 and PAVS2). Patients diagnosed with IM and whose nodules regress over time are classified within the PAVS1 group. More complex syndromes involving multisystemic and progressive abnormalities are included in the second PAVS2 group. Nevertheless, the two categories are not mutually exclusive, and patients in the PAVS1 group remain at risk of developing potentially fatal comorbidities and especially aneurysms over time. Similarly, the appearance of myofibroma in patients within the PAVS2 entity cannot be ruled out. PAVS1 is associated with the weakly activated p.R561C germline variant and a range of somatic mutations, while more potent germline variants cause PAVS2. The molecular mechanism explaining the phenotypic heterogeneity among patients carrying a PDGFRB gain-of-function mutations remains to be fully understood.
Primary familial brain calcification
In contrast to the diseases described above, loss-of-function germline PDGFRB mutations have been identified in some patients with primary familial brain calcification (PFBC), previously called idiopathic basal ganglia calcification (IBGC) or Fahr disease (although this latter term should not be used in the context of PDGFRB variants) [82,83,84,85]. Patients with PFBC show symmetric and bilateral calcifications of hydroxyapatite in the basal ganglia and other areas of the brain, which are best diagnosed by cerebral computed tomography. These patients are often asymptomatic during childhood and young adulthood. Heterogeneous clinical symptoms appear between the ages of 30 and 50 years, including psychiatric manifestations, parkinsonism, movement and speech disorders, cognitive impairment, and seizures, among others. Movement disorder and cognitive impairment seem to be the most common clinical features. Some patients may remain asymptomatic [83, 86, 87].
Germline heterozygous PDGFRB mutations were found in three different families (p.L658P, p.D737N and p.M1?) [83, 85] and in apparently sporadic cases (p.R987W, p.R695C) (Fig. 3) [82,83,84]. Three of these mutations (p.L658P, p.R987W and p.R695C) lead to impaired PDGF-B/PDGFRβ signaling in cell culture assays [84, 88]. The p.E1071V substitution does not have a negative impact on the receptor activity, suggesting that it is not related to the disease [88].
Loss-of-function mutations can also be found in several other genes associated with PFBC. PDGF-B is mutated in several patients [89,90,91,92,93,94]. Two genes encoding phosphate transporters are also related to PFBC: SLC20A2 [95,96,97,98,99,100,101,102,103], which encodes a sodium-dependent phosphate transporter, and XPR1 [104, 105], encoding a transporter controlling phosphate efflux. Although SLC20A2 mutations explain approximately 50% of PFBC cases (IBGC type 1, MIM #213600), PDGF-B and PDGFRB appear to be involved in 20% of cases (classified as IBGC4, MIM #615007, and IBGC5, MIM #615483, respectively) [86].
The link between impaired PDGF signaling and PFBC is still unclear. First, defective PDGF-B/PDGFRβ signaling might result in reduced expression of SLC20A2, leading to accumulation of phosphate in the extracellular compartment and the formation of calcification [106], a mechanism explained by the direct regulation of SLC20A2 expression by PDGF [107, 108]. Second, since PDGF-B is very important for the recruitment of pericytes during vessel formation and because pericytes are a major component of the blood–brain barrier (BBB), disruption of the BBB, allowing the formation of calcium phosphate precipitates, is a possible explanation for PFBC development [16, 86, 109, 110]. Finally, other BBB cell types may be involved, such as astrocytes [86, 111].
Interestingly, PFBC kinase domain mutations cluster with IM, Kosaki and Penttinen syndrome variants in the structure of the PDGFRβ kinase domain, as deduced from the crystal structure of the PDGFRα intracellular part [6]. This region is very close to the ATP-binding pocket.
Mouse models of impaired PDGFR signaling
Different mouse models have been generated to study in vivo the functional impact of mutations within PDGFRα or PDGFRβ.
Olson and colleagues [112] have established two models of PDGFRα conditional knock-in mice harboring either the p.D842V variant within the activation loop of the kinase or the p.V561D mutation in the juxtamembrane part of the receptor. In the embryo, these mutations promote the proliferation and migration of mesenchymal progenitor cells in the gastrointestinal tract, leading to stromal hyperplasia and polyps. A similar phenotype is observed in adult mice with mosaic activation of PDGFRα: animals develop widespread organ fibrotic lesions due to fibroblast proliferation and extracellular matrix deposition, revealing the important role of PDGFRα in the activation of signaling pathways responsible for connective tissue formation. These animals rarely develop tumors unless they are crossed with Ink4a/Arf-deficient mice, leading to sarcoma [112]. GISTs have not been documented in these mice.
Similarly, the effects of increased PDGFRβ signaling have been investigated using different knock-in mouse models. The first one consisted of a weakly activated p.D849N (corresponding to D850N in the human sequence) knock-in mutation, producing a mild phenotype [113]. Mice expressing the PDGFRB p.D849V mutant present growth deficiency during the second week after birth and usually die by day 14 [114]. Constitutive PDGFRβ signaling leads to vascular smooth muscle cell proliferation while inhibiting their differentiation, and to pericyte activation with increased coverage around blood vessels. These changes in pericyte differentiation correlate with an immune response, autoinflammation, and lipoatrophy, a set of phenomena that seem to occur via PDGFRβ-dependent STAT1 activation [114, 115]. Nevertheless, this vascular phenotype does not reflect the human phenotype associated with germline PDGFRB mutant syndromes. These mice do not develop tumors.
The mechanism of autoinflammation through PDGFRβ-STAT1 signaling was further studied by He and colleagues [73] in several mouse models. STAT1 knockout rescues the autoinflammatory phenotype observed in mice harboring the p.D849V variant. These animals show increased lifespan but progressively develop fibrosis and skeletal overgrowth. Although these results are reminiscent of some of the features observed in Penttinen or Kosaki syndromes, these animals do not constitute convincing models of the human diseases caused by activated PDGFRB mutations. Nevertheless, these findings are in line with the observation that variants associated with Penttinen syndrome (i.e., p.V665A and p.N666S) are potent activators of STAT1 in vitro [73, 74]. It is important to note that the exact germline mutations identified in humans have not yet been tested in mice.
Mouse models indicate that both PDGFRα and PDGFRβ signaling act as negative regulators of adipogenesis and adipocyte differentiation. Conditional knock-in mice with a PDGFRA p.D842V activating mutation in adipocyte progenitors promotes the transition of these cells toward a pro-fibrotic phenotype, causing adipose fibrosis and inhibiting their differentiation in adipocytes [116]. A similar phenotype is observed in mice harboring the equivalent PDGFRB p.D849V variant [73, 114]. In contrast, mice harboring a mosaic deletion of PDGFRA or PDGFRB are marked by a significant increase in adipose tissue formation [117]. The mechanism that regulates PDGFR signaling during adipogenesis remains unclear. However, this property is reminiscent of the lipoatrophy observed in some patients with Kosaki and Penttinen syndromes, which may be due to the switch of adipocyte progenitor cells toward their pro-fibrotic phenotype while inhibiting their differentiation.
A role for PDGFRβ was also suggested in myocardium regeneration through a signaling pathway involving the phosphorylation of Akt followed by EZH2-mediated Ink4a/Arf repression. This function was further studied using an AAV9 gene therapy vector to introduce the PDGFRB p.D849V variant in cardiomyocytes, to analyze heart regeneration and systolic function in mice [23]. This study provides an elegant model to evaluate the impact of PDGFR mutations in various systems.
PDGFRβ signaling in renal mesenchymal cells is essential for kidney development. In mice, specific deletion of Pdgfrb in these cells is responsible for impaired glomerulogenesis and death one month after birth [10]. In adults, the role of renal mesenchymal PDGFRβ signaling is assumed to be minor under physiological conditions; however, receptor activation increases in kidney diseases [118]. Mice harboring specific activation of PDGFRβ in renal mesenchymal cells progressively develop primary fibrosis but without any sign of inflammation or STAT1 activation [10]. Additionally, inhibition strategies using either specific antibodies or selective tyrosine-kinase inhibitors improve kidney lesions in various renal disease models [119,120,121,122]. Altogether, these results suggest that patients with a PDGFRB mutation may be at risk of developing renal disease. Interestingly, kidney failure and transplantation were reported in a complex case with generalized IM, severe hypertension and a renal artery aneurysm [123].
In conclusion, gain-of-function mutations in PDGFRα/PDGFRβ in mice seem to greatly impair the differentiation of cells of mesenchymal origin, such as adipocyte progenitors, vascular smooth muscle cells, pericytes, and renal mesenchymal cells. This promotes tissue fibrosis and organ failure, but tumors do not develop in these mice.
As described above, heterozygous loss-of-function variants have been identified in primary familial brain calcification. However, this is not observed in heterozygous Pdgfrb knockout mice, and the homozygous deletion of the receptor is lethal. Nevertheless, Keller et al. studied mice harboring hypomorphic mutations in the gene encoding PDGF-B. The mutant mice progressively develop brain calcifications and phenotypic features highly similar to those described in humans with PFBC [90]. These results provide strong evidence for a causative role of the loss of PDGFRβ signaling in the development of brain calcifications.
Clinical implications
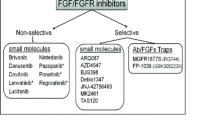
Several types of drugs target PDGF receptors, including tyrosine kinase inhibitors (TKIs), antibodies and aptamers. TKIs have been approved for the treatment of various PDGFR-driven cancers.
Imatinib is a type II TKI and the first molecule described to inhibit KIT, ABL, and PDGFRs. The compound was initially developed to treat patients with BCR-ABL1-positive leukemia and, later, KIT-mutated GIST. Moreover, good response and long-term remission have been achieved in patients affected with myeloid neoplasms associated with hypereosinophilia and PDGFR gene rearrangement, such as ETV6-PDGFRB and FIP1L1-PDGFRA [29, 124,125,126,127]. Of interest, one 36-year-old man and two children diagnosed with a multicentric form of myofibroma and harboring gain-of-function variants in PDGFRβ received imatinib monotherapy, which considerably improved their quality of life [65, 80, 81]. Finally, imatinib was administered to one boy affected with a complex syndrome related to the PDGFRB p.N666H variant [80] and in one individual diagnosed with Penttinen syndrome linked to the p.V665A variant [81]. In both cases, the treatment ameliorated several of the symptoms related to the disease. These are examples of successful application of targeted therapy using imatinib in both adults and children affected with a rare disease.
In vitro, the activity of most gain-of-function PDGFR variants is blocked when exposed to imatinib, except for PDGFRA p.D842V and its equivalent p.D850V mutation in PDGFRB, as well as p.D816V mutant in KIT. These mutations, which are located in the activation loop of the kinase domain, shift and stabilize the kinase in its active conformation. As a result, they decrease the affinity of type II TKIs, which act by occupying the ATP-binding pocket of the kinase in its inactive state.
Type I TKIs bind to the ATP-binding pocket of the kinase in the active conformation and are potentially able to counteract the resistance of mutations located within the activation loop. For example, crenolanib was able to inhibit the PDGFRA p.D842V mutation in imatinib-resistant GIST [128]. However, high activity across the human kinome associated with an increased risk of toxicity limits the routine use of this drug [129]. Avapritinib is another compound able to inhibit PDGFRA p.D842V activation loop mutants [129]. The drug was approved by the European Medicine Agency (EMA) and the Food and Drug Administration (FDA) for patients with GIST harboring the PDGFRA p.D842V mutation.
Neutralizing antibodies such as olarutumab and rinucumab target PDGFRα or PDGFRβ, respectively [130, 131]. Phase Ib and II clinical trials initially showed promising results for olarutumab in combination with doxorubicin to treat patients with soft-tissue sarcoma [132]. However, a recent phase III study did not confirm any patient benefit [133]. An anti-PDGFRβ antibody was tested in combination with an anti-VEGF in the treatment of neovascular age-related macular degeneration. Despite promising results in vitro and in vivo, recent clinical trials failed to provide visual and anatomical improvements [130, 134]. Taken together, using anti-PDGFR antibodies in therapy as a single agent or in combination did not lead to significant improvement in clinical outcome.
PDR3 and Gint4.T are nuclease-resistant RNA aptamers designed to inhibit PDGFRα and PDGFRβ, respectively [135, 136]. These molecules act by binding the ectodomain of the receptor, preventing ligand-dependent receptor activation and signaling. Both aptamers show antitumor effects in glioblastoma cell culture. Moreover, in vitro assays show that when coupled to the receptor, Gint4.T-PDGFRB and PDR3-PDGFRA complexes are rapidly internalized and targeted to the endolysosomal system. This feature makes these two aptamers good carriers to deliver small therapeutic molecules in specific cells [137]. However, these molecules have not yet reached the clinic.
Conclusions
Different types of cancers are driven by somatic gain-of-function mutations in PDGFRA and PDGFRB, which act as classical proto-oncogenes. In addition, PDGFRB variants were associated with a number of rare noncancerous diseases. Advances in deep sequencing have considerably increased the catalog of variants identified within PDGFRB. Germline loss-of-function mutations in PDGFRB have been reported in patients with primary familial brain calcification, while PDGFRB-activating mutations are associated with several disorders, namely, infantile myofibromatosis, Kosaki and Penttinen syndromes. More recent findings also suggest a role of somatic PDGFRB gain-of-function mutations in fusiform aneurysms. Some patients show a mixed phenotype overlapping with different conditions. These clinical observations shed light on the precise role of PDGFRB in human pathophysiology and highlight a number of differences with data collected from animal models. Most PDGFRB gain-of-function mutations remain sensitive to TKIs, such as imatinib, which was successfully administered to patients affected with IM or syndromic disease. However, novel therapeutic strategies are needed for several conditions described in this review, such as PFBC or glioblastoma. Many questions remain regarding the mechanisms whereby PDGFRB mutations cause such a variety of symptoms. Future experiments should focus on the identification of activated signaling pathways specific to some variants and the cell lineage-specific effects of those mutants.
References
Demoulin JB, Essaghir A (2014) PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev 25(3):273–283. https://doi.org/10.1016/j.cytogfr.2014.03.003
Andrae J, Gallini R, Betsholtz C (2008) Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22(10):1276–1312. https://doi.org/10.1101/gad.1653708
Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79(4):1283–1316
Hoch RV, Soriano P (2003) Roles of PDGF in animal development. Development 130(20):4769–4784. https://doi.org/10.1242/dev.00721
Verstraete K, Savvides SN (2012) Extracellular assembly and activation principles of oncogenic class III receptor tyrosine kinases. Nat Rev Cancer 12(11):753–766. https://doi.org/10.1038/nrc3371
Liang L, Yan X-E, Yin Y, Yun C-H (2016) Structural and biochemical studies of the PDGFRA kinase domain. Biochem Biophys Res Commun 477(4):667–672. https://doi.org/10.1016/j.bbrc.2016.06.117
Hye-Ryong Shim A, Liu H, Focia PJ, Chen X, Lin PC, He X (2010) Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc Natl Acad Sci 107(25):11307. https://doi.org/10.1073/pnas.1000806107
Chen PH, Unger V, He X (2015) Structure of full-length human PDGFRbeta bound to its activating ligand PDGF-B as determined by negative-stain electron microscopy. J Mol Biol 427(24):3921–3934. https://doi.org/10.1016/j.jmb.2015.10.003
Beenstock J, Mooshayef N, Engelberg D (2016) How do protein kinases take a selfie (autophosphorylate)? Trends Biochem Sci 41(11):938–953. https://doi.org/10.1016/j.tibs.2016.08.006
Buhl EM, Djudjaj S, Klinkhammer BM, Ermert K, Puelles VG, Lindenmeyer MT, Cohen CD, He C, Borkham-Kamphorst E, Weiskirchen R, Denecke B, Trairatphisan P, Saez-Rodriguez J, Huber TB, Olson LE, Floege J, Boor P (2020) Dysregulated mesenchymal PDGFR-beta drives kidney fibrosis. EMBO Mol Med 12(3):e11021. https://doi.org/10.15252/emmm.201911021
Zhang J, Yang PL, Gray NS (2009) Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9(1):28–39. https://doi.org/10.1038/nrc2559
Heldin C-H, Lennartsson J (2013) Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb Perspect Biol 5(8):a009100–a009100. https://doi.org/10.1101/cshperspect.a009100
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134. https://doi.org/10.1016/j.cell.2010.06.011
Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O’Rourke M, Koh GY, Soriano P, Betsholtz C, Hart TC, Marazita ML, Field LL, Tam PP, Nagy A (2004) A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet 36(10):1111–1116. https://doi.org/10.1038/ng1415
Soriano P (1997) The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124(14):2691–2700
Lindahl P, Johansson BR, Leveen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277(5323):242–245
Soriano P (1994) Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev 8(16):1888–1896
Gladh H, Folestad EB, Muhl L, Ehnman M, Tannenberg P, Lawrence AL, Betsholtz C, Eriksson U (2016) Mice lacking platelet-derived growth factor D display a mild vascular phenotype. PLoS ONE 11(3):e0152276. https://doi.org/10.1371/journal.pone.0152276
Lynch SE, Nixon JC, Colvin RB, Antoniades HN (1987) Role of platelet-derived growth factor in wound healing: synergistic effects with other growth factors. Proc Natl Acad Sci U S A 84(21):7696–7700. https://doi.org/10.1073/pnas.84.21.7696
Pierce GF, Mustoe TA, Altrock BW, Deuel TF, Thomason A (1991) Role of platelet-derived growth factor in wound healing. J Cell Biochem 45(4):319–326. https://doi.org/10.1002/jcb.240450403
Böhm AM, Dirckx N, Tower RJ, Peredo N, Vanuytven S, Theunis K, Nefyodova E, Cardoen R, Lindner V, Voet T, Van Hul M, Maes C (2019) Activation of skeletal stem and progenitor cells for bone regeneration is driven by PDGFRβ signaling. Dev Cell 51(2):236-254.e212. https://doi.org/10.1016/j.devcel.2019.08.013
Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, Gupta RK (2016) Pdgfrβ+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab 23(2):350–359. https://doi.org/10.1016/j.cmet.2015.10.018
Yue Z, Chen J, Lian H, Pei J, Li Y, Chen X, Song S, Xia J, Zhou B, Feng J, Zhang X, Hu S, Nie Y (2019) PDGFR-β signaling regulates cardiomyocyte proliferation and myocardial regeneration. Cell Rep 28(4):966-978.e964. https://doi.org/10.1016/j.celrep.2019.06.065
Gullberg D, Tingström A, Thuresson A-C, Olsson L, Terracio L, Borg TK, Rubin K (1990) β1 Integrin-mediated collagen gel contraction is stimulated by PDGF. Exp Cell Res 186(2):264–272. https://doi.org/10.1016/0014-4827(90)90305-T
Heuchel R, Berg A, Tallquist M, Åhlén K, Reed RK, Rubin K, Claesson-Welsh L, Heldin C-H, Soriano P (1999) Platelet-derived growth factor β receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3′ kinase signaling. Proc Natl Acad Sci 96(20):11410–11415. https://doi.org/10.1073/pnas.96.20.11410
Millot F, Guilhot J, Baruchel A, Petit A, Leblanc T, Bertrand Y, Mazingue F, Lutz P, Vérité C, Berthou C, Galambrun C, Sirvent N, Yacouben K, Chastagner P, Gandemer V, Reguerre Y, Couillault G, Khalifeh T, Rialland F (2014) Growth deceleration in children treated with imatinib for chronic myeloid leukaemia. Eur J Cancer 50(18):3206–3211. https://doi.org/10.1016/j.ejca.2014.10.007
Appiah-Kubi K, Lan T, Wang Y, Qian H, Wu M, Yao X, Wu Y, Chen Y (2017) Platelet-derived growth factor receptors (PDGFRs) fusion genes involvement in hematological malignancies. Crit Rev Oncol/Hematol 109:20–34. https://doi.org/10.1016/j.critrevonc.2016.11.008
Havelange V, Demoulin JB (2013) Review of current classification, molecular alterations, and tyrosine kinase inhibitor therapies in myeloproliferative disorders with hypereosinophilia. J Blood Med 4:111–121. https://doi.org/10.2147/JBM.S33142
Medves S, Demoulin JB (2012) Tyrosine kinase gene fusions in cancer: translating mechanisms into targeted therapies. J Cell Mol Med 16(2):237–248. https://doi.org/10.1111/j.1582-4934.2011.01415.x
Toffalini F, Demoulin J-B (2010) New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood 116(14):2429–2437. https://doi.org/10.1182/blood-2010-04-279752
Demoulin JB, Montano-Almendras CP (2012) Platelet-derived growth factors and their receptors in normal and malignant hematopoiesis. Am J Blood Res 2(1):44–56
Schwab C, Ryan SL, Chilton L, Elliott A, Murray J, Richardson S, Wragg C, Moppett J, Cummins M, Tunstall O, Parker CA, Saha V, Goulden N, Vora A, Moorman AV, Harrison CJ (2016) EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood 127(18):2214–2218. https://doi.org/10.1182/blood-2015-09-670166
Elling C, Erben P, Walz C, Frickenhaus M, Schemionek M, Stehling M, Serve H, Cross NC, Hochhaus A, Hofmann WK, Berdel WE, Muller-Tidow C, Reiter A, Koschmieder S (2011) Novel imatinib-sensitive PDGFRA-activating point mutations in hypereosinophilic syndrome induce growth factor independence and leukemia-like disease. Blood 117(10):2935–2943. https://doi.org/10.1182/blood-2010-05-286757
Li Z, Lan X, Li C, Zhang Y, Wang Y, Xue W, Lu L, Jin M, Zhou Z, Wang X, Li L, Zhang L, Li X, Fu X, Sun Z, Wu J, Zhang X, Yu H, Nan F, Chang Y, Yan J, Wu X, Wang G, Zhang D, Zhang Y, Young KH, Zhang M (2019) Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia 33(4):1035–1038. https://doi.org/10.1038/s41375-018-0323-6
Onoufriadis A, Boulouadnine B, Dachy G, Higashino T, Huang HY, Hsu CK, Simpson MA, Bork K, Demoulin JB, McGrath J (2021) Germline mutation in PDGFRB may be implicated in hereditary progressive mucinous histiocytosis. Br J Dermatol (In press)
Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC (2005) PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 23(23):5357–5364. https://doi.org/10.1200/JCO.2005.14.068
Lasota J, Stachura J, Miettinen M (2006) GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Laboratory investigation; a journal of technical methods and pathology 86 (1):94–100. doi:https://doi.org/10.1038/labinvest.3700360
Huss S, Wardelmann E, Goltz D, Binot E, Hartmann W, Merkelbach-Bruse S, Buttner R, Schildhaus HU (2012) Activating PDGFRA mutations in inflammatory fibroid polyps occur in exons 12, 14 and 18 and are associated with tumour localization. Histopathology 61(1):59–68. https://doi.org/10.1111/j.1365-2559.2012.04203.x
Lasota J, Wang ZF, Sobin LH, Miettinen M (2009) Gain-of-function PDGFRA mutations, earlier reported in gastrointestinal stromal tumors, are common in small intestinal inflammatory fibroid polyps. A study of 60 cases. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc 22 (8):1049–1056. doi:https://doi.org/10.1038/modpathol.2009.62
Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, Lenoir GM, Bressac-De Paillerets B (2004) PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 126(1):318–321
Ricci R, Martini M, Cenci T, Carbone A, Lanza P, Biondi A, Rindi G, Cassano A, Larghi A, Persiani R, Larocca LM (2015) PDGFRA-mutant syndrome. Modern Pathology: an official journal of the United States and Canadian Academy of Pathology, Inc 28 (7):954–964. doi:https://doi.org/10.1038/modpathol.2015.56
Carney JA, Stratakis CA (2008) Stromal, fibrous, and fatty gastrointestinal tumors in a patient with a PDGFRA gene mutation. Am J Surg Pathol 32(9):1412–1420. https://doi.org/10.1097/PAS.0b013e31816250ce
Ricci R, Martini M, Cenci T, Riccioni ME, Maria G, Cassano A, Larocca LM (2016) Divergent gastrointestinal stromal tumors in syndromic settings. Cancer Genetics 209(7–8):354–358. https://doi.org/10.1016/j.cancergen.2016.05.073
Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, Huse JT, Pedraza A, Utsuki S, Yasui Y, Tandon A, Fomchenko EI, Oka H, Levine RL, Fujii K, Ladanyi M, Holland EC (2010) PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev 24(19):2205–2218. https://doi.org/10.1101/gad.1972310
Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, Broniscer A, Wetmore C, Zhang J, Jones C, Ellison DW, Baker SJ (2013) Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 73(20):6219–6229. https://doi.org/10.1158/0008-5472.CAN-13-1491
Ip CKM, Ng PKS, Jeong KJ, Shao SH, Ju Z, Leonard PG, Hua X, Vellano CP, Woessner R, Sahni N, Scott KL, Mills GB (2018) Neomorphic PDGFRA extracellular domain driver mutations are resistant to PDGFRA targeted therapies. Nat Commun 9(1):4583. https://doi.org/10.1038/s41467-018-06949-w
Velghe AI, Van Cauwenberghe S, Polyansky AA, Chand D, Montano-Almendras CP, Charni S, Hallberg B, Essaghir A, Demoulin JB (2014) PDGFRA alterations in cancer: characterization of a gain-of-function V536E transmembrane mutant as well as loss-of-function and passenger mutations. Oncogene 33(20):2568–2576. https://doi.org/10.1038/onc.2013.218
Lapin DH, Tsoli M, Ziegler DS (2017) Genomic insights into diffuse intrinsic pontine glioma. Front Oncol 7:57–57. https://doi.org/10.3389/fonc.2017.00057
Chiang JCH, Harreld JH, Tanaka R, Li X, Wen J, Zhang C, Boué DR, Rauch TM, Boyd JT, Chen J, Corbo JC, Bouldin TW, Elton SW, Liu LL, Schofield D, Lee SC, Bouffard JP, Georgescu MM, Dossani RH, Aguiar MA, Sances RA, Saad AG, Boop FA, Qaddoumi I, Ellison DW (2019) Septal dysembryoplastic neuroepithelial tumor: a comprehensive clinical, imaging, histopathologic, and molecular analysis. Neuro Oncol 21(6):800–808. https://doi.org/10.1093/neuonc/noz037
Lucas CG, Villanueva-Meyer JE, Whipple N, Oberheim Bush NA, Cooney T, Chang S, McDermott M, Berger M, Cham E, Sun PP, Putnam A, Zhou H, Bollo R, Cheshier S, Poppe MM, Fung KM, Sung S, Glenn C, Fan X, Bannykh S, Hu J, Danielpour M, Li R, Alva E, Johnston J, Van Ziffle J, Onodera C, Devine P, Grenert JP, Lee JC, Pekmezci M, Tihan T, Bollen AW, Perry A, Solomon DA (2020) Myxoid glioneuronal tumor, PDGFRA p.K385-mutant: clinical, radiologic, and histopathologic features. Brain pathology 30 (3):479–494. doi:https://doi.org/10.1111/bpa.12797
Solomon DA, Korshunov A, Sill M, Jones DTW, Kool M, Pfister SM, Fan X, Bannykh S, Hu J, Danielpour M, Li R, Johnston J, Cham E, Cooney T, Sun PP, Oberheim Bush NA, McDermott M, Van Ziffle J, Onodera C, Grenert JP, Bastian BC, Villanueva-Meyer JE, Pekmezci M, Bollen AW, Perry A (2018) Myxoid glioneuronal tumor of the septum pellucidum and lateral ventricle is defined by a recurrent PDGFRA p.K385 mutation and DNT-like methylation profile. Acta Neuropathol 136 (2):339–343. doi:https://doi.org/10.1007/s00401-018-1883-2
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529(7584):110–114. https://doi.org/10.1038/nature16490
Mashiah J, Hadj-Rabia S, Dompmartin A, Harroche A, Laloum-Grynberg E, Wolter M, Amoric J-C, Hamel-Teillac D, Guero S, Fraitag S, Bodemer C (2014) Infantile myofibromatosis: a series of 28 cases. J Am Acad Dermatol 71(2):264–270. https://doi.org/10.1016/j.jaad.2014.03.035
Cheung YH, Gayden T, Campeau PM, LeDuc CA, Russo D, Nguyen VH, Guo J, Qi M, Guan Y, Albrecht S, Moroz B, Eldin KW, Lu JT, Schwartzentruber J, Malkin D, Berghuis AM, Emil S, Gibbs RA, Burk DL, Vanstone M, Lee BH, Orchard D, Boycott KM, Chung WK, Jabado N (2013) A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92(6):996–1000. https://doi.org/10.1016/j.ajhg.2013.04.026
Ito N, Watanabe S, Mishima H, Kinoshita A, Okada M, Moriuchi H, Yoshiura K-i (2019) A mutation in PDGFRB in a family with infantile myofibromatosis. Acta Medica Nagasakiensia 63(1):49–53. https://doi.org/10.11343/amn.63.49
Lepelletier C, Al-Sarraj Y, Bodemer C, Shaath H, Fraitag S, Kambouris M, Hamel-Teillac D, El Shanti H, Hadj-Rabia S (2017) Heterozygous PDGFRB mutation in a three-generation family with autosomal dominant infantile myofibromatosis. Acta Derm Venereol 97(7):858–859. https://doi.org/10.2340/00015555-2671
Linhares ND, Freire MC, Cardenas RG, Bahia M, Puzenat E, Aubin F, Pena SD (2014) Modulation of expressivity in PDGFRB-related infantile myofibromatosis: a role for PTPRG? Genet Mol Res 13(3):6287–6292. https://doi.org/10.4238/2014.August.15.11
Martignetti JA, Tian L, Li D, Ramirez MC, Camacho-Vanegas O, Camacho SC, Guo Y, Zand DJ, Bernstein AM, Masur SK, Kim CE, Otieno FG, Hou C, Abdel-Magid N, Tweddale B, Metry D, Fournet JC, Papp E, McPherson EW, Zabel C, Vaksmann G, Morisot C, Keating B, Sleiman PM, Cleveland JA, Everman DB, Zackai E, Hakonarson H (2013) Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet 92(6):1001–1007. https://doi.org/10.1016/j.ajhg.2013.04.024
Murray N, Hanna B, Graf N, Fu H, Mylene V, Campeau PM, Ronan A (2017) The spectrum of infantile myofibromatosis includes both non-penetrance and adult recurrence. Eur J Med Genet 60(7):353–358. https://doi.org/10.1016/j.ejmg.2017.02.005
Arts FA, Chand D, Pecquet C, Velghe AI, Constantinescu S, Hallberg B, Demoulin JB (2016) PDGFRB mutants found in patients with familial infantile myofibromatosis or overgrowth syndrome are oncogenic and sensitive to imatinib. Oncogene 35(25):3239–3248. https://doi.org/10.1038/onc.2015.383
Arts FA, Sciot R, Brichard B, Renard M, de Rocca SA, Dachy G, Noel LA, Velghe AI, Galant C, Debiec-Rychter M, Van Damme A, Vikkula M, Helaers R, Limaye N, Poirel HA, Demoulin JB (2017) PDGFRB gain-of-function mutations in sporadic infantile myofibromatosis. Hum Mol Genet 26(10):1801–1810. https://doi.org/10.1093/hmg/ddx081
Dachy G, de Krijger RR, Fraitag S, Theate I, Brichard B, Hoffman SB, Libbrecht L, Arts FA, Brouillard P, Vikkula M, Limaye N, Demoulin JB (2019) Association of PDGFRB mutations with pediatric myofibroma and myofibromatosis. JAMA Dermatol. https://doi.org/10.1001/jamadermatol.2019.0114
Agaimy A, Bieg M, Michal M, Geddert H, Markl B, Seitz J, Moskalev EA, Schlesner M, Metzler M, Hartmann A, Wiemann S, Michal M, Mentzel T, Haller F (2017) Recurrent somatic PDGFRB mutations in sporadic infantile/solitary adult myofibromas but not in angioleiomyomas and myopericytomas. Am J Surg Pathol 41(2):195–203. https://doi.org/10.1097/pas.0000000000000752
Al Qawahmed R, Sawyer SL, Vassilyadi M, Qin W, Boycott KM, Michaud J (2019) Infantile myofibromatosis with intracranial extradural involvement and PDGFRB mutation: a case report and review of the literature. Pediatr Dev Pathol 22(3):258–264. https://doi.org/10.1177/1093526618787736
Weller JM, Keil VC, Gielen GH, Herrlinger U, Schafer N (2019) PDGRFB mutation-associated myofibromatosis: response to targeted therapy with imatinib. Am J Med Genet A 179(9):1895–1897. https://doi.org/10.1002/ajmg.a.61283
Takenouchi T, Yamaguchi Y, Tanikawa A, Kosaki R, Okano H, Kosaki K (2015) Novel overgrowth syndrome phenotype due to recurrent De Novo PDGFRB mutation. J Pediatr 166(2):483–486. https://doi.org/10.1016/j.jpeds.2014.10.015
Foster A, Chalot B, Antoniadi T, Schaefer E, Keelagher R, Ryan G, Thomas Q, Philippe C, Bruel AL, Sorlin A, Thauvin-Robinet C, Bardou M, Luu M, Quenardelle V, Wolff V, Woodley J, Vabres P, Lim D, Igbokwe R, Joseph A, Walker H, Jester A, Ellenbogen J, Johnson D, Rooke B, Moss C, Cole T, Faivre L (2020) Kosaki overgrowth syndrome: a novel pathogenic variant in PDGFRB and expansion of the phenotype including cerebrovascular complications. Clin Genet. https://doi.org/10.1111/cge.13752
Gawlinski P, Pelc M, Ciara E, Jhangiani S, Jurkiewicz E, Gambin T, Rozdzynska-Swiatkowska A, Dawidziuk M, Coban-Akdemir ZH, Guilbride DL, Muzny D, Lupski JR, Krajewska-Walasek M (2018) Phenotype expansion and development in Kosaki overgrowth syndrome. Clin Genet 93(4):919–924. https://doi.org/10.1111/cge.13192
Minatogawa M, Takenouchi T, Tsuyusaki Y, Iwasaki F, Uehara T, Kurosawa K, Kosaki K, Curry CJ (2017) Expansion of the phenotype of Kosaki overgrowth syndrome. Am J Med Genet A 173(9):2422–2427. https://doi.org/10.1002/ajmg.a.38310
Zarate YA, Boccuto L, Srikanth S, Pauly R, Ocal E, Balmakund T, Hinkle K, Stefans V, Schaefer GB, Collins RT 2nd (2019) Constitutive activation of the PI3K-AKT pathway and cardiovascular abnormalities in an individual with Kosaki overgrowth syndrome. Am J Med Genet A 179(6):1047–1052. https://doi.org/10.1002/ajmg.a.61145
Penttinen M, Niemi K-M, Vinkka-Puhakka H, Johansson R, Aula P (1997) New progeroid disorder. Am J Med Genet 69(2):182–187. https://doi.org/10.1002/(sici)1096-8628(19970317)69:2%3c182::Aid-ajmg13%3e3.0.Co;2-h
Johnston JJ, Sanchez-Contreras MY, Keppler-Noreuil KM, Sapp J, Crenshaw M, Finch NA, Cormier-Daire V, Rademakers R, Sybert VP, Biesecker LG (2015) A point mutation in PDGFRB causes autosomal-dominant penttinen syndrome. Am J Hum Genet 97(3):465–474. https://doi.org/10.1016/j.ajhg.2015.07.009
He C, Medley SC, Kim J, Sun C, Kwon HR, Sakashita H, Pincu Y, Yao L, Eppard D, Dai B, Berry WL, Griffin TM, Olson LE (2017) STAT1 modulates tissue wasting or overgrowth downstream from PDGFRβ. Genes Dev 31(16):1666–1678. https://doi.org/10.1101/gad.300384.117
Bredrup C, Stokowy T, McGaughran J, Lee S, Sapkota D, Cristea I, Xu L, Tveit KS, Hovding G, Steen VM, Rodahl E, Bruland O, Houge G (2019) A tyrosine kinase-activating variant Asn666Ser in PDGFRB causes a progeria-like condition in the severe end of Penttinen syndrome. Eur J Hum Genet 27(4):574–581. https://doi.org/10.1038/s41431-018-0323-z
Brasseur B, Chantrain CF, Godefroid N, Sluysmans T, Anslot C, Menten R, Clapuyt P, Dupont S, Vermylen C, Brichard B (2010) Development of renal and iliac aneurysms in a child with generalized infantile myofibromatosis. Pediatr Nephrol 25(5):983–986. https://doi.org/10.1007/s00467-009-1393-5
Wright C, Corbally MT, Hayes R, McDermott MB (2004) Multifocal infantile myofibromatosis and generalized fibromuscular dysplasia in a child: evidence for a common pathologic process? Pediatr Dev Pathol 7(4):385–390. https://doi.org/10.1007/s10024-003-0107-4
Karasozen Y, Osbun J, Parada C, Busald T, Tatman P, Gonzalez-Cuyar L, Hale C, Alcantara D, O’Driscoll M, Dobyns W, Murray M, Kim L, Byers P, Dorschner M, Ferreira M (2019) Somatic PDGFRB activating variants in fusiform cerebral aneurysms. Am J Human Genetics. https://doi.org/10.1016/j.ajhg.2019.03.014
Zufferey F, Hadj-Rabia S, De Sandre-Giovannoli A, Dufier JL, Leheup B, Schweitze C, Bodemer C, Cormier-Daire V, Le Merrer M (2013) Acro-osteolysis, keloid like-lesions, distinctive facial features, and overgrowth: two newly recognized patients with premature aging syndrome, Penttinen type. Am J Med Genet A 161A(7):1786–1791. https://doi.org/10.1002/ajmg.a.35984
Guimier A, Gordon CT, Hully M, Blauwblomme T, Minard-Colin V, Bole-Feysot C, Nitschke P, Oufadem M, Boddaert N, Sarnacki S, Amiel J (2019) A novel de novo PDGFRB variant in a child with severe cerebral malformations, intracerebral calcifications, and infantile myofibromatosis. Am J Med Genet A 179(7):1304–1309. https://doi.org/10.1002/ajmg.a.61151
Pond D, Arts FA, Mendelsohn NJ, Demoulin JB, Scharer G, Messinger Y (2018) A patient with germ-line gain-of-function PDGFRB p.N666H mutation and marked clinical response to imatinib. Genet Med 20 (1):142–150. doi:https://doi.org/10.1038/gim.2017.104
Wenger TL, Bly RA, Wu N, Albert CM, Park J, Shieh J, Chenbhanich J, Heike CL, Adam MP, Chang I, Sun A, Miller DE, Beck AE, Gupta D, Boos MD, Zackai EH, Everman D, Ganapathi S, Wilson M, Christodoulou J, Zarate YA, Curry C, Li D, Guimier A, Amiel J, Hakonarson H, Webster R, Bhoj EJ, Perkins JA, Dahl JP, Dobyns WB (2020) Activating variants in PDGFRB result in a spectrum of disorders responsive to imatinib monotherapy. Am J Med Genet A 182(7):1576–1591. https://doi.org/10.1002/ajmg.a.61615
Nicolas G, Pottier C, Charbonnier C, Guyant-Marechal L, Le Ber I, Pariente J, Labauge P, Ayrignac X, Defebvre L, Maltete D, Martinaud O, Lefaucheur R, Guillin O, Wallon D, Chaumette B, Rondepierre P, Derache N, Fromager G, Schaeffer S, Krystkowiak P, Verny C, Jurici S, Sauvee M, Verin M, Lebouvier T, Rouaud O, Thauvin-Robinet C, Rousseau S, Rovelet-Lecrux A, Frebourg T, Campion D, Hannequin D, French ISG (2013) Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain J Neurol 136(Pt 11):3395–3407. https://doi.org/10.1093/brain/awt255
Nicolas G, Pottier C, Maltete D, Coutant S, Rovelet-Lecrux A, Legallic S, Rousseau S, Vaschalde Y, Guyant-Marechal L, Augustin J, Martinaud O, Defebvre L, Krystkowiak P, Pariente J, Clanet M, Labauge P, Ayrignac X, Lefaucheur R, Le Ber I, Frebourg T, Hannequin D, Campion D (2013) Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80(2):181–187. https://doi.org/10.1212/WNL.0b013e31827ccf34
Sanchez-Contreras M, Baker MC, Finch NA, Nicholson A, Wojtas A, Wszolek ZK, Ross OA, Dickson DW, Rademakers R (2014) Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology. Hum Mutat 35(8):964–971. https://doi.org/10.1002/humu.22582
Wang C, Yao XP, Chen HT, Lai JH, Guo XX, Su HZ, Dong EL, Zhang QJ, Wang N, Chen WJ (2017) Novel mutations of PDGFRB cause primary familial brain calcification in Chinese families. J Hum Genet. https://doi.org/10.1038/jhg.2017.25
Betsholtz C, Keller A (2014) PDGF, pericytes and the pathogenesis of idiopathic basal ganglia calcification (IBGC). Brain Pathol 24(4):387–395. https://doi.org/10.1111/bpa.12158
Manyam BV (2005) What is and what is not “Fahr’s disease.” Parkinsonism Relat Disord 11(2):73–80. https://doi.org/10.1016/j.parkreldis.2004.12.001
Arts FA, Velghe AI, Stevens M, Renauld JC, Essaghir A, Demoulin JB (2015) Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J Cell Mol Med 19(1):239–248. https://doi.org/10.1111/jcmm.12443
Biancheri R, Severino M, Robbiano A, Iacomino M, Del Sette M, Minetti C, Cervasio M, Caro DBD, M, Striano P, Zara F, (2016) White matter involvement in a family with a novel PDGFB mutation. Neurol Genetics 2(3):e77. https://doi.org/10.1212/NXG.0000000000000077
Keller A, Westenberger A, Sobrido MJ, Garcia-Murias M, Domingo A, Sears RL, Lemos RR, Ordonez-Ugalde A, Nicolas G, da Cunha JE, Rushing EJ, Hugelshofer M, Wurnig MC, Kaech A, Reimann R, Lohmann K, Dobricic V, Carracedo A, Petrovic I, Miyasaki JM, Abakumova I, Mae MA, Raschperger E, Zatz M, Zschiedrich K, Klepper J, Spiteri E, Prieto JM, Navas I, Preuss M, Dering C, Jankovic M, Paucar M, Svenningsson P, Saliminejad K, Khorshid HR, Novakovic I, Aguzzi A, Boss A, Le Ber I, Defer G, Hannequin D, Kostic VS, Campion D, Geschwind DH, Coppola G, Betsholtz C, Klein C, Oliveira JR (2013) Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 45:1077–1082. https://doi.org/10.1038/ng.2723
Keogh MJ, Pyle A, Daud D, Griffin H, Douroudis K, Eglon G, Miller J, Horvath R, Chinnery PF (2015) Clinical heterogeneity of primary familial brain calcification due to a novel mutation in PDGFB. Neurology 84(17):1818–1820. https://doi.org/10.1212/WNL.0000000000001517
Nicolas G, Jacquin A, Thauvin-Robinet C, Rovelet-Lecrux A, Rouaud O, Pottier C, Aubriot-Lorton MH, Rousseau S, Wallon D, Duvillard C, Bejot Y, Frebourg T, Giroud M, Campion D, Hannequin D (2014) A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur J Hum Genet. https://doi.org/10.1038/ejhg.2014.9
Nicolas G, Rovelet-Lecrux A, Pottier C, Martinaud O, Wallon D, Vernier L, Landemore G, Chapon F, Prieto-Morin C, Tournier-Lasserve E, Frebourg T, Campion D, Hannequin D (2014) PDGFB partial deletion: a new, rare mechanism causing brain calcification with leukoencephalopathy. J Mol Neurosci 53(2):171–175. https://doi.org/10.1007/s12031-014-0265-z
Yao XP, Wang C, Su HZ, Guo XX, Lu YQ, Zhao M, Liu YB, Lai JH, Chen HT, Wang N, Chen WJ (2016) Mutation screening of PDGFB gene in Chinese population with primary familial brain calcification. Gene. https://doi.org/10.1016/j.gene.2016.10.037
Chen WJ, Yao XP, Zhang QJ, Ni W, He J, Li HF, Liu XY, Zhao GX, Murong SX, Wang N, Wu ZY (2013) Novel SLC20A2 mutations identified in southern Chinese patients with idiopathic basal ganglia calcification. Gene 529(1):159–162. https://doi.org/10.1016/j.gene.2013.07.071
David S, Ferreira J, Quenez O, Rovelet-Lecrux A, Richard AC, Verin M, Jurici S, Le Ber I, Boland A, Deleuze JF, Frebourg T, Mendes de Oliveira JR, Hannequin D, Campion D, Nicolas G (2016) Identification of partial SLC20A2 deletions in primary brain calcification using whole-exome sequencing. Eur J Hum Genet 24(11):1630–1634. https://doi.org/10.1038/ejhg.2016.50
Gagliardi M, Morelli M, Annesi G, Nicoletti G, Perrotta P, Pustorino G, Iannello G, Tarantino P, Gambardella A, Quattrone A (2015) A new SLC20A2 mutation identified in southern Italy family with primary familial brain calcification. Gene 568(1):109–111. https://doi.org/10.1016/j.gene.2015.05.005
Gagliardi M, Morelli M, Iannello G, Colica C, Annesi G, Quattrone A (2017) A SLC20A2 mutation identified in an asymptomatic patient with brain calcification. J Neurol Sci 372:70–72. https://doi.org/10.1016/j.jns.2016.11.038
Hsu SC, Sears RL, Lemos RR, Quintans B, Huang A, Spiteri E, Nevarez L, Mamah C, Zatz M, Pierce KD, Fullerton JM, Adair JC, Berner JE, Bower M, Brodaty H, Carmona O, Dobricic V, Fogel BL, Garcia-Estevez D, Goldman J, Goudreau JL, Hopfer S, Jankovic M, Jauma S, Jen JC, Kirdlarp S, Klepper J, Kostic V, Lang AE, Linglart A, Maisenbacher MK, Manyam BV, Mazzoni P, Miedzybrodzka Z, Mitarnun W, Mitchell PB, Mueller J, Novakovic I, Paucar M, Paulson H, Simpson SA, Svenningsson P, Tuite P, Vitek J, Wetchaphanphesat S, Williams C, Yang M, Schofield PR, de Oliveira JR, Sobrido MJ, Geschwind DH, Coppola G (2013) Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 14(1):11–22. https://doi.org/10.1007/s10048-012-0349-2
Lemos RR, Ramos EM, Legati A, Nicolas G, Jenkinson EM, Livingston JH, Crow YJ, Campion D, Coppola G, Oliveira JR (2015) Update and mutational analysis of SLC20A2: a major cause of primary familial brain calcification. Hum Mutat 36(5):489–495. https://doi.org/10.1002/humu.22778
Pasanen P, Makinen J, Myllykangas L, Guerreiro R, Bras J, Valori M, Viitanen M, Baumann M, Tienari PJ, Poyhonen M, Baumann P (2016) Primary familial brain calcification linked to deletion of 5’ noncoding region of SLC20A2. Acta Neurol Scand. https://doi.org/10.1111/ane.12697
Rubino E, Giorgio E, Godani M, Grosso E, Zibetti M, Lopiano L, Ferrero P, Duca S, Moretti L, Gallone S, Rainero I, Brusco A (2017) Three novel missense mutations in SLC20A2 associated with idiopathic basal ganglia calcification. J Neurol Sci 377:62–64. https://doi.org/10.1016/j.jns.2017.03.053
Wang C, Li Y, Shi L, Ren J, Patti M, Wang T, de Oliveira JR, Sobrido MJ, Quintans B, Baquero M, Cui X, Zhang XY, Wang L, Xu H, Wang J, Yao J, Dai X, Liu J, Zhang L, Ma H, Gao Y, Ma X, Feng S, Liu M, Wang QK, Forster IC, Zhang X, Liu JY (2012) Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 44(3):254–256. https://doi.org/10.1038/ng.1077
Anheim M, Lopez-Sanchez U, Giovannini D, Richard AC, Touhami J, N’Guyen L, Rudolf G, Thibault-Stoll A, Frebourg T, Hannequin D, Campion D, Battini JL, Sitbon M, Nicolas G (2016) XPR1 mutations are a rare cause of primary familial brain calcification. J Neurol 263(8):1559–1564. https://doi.org/10.1007/s00415-016-8166-4
Legati A, Giovannini D, Nicolas G, Lopez-Sanchez U, Quintans B, Oliveira JR, Sears RL, Ramos EM, Spiteri E, Sobrido MJ, Carracedo A, Castro-Fernandez C, Cubizolle S, Fogel BL, Goizet C, Jen JC, Kirdlarp S, Lang AE, Miedzybrodzka Z, Mitarnun W, Paucar M, Paulson H, Pariente J, Richard AC, Salins NS, Simpson SA, Striano P, Svenningsson P, Tison F, Unni VK, Vanakker O, Wessels MW, Wetchaphanphesat S, Yang M, Boller F, Campion D, Hannequin D, Sitbon M, Geschwind DH, Battini JL, Coppola G (2015) Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 47(6):579–581. https://doi.org/10.1038/ng.3289
Jensen N, Schroder HD, Hejbol EK, Fuchtbauer EM, de Oliveira JR, Pedersen L (2013) Loss of function of Slc20a2 associated with familial idiopathic Basal Ganglia calcification in humans causes brain calcifications in mice. J Mol Neurosci 51(3):994–999. https://doi.org/10.1007/s12031-013-0085-6
Demoulin JB, Ericsson J, Kallin A, Rorsman C, Ronnstrand L, Heldin CH (2004) Platelet-derived growth factor stimulates membrane lipid synthesis through activation of phosphatidylinositol 3-kinase and sterol regulatory element-binding proteins. J Biol Chem 279(34):35392–35402. https://doi.org/10.1074/jbc.M405924200
Kakita A, Suzuki A, Nishiwaki K, Ono Y, Kotake M, Ariyoshi Y, Miura Y, Ltoh M, Oiso Y (2004) Stimulation of Na-dependent phosphate transport by platelet-derived growth factor in rat aortic smooth muscle cells. Atherosclerosis 174(1):17–24. https://doi.org/10.1016/j.atherosclerosis.2003.12.039
Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468(7323):562–566. https://doi.org/10.1038/nature09513
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV (2015) Establishment and dysfunction of the blood-brain barrier. Cell 163(5):1064–1078. https://doi.org/10.1016/j.cell.2015.10.067
Vanlandewijck M, Lebouvier T, Andaloussi Mäe M, Nahar K, Hornemann S, Kenkel D, Cunha SI, Lennartsson J, Boss A, Heldin C-H, Keller A, Betsholtz C (2015) Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS ONE 10(11):e0143407–e0143407. https://doi.org/10.1371/journal.pone.0143407
Olson LE, Soriano P (2009) Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 16(2):303–313. https://doi.org/10.1016/j.devcel.2008.12.003
Suzuki S, Heldin CH, Heuchel RL (2007) Platelet-derived growth factor receptor-beta, carrying the activating mutation D849N, accelerates the establishment of B16 melanoma. BMC Cancer 7:224. https://doi.org/10.1186/1471-2407-7-224
Olson LE, Soriano P (2011) PDGFRbeta signaling regulates mural cell plasticity and inhibits fat development. Dev Cell 20(6):815–826. https://doi.org/10.1016/j.devcel.2011.04.019
He C, Medley SC, Hu T, Hinsdale ME, Lupu F, Virmani R, Olson LE (2015) PDGFRβ signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat Commun 6(1):7770. https://doi.org/10.1038/ncomms8770
Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, Olson LE (2015) PDGFRα signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev 29(11):1106–1119. https://doi.org/10.1101/gad.260554.115
Sun C, Sakashita H, Kim J, Tang Z, Upchurch GM, Yao L, Berry WL, Griffin TM, Olson LE (2020) Mosaic mutant analysis identifies PDGFRalpha/PDGFRbeta as negative regulators of adipogenesis. Cell Stem Cell 26 (5):707–721 e705. doi:https://doi.org/10.1016/j.stem.2020.03.004
Liao X, Escobedo JA, Williams LT (1996) Viability of transgenic mice expressing a platelet derived growth factor (PDGF) antagonist in plasma. J Investig Med 44(4):139–143
Floege J, Eitner F, Alpers CE (2008) A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol 19(1):12–23. https://doi.org/10.1681/asn.2007050532
Lassila M, Jandeleit-Dahm K, Seah KK, Smith CM, Calkin AC, Allen TJ, Cooper ME (2005) Imatinib attenuates diabetic nephropathy in apolipoprotein E-knockout mice. J Am Soc Nephrol 16(2):363–373. https://doi.org/10.1681/asn.2004050392
Wang S, Wilkes MC, Leof EB, Hirschberg R (2005) Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. Faseb j 19(1):1–11. https://doi.org/10.1096/fj.04-2370com
Zoja C, Corna D, Rottoli D, Zanchi C, Abbate M, Remuzzi G (2006) Imatinib ameliorates renal disease and survival in murine lupus autoimmune disease. Kidney Int 70(1):97–103. https://doi.org/10.1038/sj.ki.5001528
Lundby A, Franciosa G, Emdal KB, Refsgaard JC, Gnosa SP, Bekker-Jensen DB, Secher A, Maurya SR, Paul I, Mendez BL, Kelstrup CD, Francavilla C, Kveiborg M, Montoya G, Jensen LJ, Olsen JV (2019) Oncogenic mutations rewire signaling pathways by switching protein recruitment to phosphotyrosine sites. Cell 179(2):543-560.e526. https://doi.org/10.1016/j.cell.2019.09.008
Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, Chase A, Chessells JM, Colombat M, Dearden CE, Dimitrijevic S, Mahon FX, Marin D, Nikolova Z, Olavarria E, Silberman S, Schultheis B, Cross NC, Goldman JM (2002) Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med 347(7):481–487. https://doi.org/10.1056/NEJMoa020150
Cheah CY, Burbury K, Apperley JF, Huguet F, Pitini V, Gardembas M, Ross DM, Forrest D, Genet P, Rousselot P, Patton N, Smith G, Dunbar CE, Ito S, Aguiar RCT, Odenike O, Gimelfarb A, Cross NCP, Seymour JF (2014) Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood 123(23):3574–3577. https://doi.org/10.1182/blood-2014-02-555607
Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, Malone J, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P, Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P, Coutre SE, Stone R, Gilliland DG (2003) A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 348(13):1201–1214. https://doi.org/10.1056/NEJMoa025217
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H (2002) Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347(7):472–480. https://doi.org/10.1056/NEJMoa020461
Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, Debiec-Rychter M (2012) Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res 18(16):4375–4384. https://doi.org/10.1158/1078-0432.Ccr-12-0625
Evans EK, Gardino AK, Kim JL, Hodous BL, Shutes A, Davis A, Zhu XJ, Schmidt-Kittler O, Wilson D, Wilson K, DiPietro L, Zhang Y, Brooijmans N, LaBranche TP, Wozniak A, Gebreyohannes YK, Schöffski P, Heinrich MC, DeAngelo DJ, Miller S, Wolf B, Kohl N, Guzi T, Lydon N, Boral A, Lengauer C (2017) A precision therapy against cancers driven by <em>KIT/PDGFRA</em> mutations. Sci Transl Med 9 (414):eaao1690. doi:https://doi.org/10.1126/scitranslmed.aao1690
Cheung E, Lobov IB, Cao J, Yancopaulos G, Romano C, Wiegand SJ (2015) Effects of combined inhibition of VEGF and PDGFRβ using aflibercept (VEGF Trap) and anti-PDGFRβ antibody on developing retinal angiogenesis in mice. Invest Ophthalmol Vis Sci 56(7):2314–2314
Loizos N, Xu Y, Huber J, Liu M, Lu D, Finnerty B, Rolser R, Malikzay A, Persaud A, Corcoran E, Deevi DS, Balderes P, Bassi R, Jimenez X, Joynes CJ, Mangalampalli VRM, Steiner P, Tonra JR, Wu Y, Pereira DS, Zhu Z, Ludwig DL, Hicklin DJ, Bohlen P, Witte L, Kussie P (2005) Targeting the platelet-derived growth factor receptor α with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: Implications as a potential therapeutic target. Mol Cancer Ther 4(3):369–379. https://doi.org/10.1158/1535-7163.Mct-04-0114
Tap WD, Jones RL, Van Tine BA, Chmielowski B, Elias AD, Adkins D, Agulnik M, Cooney MM, Livingston MB, Pennock G, Hameed MR, Shah GD, Qin A, Shahir A, Cronier DM, Ilaria R Jr, Conti I, Cosaert J, Schwartz GK (2016) Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 388(10043):488–497. https://doi.org/10.1016/s0140-6736(16)30587-6
Cornillie J, Wozniak A, Van Renterghem B, Van Winkel N, Wellens J, Gebreyohannes YK, Debiec-Rychter M, Sciot R, Hompes D, Schöffski P (2019) Assessment of the platelet-derived growth factor receptor alpha antibody olaratumab in a panel of patient-derived soft tissue sarcoma xenografts. BMC Cancer 19(1):724. https://doi.org/10.1186/s12885-019-5872-1
Heier JS, Wykoff CC, Waheed NK, Kitchens JW, Patel SS, Vitti R, Perlee L, Chu KW, Leal S, Asmus F, Son V, Schmelter T, Brown DM (2020) Intravitreal combined aflibercept + anti platelet-derived growth factor receptor b for neovascular age-related macular degeneration: results of the phase 2 CAPELLA trial. Ophthalmology 127(2):211–220. https://doi.org/10.1016/j.ophtha.2019.09.021
Camorani S, Esposito CL, Rienzo A, Catuogno S, Iaboni M, Condorelli G, de Franciscis V, Cerchia L (2014) Inhibition of receptor signaling and of glioblastoma-derived tumor growth by a novel PDGFRbeta aptamer. Mol Ther 22(4):828–841. https://doi.org/10.1038/mt.2013.300
Yoon S, Wu X, Armstrong B, Habib N, Rossi JJ (2019) An RNA aptamer targeting the receptor tyrosine kinase PDGFRalpha induces anti-tumor effects through STAT3 and p53 in glioblastoma. Mol Ther Nucleic Acids 14:131–141. https://doi.org/10.1016/j.omtn.2018.11.012
Romanelli A, Affinito A, Avitabile C, Catuogno S, Ceriotti P, Iaboni M, Modica J, Condorelli G, Catalucci D (2018) An anti-PDGFRβ aptamer for selective delivery of small therapeutic peptide to cardiac cells. PLoS ONE 13(3):e0193392. https://doi.org/10.1371/journal.pone.0193392
Acknowledgements
The authors were supported by grants from the Foundation against Cancer and King-Baudouin Foundation (Belgium). E.G., F.A. and G.D. are recipients of fellowships from National Fund for Scientific Research (FNRS) (Belgium).
Author information
Authors and Affiliations
Contributions
EG, FA, GD, BB and JBD wrote the manuscript. EG drew the figures.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guérit, E., Arts, F., Dachy, G. et al. PDGF receptor mutations in human diseases. Cell. Mol. Life Sci. 78, 3867–3881 (2021). https://doi.org/10.1007/s00018-020-03753-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03753-y