
Abstract
The POU (Pit-Oct-Unc) protein family is an evolutionary ancient group of transcription factors (TFs) that bind specific DNA sequences to direct gene expression programs. The fundamental importance of POU TFs to orchestrate embryonic development and to direct cellular fate decisions is well established, but the molecular basis for this activity is insufficiently understood. POU TFs possess a bipartite ‘two-in-one’ DNA binding domain consisting of two independently folding structural units connected by a poorly conserved and flexible linker. Therefore, they represent a paradigmatic example to study the molecular basis for the functional versatility of TFs. Their modular architecture endows POU TFs with the capacity to accommodate alternative composite DNA sequences by adopting different quaternary structures. Moreover, associations with partner proteins crucially influence the selection of their DNA binding sites. The plentitude of DNA binding modes confers the ability to POU TFs to regulate distinct genes in the context of different cellular environments. Likewise, different binding modes of POU proteins to DNA could trigger alternative regulatory responses in the context of different genomic locations of the same cell. Prominent POU TFs such as Oct4, Brn2, Oct6 and Brn4 are not only essential regulators of development but have also been successfully employed to reprogram somatic cells to pluripotency and neural lineages. Here we review biochemical, structural, genomic and cellular reprogramming studies to examine how the ability of POU TFs to select regulatory DNA, alone or with partner factors, is tied to their capacity to epigenetically remodel chromatin and drive specific regulatory programs that give cells their identities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The octameric DNA element (ATGCAAAT) was originally discovered in 1984 as a conserved nucleotide sequence upstream of immunoglobulin genes [1]. Functional DNA elements related to the octamer sequence were subsequently found in promoters of the ubiquitously expressed small nucleolar RNA and histone 2B genes, enhancers of the SV40 virus as well as in the adenovirus origin of replication [2,3,4,5]. Oct1 and Oct2 (initially termed NF-A1 and NF-A2 [6], OTF-1 and OTF-2 [7,8,9], OBP100 [5] or NFIII [10]) were simultaneously discovered by various labs in the search for the trans-acting protein factors utilizing octamer DNA to regulate gene expression [11,12,13,14]. At the same time a factor was discovered as transcriptional activator of growth hormones and prolactin genes in the pituitary gland and termed Pit1 [15, 16]. Pit1, Oct1 and Oct2 contain two sequence motifs spanning a total of about 160 amino acids with homology to the C. elegans unc-86 gene [17]. This novel bipartite domain was, therefore, designated with the acronym POU (pronounced ‘pow’) after its founding family members Pit, Oct and Unc-86 [18]. One of the two sequence motifs is a 60 amino acids region with homology to the homeodomain initially discovered in the fruit fly Drosophila [19, 20] and, is therefore, termed POU-homeodomain (POUHD). The second sequence motif, unknown by the time POU was discovered, is composed of 75 amino acids located N-terminally to the POUHD that is specific for these proteins and is, therefore, named POU-specific (POUS) domain [18, 21]. A linker of variable length and sequence joins the POUS and POUHD. Mouse and human genomes encode 15 POU genes (Fig. 1a). POU TFs have essential roles in a wide array of cellular processes and gained particular prominence in studies demonstrating the reprogramming of somatic cells to pluripotency and the directed lineage reprogramming by the trans-differentiation to neural precursor cells (NPCs) or post-mitotic neurons. The bipartite architecture of the POU endows these TFs with the ability to associate with DNA in structurally diverse configurations. Several excellent reviews have discussed DNA binding by POU TFs and their function in mammalian development [22,23,24,25,26,27,28,29]. Here we provide an update about our current understanding on the molecular basis for the selective DNA recognition and context-dependent gene regulation by POU TFs and discuss these properties in the functional context of cellular reprogramming and chromatin remodeling.

Classification and expression of POU TFs. a Dendrogram generated using full-length mouse POU protein sequences with T-Coffee (http://www.tcoffee.org/). Systematic names and commonly used synonyms are indicated for each of the 15 factors. The six classes are represented in different colors and the traditionally used separation into octamer and non-octamer binding factors is highlighted. b Expression of POU genes in eight developmental domains represented as mean Z-scores of expression for all cell types in a domain [34]. POU factors are color coded according to class membership. c Expression of POU genes in selected mouse cell and tissue types represented as log2 transformation of GC-normalised RNA-seq read count data per gene. Domain assignments of datasets are indicated and color-coded. EM embryonic, SE surface ectoderm, MS mesoderm, NC neural crest, GC germ cells, EN endoderm, NR neuroectoderm, BM blood mesoderm. Pou4f3 is absent from the expression table as it was barely detectable in any of the 272 analyzed cell and tissue types
Classification, nomenclature and expression
As most other major transcription factor (TF) families POU genes are present in the genomes of all metazoans designating them as basic molecular toolkit driving animal evolution [30]. However, they are absent in plants and fungi. Many POU genes were identified in the late 1980s and early 1990s in parallel by several laboratories using different model organisms. POU factors were discovered either genetically or biochemically using gel shift assays with octamer DNA as probe and subsequent cloning of the associated gene. As a consequence, the nomenclature has been somewhat confusing with alternative names for many family members. POU genes were grouped into six classes based on sequence similarities denoted with roman numbers I–VI [29, 31]. Individual genes are now unambiguously referred to with prefixes designating class membership from POU1 to POU6 (Pou1–Pou6 for mouse factors) [32]. However, the classical synonyms are still more common in the literature and will thus also be used in this review (Fig. 1a). POU classes I, III, IV and VI can be found in genomes of sponges and eumetazoans and are, therefore, present in the common ancestors of all living animals. The POU class II group evolved later in bilaterian evolution whilst class V is constrained to vertebrates [30]. Traditionally, POU genes were grouped based on their biochemical activity as octamer DNA binding (classes II, III, and V) and non-octamer binding (classes I, IV, VI) (Fig. 1a, [27]). Octamer binding factors were numbered based on the position of retarded DNA probes in electrophoretic mobility shift assays from Oct1 to Oct11 [33].
POU factors function pleiotropically in a wide range of cell types. To illustrate expression patterns of POU genes, we used a recently compiled collection of RNA sequencing (RNA-seq) data from 272 mouse cell and tissue types that were assigned to 8 broad developmental domains (Fig. 1b, c) [34]. In Fig. 1c, a selection of these cell and tissue types is shown where individual POU genes are strongly expressed. The first isolated family members belong to classes I (Pit1) or II (Oct1, Oct2 and the later discovered Oct11). Pit1 has been intensely studied for its function in the anterior pituitary gland [15, 16] but is also widely expressed in the blood mesoderm (Fig. 1b, c). Oct1 is ubiquitously expressed and can be detected in almost any cell type [5, 7, 35]. In accordance with early studies, Oct2 is strongly expressed in the blood mesoderm in particular B-lymphocytes and can also be found in cells of the neuroectoderm [6, 9, 13, 14, 36, 37]. Oct11 has the most restricted expression of class II POU genes and predominates in cells of the surface ectoderm such as the epidermis, skin and taste buds [38, 39]. Some members of classes III (Brn1, Brn2 and Brn4), IV (Brn3) and VI (Brn5) were originally discovered in adult brain tissue and hence designated Brn1-5 [40,41,42,43]. A fourth POU III class gene, Oct6, was initially detected in the rat testes (termed Tst-1) [41] and in glial cells (termed SCIP for suppressed cAMP inducible POU) [44] but was later found to be also expressed in the blastocyst and other lineages of the early embryo such as developing brain and skin [38, 45]. Class III POU genes are devoid of any introns. Brn2 is expressed in the neuroendocrine hypothalamus and pituitary [46, 47], Brn1 in the developing nervous system, hypothalamus and kidney [48, 49] while Brn4 is expressed in developing nervous system, hypothalamus, pituitary gland and inner ear [48, 49]. Genome-wide profiling underlines the prominent and specific expression of the four class III POU genes in many neuroectodermal cell types (Fig. 1b, c). Class IV has the closest homology to the C. elegans gene unc-86 and contains three members termed Brn3a [41, 50], Brn3b [51,52,53] and Brn3c [50, 54]. Although they were initially detected in neuroectodermal cell types, Brn3a is also expressed in the embryonic domain (Fig. 1c). Class VI comprises two genes that are either broadly expressed (Pou6f1/Brn5) or exhibit an overall rather weak expression (Pou6f2/RPF-1). Brn5 was initially discovered in the neocortex [40] and Pou6f2 in human retina cDNA libraries, thus called retina-derived POU-domain factor-1 (RPF-1) [55]. Class V POU TFs consists of Pou5f1 (Oct4) as well as Pou5f2 (Sprm1). Oct4 is possibly the most prominent POU family member and was identified in embryonic and endometrial cancer cell lines for its activity to bind octamer DNA in electrophoretic mobility shift assays (EMSA) [48, 56, 57]. Several labs independently discovered Oct4 and used diverging numbering conventions initially leading to the parallel use of the designations Oct3 or Oct4 (in some studies converging to Oct3/4) [48, 56,57,58]. Oct4 is a hallmark factor regulating the pluripotency of embryonic stem cells and is involved in the earliest cell fate decisions in mammalian development [59,60,61]. The expression of Oct4 is largely restricted to the embryonic domain and can barely be detected in somatic cell types (Fig. 1b, c). Several transcript isoforms of Oct4 were reported in human [62] and mouse [63]. The full-length Oct4 is termed Oct4A and a truncated isoform was named Oct4B [64]. The two versions of Oct4 have different expression patterns [65], gene regulation and self-renewal potential [64]. The second member of class V, Sprm1/Pou5f2, is transiently expressed in the testis [66] and is required for the development of male germ cells [67]. Class V has a complex evolutionary history with initially three members (Pou5f1, Pou5f2 and Pouf5f3) present in different combinations in vertebrate clades [68]. A zebrafish homologue was initially named Pou2 and classified as Pou5f1 orthologue [68, 69]. This was recently reinterpreted and Pou2 is now considered to be a Pou5f3 orthologue that is present in marsupials, monotremes, birds, xenopus, salamanders and teleost fishes but unlike Pou5f1 and Pou5f2, it is absent in eutherians.
POU-erful reprogramming factors
The capacity of TFs to interconvert the state of somatic cells has first been demonstrated when the overexpression of the helix–loop–helix (HLH) factor MyoD alone could convert fibroblasts into myocytes [70]. Subsequently, the overexpression of C/EBPα or C/EBPβ was found to be able to convert differentiated B-cells into macrophages [71]. Likewise, several POU factors are potent mediators of cellular reprogramming. This has been demonstrated when transcription factor cocktails were overexpressed in somatic cells leading to the reprogramming into pluripotent stem cells [72,73,74], proliferative neural precursor cells [75,76,77,78,79,80] and post-mitotic neurons [81,82,83,84,85] (Fig. 2a–d). However, the reprogramming trajectory and outcome depend on the identity of the POU factor used in these assays as well as on the external cues applied during reprogramming.

POU factors used in pluripotency and lineage reprogramming cocktails. POU factor-containing cocktails used in pluripotency (a, b) or direct lineage (c, d) reprogramming of mouse (a, c) or human (b, d) cells. The starting cell type is represented in the center as sphere or box. The various reprogramming cocktail are shown besides the arrow. The size of cell cluster in a and b schematically represents efficiency. The reprogramming cocktail in each section (a–d) is numbered and abbreviations and references are given below. a Mouse pluripotency reprogramming cocktails: 1 OSKM (O: Oct4; S: Sox2; K: Klf4, M: c-Myc) [73], 2 OKM + S1/S3 (Sox1/Sox3) [86], 3 OSM + K1/K2/K5 (Klf1/Klf2/Klf5) [86], 4 OSK + N-Myc/C-Myc/L-Myc [86], 5 OSK + Sall4 [237], 6 OSK + Glis1 [238], 7 OSK [86, 239], 8 OS + Esrrb [240], 9 OKM + Alk5 inhibitor [241], 10 OSM + Kenpaullone [242], 11 OS [243], 12 O + Bmp4 [244], 13 O + Bmi1 [245], 14 OK + BIX01294 + BayK8644 [246], 15 OK + BIX01294 + RG108 [246], 16 OK + BIX01294 [246], 17 O4 + VC6T [VC6T: (valproic acid, CHIR99021, 616452, tranylcypromine)] [247], 18 O + oxysterol and/or puromorphine [245], 19 O4 + Shh [245], 20 *Oct6 (engineered Oct6) [87] /Brn4 [75], 21 DP (dermal papilla): OKM, OK, O [248, 249], 22 HPC (hematopoietic stem cells): OSKM [250], 23 ADC (adipose derived stem cells): OSKM [251], 24 TSC (trophoblast stem cells): OSKM [252], 25 NSC (neural stem cells): OKM [250], OK + BIX01294 [253], OK + 2i (2i denotes PD0325901 and CHIR99021) [254], OK [255], O [89], 26 melanocytes and melanoma cells: OKM [256], 27 hepatic EN (endoderm): [257], 28 B and T cells: [250, 258], 29 pancreatic B (beta) cells: [259], 30 myeloid PC (progenitor cells): [250], 31 skeletal MSC (muscle stem cells): [260], 32 Oct6-KSM [86], 33 Oct1-KSM [86]. b Human pluripotency reprogramming cocktails: 1 OSKM [72, 261, 262], 2 OSNL (N: Nanog; L: Lin28a) [74], 3 OSK + Sall4 [237], 4 OSK + Utf1 [263], 5 OSK + Glis1 [238], 6 OSK, OS [243], 7 OS + VPA (valproic acid) [264], 8 urine (renal epithelium cells): OSKM [95], 9 keratinocytes: OSKM [265], OSK [266], 10 MSC/dental (mesenchyme-like stem cells of dental origin): OSNL [267], 11 NSC: O [88], 12 Am-DC (amnion derived cells): OSN [268, 269], 13 SMA (spinal muscular atrophy): OSNL [90], 14 FD (familial dysautonomia): OSKM [91], 15 ALS (amylotropic lateral sclerosis): OSKM, [92], 16 Parkinson’s: OSKM, OSK [93], 17 Genetic diseases: OSKM, OSK: [94]. 18 CB (cord blood): OSNL [270], OSKM, OSK, OS [271], 19 hepatocytes: OSKM [272], 20 ADC (adipose derived cells): OSKM [251], OSK, [273], 21 M and M (melanocytes and melanoma cells): OKM [256], 22 pancreatic beta cells: OSKM [274]. c Mouse lineage reprogramming cocktails: 1 BAM (Brn2, Ascl1, Myt1l) [105], 2 BAM [83, 85], 3 NPCs [77, 79], 4 pancreatic lineages [99], 5 cardiomyocytes [101], 6 cardiomyocytes (Oct4 alone) [84], 7 BKSM/OSKM [75], 8 BKSM + E47/Tcf3 [76], 9 Brn2 + Foxg1 + Sox2) [78]. d Human lineage reprogramming cocktails: 1 endothelial cells [102], 2 blood [103], 3 hepatic cells [100], 4 iNSC (induced neural stem cell) [80], 5 Brn2 + Neurod1(β2) [82], 6 BAM or BAM + β2 or BM + β2 (β2: Neurod1) [82], 7 BAM + Lmx1a +Foxa2 [108], 8 Brn2 + Mytl1 + miR-124 [81]
Somatic cell reprogramming to pluripotent stem cells
A hallmark accomplishment was the demonstration that self-renewing induced pluripotent stem cells (iPSCs) can be generated by de-differentiating fibroblasts through the forced expression of exogenously provided TF cocktails (Fig. 2a, b). To accomplish this feat, 24 factors were screened and a minimal set of four factors comprising Oct4, Sox2, Klf4 and c-Myc (OSKM) was identified [73]. The same cocktail was also able to achieve pluripotency reprogramming of human somatic cells [72]. The same year, a modified human iPSCs generating cocktail was reported that also contained Sox2/Oct4 but Nanog/Lin28 replaced Klf4/c-Myc [74]. Oct4 could not be replaced by Oct1 or Oct6 to induce pluripotency despite profound sequence conservation emphasizing its uniqueness [86]. However, recently Oct6 could be converted into an iPSCs inducer using rational protein engineering [87]. Similarly, the neural factor Brn4 was reported to allow iPSCs generation as part of an inducible polycistronic cassette consisting of Brn4, Sox2, Klf4, c-Myc (BSKM) [75]. However, both engineered Oct6 and Brn4 induce iPSCs with very low efficiency (Fig. 2a). Therefore, what endows Oct4 with its potent reprogramming activity is still an open question. Besides fibroblasts, Oct4 also reprograms other cell types (summarized in Fig. 2). Oct4 alone could induce pluripotency in mouse and human NPCs [88, 89]. This is possible because the high expression of Sox2 obviates the need for its exogenous supply. A number of studies explored alternative reprogramming strategies centered on Oct4 where the SKM factors were replaced by small molecules or other factors (Fig. 2). The iPS technology has broad clinical applications in particular in disease modeling and iPSCs could be derived from patients affected by spinal muscular atrophy (SMA) [90], familial dysautonomia (FD) [91], amylotropic lateral sclerosis (ALS) [92], Parkinson’s disease [93] and variety of genetic diseases [94]. Oct4 has also been deployed to convert the renal epithelium cells from urine in a quick and efficient way, omitting the need of invasive techniques for obtaining patient samples [95]. In sum, Oct4 is a stalwart component of cocktails directing the induction of pluripotency in large variety of reprogramming systems and technologies.
Lineage reprogramming to directly trans-differentiate somatic cells
Neural precursors cells (NPCs, here used to jointly include neural stem cells and neural progenitor cells following [96]) are proliferative and can, therefore, be expanded in culture and possess the capacity to differentiate into mature post-mitotic cells. If NPCs are tripotent they can form neurons as well as the non-neural glia including oligodendrocytes and astrocytes. Induced NPCs (iNPCs) have initially been generated using the otherwise iPSCs generating OSKM cocktail but with modified culture conditions favoring neural lineages [77] (Fig. 2c, d). OSKM-driven production of iNPCs could also be achieved by limiting the exposure of reprogramming cells to Oct4 to the early stages [79]. Alternatively, modified cocktails were used to produce iNPCs where the pluripotency factor Oct4 was replaced with Brn4 or Brn2. First, Brn4 was used alongside Sox2, Klf4 and c-Myc and the reprogramming efficiency could be further improved by the addition of E47/Tcf3 [76]. Second, tripotent iNPCs could be generated using a Brn2/Sox2/FoxG1 cocktail [78]. This suggested that the reprogramming of fibroblasts takes fundamentally different routes depending on whether Oct4 or class III POU TFs are forcibly expressed in the starting cells. Apparently, POU TFs have profoundly different activities in an identical nuclear environment despite a high degree of sequence homology. However, recent lineage tracing experiments challenged this view and indicated that both OSKM and BSKM cocktails induce a pluripotent state [75]. Therefore, iNPCs may in fact transit through an intermediate pluripotent state rather than being directly reprogrammed. This also implied that exogenous Oct4 and class III POU TFs engage the fibroblast genome similarly to initiate reprogramming and the direction of the cell state conversion relies on signaling provided at later stages. To resolve this question the genome engagement and associated epigenetic changes driven by Oct4 and class III POU TFs should be profiled side-by-side.
With the objective to generate differentiated lineages, Oct4-containing cocktails have been deployed in a strategy called cell activation and signaling directed (CASD) lineage reprogramming [97, 98]. Typically, a pulse of OSKM expression epigenetically de-differentiates the donor cells and induces a plastic state where cells are receptive for differentiation cues provided by small molecules or growth factors [99]. This strategy has been used to convert fibroblast cells into hepatic [100], cardiac [84, 101], neural [77], endothelial [102], pancreatic [99] and blood lineages [103]. However, lineage-tracing experiments have put into question whether this approach truly circumvents the pluripotent state [75, 104].
Besides the generation of iNPCs, class III POU TFs have also been deployed to produce post-mitotic neurons. Here, Brn2 along with Ascl1 and Myt1l (BAM, Fig. 2c, d) could convert dermal fibroblasts into neurons with surprisingly high efficiency and speed [83, 85]. Besides fibroblasts, the same cocktail could also reprogram endodermal hepatocytes [105] (Fig. 2c). Brn2 containing cocktails were also used to induce neurons from human cells using analogous strategies [81, 82, 106,107,108] (Fig. 2d). However, the efficiency in the human system is much lower and additional factors are required. Collectively, these studies demonstrate that the POU TF scaffold enables the potent interconversion of cellular states. However, individual POU TFs function non-redundantly and are mostly irreplaceable by other family members. Reprogramming experiments provide a powerful assay to delineate the sequence-function relationships defining the regulatory programs driven by POU factors.
DNA recognition
Monomeric binding and cross talk of POUS and POUHD
After the discovery of octamer DNA in a selected set of regulatory sequences, it was verified as the dominant recognition sequence for many POU TFs by several unbiased assays. Initially, the octamer was recovered as the preferred binding sequence for Oct1 in a random oligonucleotide selection study using the POU domain of Oct1 [109]. The relevance of octamer DNA to recruit POU TFs to chromatin was, for example, verified in a ChIP-seq study for Oct2 in splenic B cells [110]. To understand the structural basis for the recognition of the octamer, the structures of the 75 amino acid POUS [111, 112] and the 60 amino acid POUHD [113, 114] were initially analyzed separately by nuclear magnetic resonance (NMR) in the absence of DNA. The crystal structure of a whole POU could first be solved for Oct1 bound to a classical octamer DNA element derived from the histone 2B promoter (Fig. 3a) [115]. These studies showed that the POU domain belongs to the class of all-alpha domains with four helices in the POUS and three helices in the POUHD. Helices 2 and 3 of the POUS adopt a helix-turn-helix (HTH) fold and bind DNA using amino acid–DNA base interactions nearly identical to HTH prototypes found in repressor proteins of the bacteriophages 434 and λ [111, 115]. The four amino acids used to make base specific contacts are conserved in the POU family and in phage repressors indicating that the fold is evolutionarily ancient. Analogously, helices 2 and 3 of the POUHD form a HTH unit. For both the POUS and the POUHD, helix 3 is inserted into the major groove of the DNA and contributes most of the residues involved in base-specific interactions (Fig. 3a–e). The overall fold of the POUHD and residues engaged in base-specific contacts are very similar to classical homeodomains including the invariant Asn51 typically making a bi-dentate H-bonds with an adenine in the recognition sequence. Cys50 of the POUHD is a sequence variation characteristic for the POU family (Fig. 3b). The POUHD contains an extended Arg-Lys rich N-terminal arm mediating minor groove contacts. Surprisingly, the POUHD and the POUS are not making any direct protein–protein contacts when binding the canonical octamer DNA and the two domains lie on opposite sides of the DNA. This suggested that both units are in fact independently acting domains with autonomous DNA binding activity. Consistently, the isolated POUS and POUHD can bind DNA sequence-specifically with the POUs selecting a [A/G]TAATNA and the POUHD a GAATAT[T/G]C) consensus [109]. However, the affinity of the separated domains for these elements is lower than that of the intact POU for the full octamer. In the context of an intact POU-bound octameric DNA, the POUs binds the ATGC half-site and the POUHD the AAAT half-site [109, 115]. In particular, the POUS has only a moderate affinity for DNA by itself, but is highly sequence-specific. Further studies showed that two-nucleotide spacers between ATGC and AAAT half-sites are permitted [116]. Despite the lack of direct interactions, POUS and POUHD mutually influence each other’s DNA binding by a mechanism termed DNA mediated cooperativity. Here, allosteric changes to the structure of the DNA indirectly facilitate the recruitment of partner factors. This mechanism has been reported for a range of dimeric TF associations [117,118,119] and the POU TFs present an intriguing example of this binding mode for covalently coupled domains [120]. Consistently, high-throughput protein-binding-microarrays (PBMs) verified the full octamer as well as the POUHD half-site as being part of the binding landscape of Pit1 and Oct1 even in the context of the bipartite POU [121, 122]. The authors highlighted Oct1 as an example of a TF that is able to associate with DNA in multiple binding modes leading to primary, secondary and tertiary DNA binding motifs. TFs with several structural units such as the POU appear to be particularly well suited to accommodate alternative DNA sequences. NMR studies led to an interesting proposal of how the modular make-up of the POU TFs facilitates their search for functional target sites in the genome. These studies demonstrated that the POUS and the POUHD of Oct1 could bind non-consecutive DNA elements independently and thereby tether unlinked DNA molecules [123]. This observation inspired a model where the POU scans the genome with the more tightly bound POUHD remaining DNA associated whilst the detached POUS acts as a molecular antenna and samples proximal binding sites. If suitable alternative sites are encountered a process termed intersegment transfer via intermolecular translocation ensues.

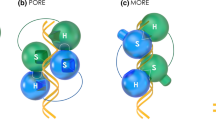
Structural basis for monomeric and dimeric DNA recognition. a Structure of the Oct1-POU bound as monomer to octamer DNA (PDB ID: 1OCT [115]). The POU-specific (POUS) domain is colored green, the POU-homeodomain (POUHD) orange and the linker magenta. b Sequence logo representing the POUS (upper panel) and the POUHD (lower panel) of all 15 mouse POU proteins generated using weblogo (http://weblogo.berkeley.edu/). DNA contact residues, interaction interfaces on MORE and PORE DNA and selected phosphorylation sites are indicated. The two asterisks mark the residue 22 of the POUHD shown to switch the function of Oct1/2 and Brn3a/Brn3b and the residue 59 of the POUHD switching the preference of Oct4/Oct6 for MORE DNA, respectively [227, 229]. c Sequences of the hypervariable linker connecting the POUS and the POUHD. d Oct4 homodimer on PORE DNA element (3L1P [186]) and e of Oct6 bound to MORE DNA (2XSD, [131]. The POU-specific (POUS) and the POU-homeodomain (POUHD) are colored as in a with lighter shades for molecule 1 (M1) and darker shades for molecule 2 (M2) of the POU homodimers. N and C termini are marked
DNA dependent partnerships
POU TFs engage in protein interaction networks that likely change in the context of the bound DNA sequences leading to the recognition of variable sequence signature in chromatin (Fig. 4a–c). These interactions are expected to be critical for the cell-context specific function of POU TFs. The protein interactions are not restricted to the nuclear compartment but also influence other important processes such as intracellular transport and targeted degradation. Here we focus on the protein interactions that are supported by dedicated biochemical experiments or structural analysis and influence DNA recognition and target gene selection.

DNA motifs from genome-wide studies. Position weight matrices (PWMs) resembling a the octamer, b MORE and c canonical/compressed SoxOct elements. PWMs were downloaded from the HOMER database (http://homer.ucsd.edu/homer/index.html) originating from ChIP-seq studies for Oct2 (GSE21512); Oct4 (GSE11431); Oct6, Brn1, Brn2 (GSE35496); Pit1 (GSE58009); canonical SoxOct (GSE11431); compressed SoxOct (GSE44553). Cartoons depict the expected configurations of POUs bound to these sequences. Cartoons for the PORE and SoxOct + 3 bp configuration are shown as reference but the corresponding PWMs are not represented in the database. Selected genes regulated by variant SoxOct elements are listed. The Sox2-HMG is brown and the Sox17-HMG red
Homotypic dimerization
We refer to homotypic interactions for homodimers or heterodimers between paralogous POU TFs. POU factors are a relatively rare example of proteins forming facultative DNA dependent dimers in a versatile range of configurations mediated by their DNA binding domain. Other TF families such as basic helix–loop–helix factors (bHLH, i.e., Myc, Ascl1) or leucine zipper form obligate DNA independent dimers. In early studies cooperative homodimeric binding of Oct1 and Oct2 was observed on composite sequences with a low-affinity heptameric sequence preceding the classical octamer separated by a 2 bp spacer (CTCATGAATATGCAAAT) [124,125,126]. If the heptamer was replaced by a second octamer in the reverse orientation cooperative dimerization persisted [125]. Using random oligonucleotide selection, Brn2 of the POU III class, was found to bind a sequence with flexible spacing between the palindromic half-sites [127]. Brn2 as well as Brn3a (Pou4f1, in [127] termed Brn-3.0) were found to homodimerise on this sequence in a highly cooperative fashion. A structural basis for the homotypic POU dimerization was first provided for Pit1 bound to a PitD DNA element (ATGTATATACAT) [128]. In an effort to test whether the configuration of Pit1 can also be adopted by octamer binding POU TFs, Tomilin et al. converted this element into a perfect palindrome by introducing nucleotides favored by the POUS on octamer DNA [129]. This led to the identification of the More palindromic Oct factor Recognition Element (MORE) with a consensus ATG(C/A)AT(A/T)0–2AT(G/T)CAT that resembles both PitD as well as the sequence preferred by Brn2 [127,128,129,130]. On MORE DNA, cooperative formation of homodimer and heterodimers are formed by Oct1, Oct2, Oct4 and Oct6 and a half-site spacing up to 2 base-pairs is tolerated [129]. Structural studies confirmed that the binding mode of Oct1 [130] and Oct6 [131] to MORE is very similar to Pit1/PitD [128, 132]. Closer inspection of the above-mentioned composite heptamer/octamer element revealed that it in fact also represents a degenerate MORE variant (ATGaATATGCAa, positions deviating from the MORE consensus in small caps) [129]. Further, POU TFs form homodimers on an imperfect palindrome termed PORE (Palindromic Octamer Recognition Element, ATTTGAAATGCAAAT) discovered in the enhancer region of the Osteopontin gene [133]. Effective Oct4 dimerization on the PORE is necessary for transactivation. Crystallographic analysis revealed that Oct1 binds MORE and PORE DNA in markedly different configurations [130]. On PORE DNA, the POU subdomains of one molecule bind major grooves on opposite sides of the DNA. By contrast, on MORE DNA the POU rearranges such that the two domains bind adjacent major grooves on the same face of the DNA (Fig. 3d, e). As a consequence, alternative protein contact interfaces of the POUS and the POUHD mediate the dimeric interactions (Fig. 3b). The functional importance of the MORE motifs in gene regulation in a chromatin context has recently been supported by ChIP-seq analysis for Pit1 [134] and class III TFs [135] (Fig. 4b). However, the PORE has been recovered only once with moderate enrichment levels in a re-analysis of ChIP-seq data of mouse embryonic stem cells (ESCs) [136] suggesting that the bona fide PORE consensus is rarely utilized in a chromatin context. Nevertheless, PORE-like dimer configurations could well be a common mode of chromatin engagement by POU factors as non-DNA binding co-factors can compensate for the degeneration of the PORE consensus. A particularly intriguing example of how sequence requirements of the POU for its DNA target elements can be relieved, is provided by the B cell specific co-factor OBF1 (OcaB, Bob1). OBF1 forms a complex with POU dimers bound to PORE DNA reducing the dissociation rate leading to a profound stabilization of the DNA bound complex [137]. Strikingly, mutations to the POU protein and to the PORE DNA recognition element that normally obstruct dimeric binding can be reversed by OBF1 leading to protein–DNA complexes indistinguishable from the wild-type situation. Apparently, OBF1 alleviates sequence requirements in both the POU domain as well as in the DNA recognition elements including its actual sequence and the half-site spacing. This mechanism adds an additional layer of control and permits the recognition of target genes controlled by DNA elements that strongly diverge from consensus POU recognition sequences that would not be bound in the absence of the co-factor.
To identify structural elements that set POU factors biochemically and functionally apart, models to quantify homodimer cooperativity factors [138] were used to compare POU homodimerisation on a canonical MORE element (ATGAATATTCAT) for factors from POU classes I (Pit1), II (Oct1), III (Brn2 and Oct6), V (Oct4) and VI (Brn5) [87]. Whilst all tested POU TFs are able to form homodimers on the MORE, the extent of the cooperativity is substantially different. All tested somatic POU TFs (Pit1, Oct1, Brn2, Oct6 and Brn5) homodimerise on the MORE with a stronger cooperativity factors than the pluripotency factor Oct4. By structural analysis, the basis for this difference could be worked out. A single amino acid at the C-terminal position 59 of the POUHD (corresponding to position 151 of the Oct4 POU) interacts with a hydrophobic pocket of the POUS located at the opposite face of the DNA (Fig. 3b). Oct4 encodes a polar serine but Oct6 a hydrophobic methionine at this position [87, 130, 131]. When these residues are swapped, the resulting Oct6M59S shows a reduction in cooperativity on MORE whereas Oct4S59M now homodimerises very efficiently reminiscent to wild-type Oct6. The Oct6M59S mutation in combination with further modification can convert Oct6 into a pluripotency reprogramming factor [87]. This suggests that the ability to associate on MORE DNA in a cooperative manner evolved to functionally distinguish POU family members and in particular Oct4. Consistently, POUHD residue 59 is part of a distinctive dipeptide that defines the branches of the POU family suggesting it could be under positive selection leading to family specific functionality [30].
High-throughput SELEX (systematic evolution of ligands by exponential enrichment) reiterated the complexity of DNA recognition by POU TFs [139, 140]. Many of the profiled POU factors were found to share partially overlapping sites that can be arranged in a tiled pattern with overlapping half-site and alternative spacing. These studies recovered a number of motifs resembling the canonical octamer as well as the dimerization promoting MORE elements. Yet, a number of the reported elements suggest potentially novel binding configuration for which the stoichiometry is not obvious. Collectively, homotypic POU dimers critically contribute to the function of these proteins and such binding configurations are more common than initially assumed.
Heterotypic dimerization
The most prominent and intensely studied heterodimer partners of the POU family are from the Sry-related box (Sox) family. The Sox family consists of 20 members and possesses a high mobility group (HMG, 79 amino acids) DNA binding domain associating with a CATTGTC consensus via the minor groove of the DNA [141,142,143,144,145,146]. A DNA-dependent interaction between Oct4 and Sox2 was first described for an enhancer in the last exon of the Fgf4 gene [61, 147, 148]. Transactivation relies on Sox2/Oct4 dimerization on a composite DNA element with a 3 base-pair spacer between Sox and octamer sites (CTTTGTTtggATGCTAAT, spacer nucleotides in small cap). The Sox2/Oct4 dimer functions non-redundantly, as paralogous POU and Sox family members could not activate Fgf4 [61, 147]. Subsequently, co-operative binding of Oct4 and Sox2 on composite SoxOct elements was found to regulate Utf1 [149], Lefty1 [150], Fbx15 [151], Nanog [152, 153] as well as to auto-regulate Sox2 [154] and Oct4 [155, 156]. However, in all these cases the 3 bp spacer found in the Fgf4 enhancer was eliminated leading to a more compact composite element.
Microarray and deep sequencing enabled the genome-wide profiling of POU TFs and pluripotent cells were intensely scrutinized using these technologies with the consequence that Oct4 is the best studied family member. Initially, Oct4 binding to transcription start site (TSS) upstream regions was profiled in human ESCs using DNA microarrays (ChIP-on-ChIP) [157]. This study revealed a high degree of co-occupancy of Sox2, Oct4 and Nanog. A year later Oct4 binding was profiled in mouse ESCs using the chromatin immunoprecipitation paired-end ditag method and de novo motif discovery revealed the composite SoxOct motif as the most enriched sequence signature [158]. This suggested that DNA dependent heterodimerisation of Oct4 with Sox2 on the enhancers of pluripotency genes is a very frequent event. The advent of the more comprehensive chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) technique again led to the identification of a composite DNA element composed of the Sox half site juxtaposed by the Octamer element (CATTGTCATGCAAAT, henceforth termed canonical SoxOct motif). This sequence signature was found to be the most enriched de novo motif not just for Sox2 and Oct4 but also for Nanog, Smad1 and p300 datasets [159] (Fig. 4c). Clearly, Sox2/Oct4 heterodimers play essential roles to assemble regulatory complexes that regulate 100s if not 1000s of genes conferring the pluripotent phenotype. The SoxOct element was also the top motif in ChIP-seq studies using human ESCs [160]. Nevertheless, the cistrome of Oct4 is poorly conserved in mouse and human indicating a high degree of rewiring of cis-regulatory networks in mammalian evolution mainly driven by transposable elements [160]. The notion that the cooperative formation of Sox2/Oct4 heterodimers is essential for pluripotency was reinforced with rationally designed mutants that do not affect the monomeric DNA binding of Sox2 or Oct4 but disrupt DNA dependent heterodimerisation. Both, mutations disrupting dimerization on the canonical SoxOct elements introduced to Sox2 [161, 162] or to Oct4 [87] prevent pluripotency reprogramming. Mutations interfering with Sox2/Oct4 heterodimerisation on the Fgf4-like sequence with 3-bp spacer reduce reprogramming efficiency but do not completely disrupt the process [162].
The genome-wide profiling of Oct4 binding at different stages of pluripotency reprogramming in mouse cells showed that the genome engagement of Oct4 is highly dynamic [163, 164]. These studies indicate that Oct4 is not hitting its binding site in pluripotency enhancers ‘on-target’ but initially binds mostly somatic enhancers and switches to pluripotency enhancers at later stages of the 1–2 week procedure. Only a small proportion of sites are constitutively bound. The correlation of Oct4 binding with histone marks revealed that enhancer activation happens in a stepwise manner whereby Oct4 binding is preceded by H3K4me1 (priming mark) and followed by H3K27ac (activation mark) [163]. The activation of early engaged and constitutively bound pluripotency enhancers require Oct4, Sox2, Klf4 (OSK) to work in concert with somatic TFs (Runx1, Fra1, Cebpa, Cebpb) that are replaced by pluripotency related TFs later on [164]. On the contrary, the co-binding of Sox2/Oct4 and Esrrb facilitates the activation of late bound pluripotency enhancers [164]. These studies indicate that intricate stage specific heterotypic partnerships of Oct4 enable differentiated cells to attain pluripotency. A single molecule imaging study proposed that chromatin engagement of Sox2 and Oct4 is hierarchically ordered with Sox2 binding first assisting the subsequent recruitment of Oct4 [165]. This suggests that Sox2 facilitates the target selection whilst Oct4 predominantly functions to stabilize the heterodimer complex extending its residence time [165]. As these observations were made in 3T3 cells it is unclear whether a similar mechanism applies to pluripotency reprogramming and whether the binding hierarchy is different for closed versus open chromatin.
A high throughput consecutive affinity-purification-SELEX (CAP-SELEX) approach enabled the profiling of TF dimerization using target sequences with 40 randomized base pairs in a scalable manner [166]. This way, a number of co-motifs for POU TFs could be identified including the canonical SoxOct composite motif. Additional heterodimers were predicted for Oct1/POU2F1 with GSC2, TBX21, EOMES, ELK1, ETV1, SOX15, HOXB13 and SOX2. The OCT1/GSC2 dimer showed a strictly constrained arrangement of the two half sites with regard to spacing and order of the TFs. It will be interesting to probe whether these dimers also occur in the context of chromatin and mediate POU functions.
Is there a SoxOct partner code?
The Sox2/Oct4 heterodimer is one of the most prominent examples of heterotypic TF partnerships for its relevance in the gene regulatory network inducing and maintaining pluripotency. The abundance and the strict physical constraint of this complex lead to its repeated discovery in enhancer sequences. A series of additional Sox/Oct heterodimers were reported giving rise to the notion that there could be a Sox/Oct partner code at the heart of gene regulatory networks in early development [167,168,169]. A Sox/Oct partner code presumes two possible scenarios. First, alternative heterodimers prefer different composite DNA motifs and hence select specific sets of target genes. Monomeric factors lack the sequence selectivity and affinity to target functionally relevant gene sets. Second, alternative dimers may retain similar preference for DNA target sites. Yet, molecular events and functional outcome of a binding event may change by turning an activating regulatory complex into a repressive one by means of direct competition. For example, a Sox10/Oct6 dimer was reported to selectively synergize in glial cells [170, 171]. Yet, the DNA sequence requirements for this partnership could not be worked out. In NPCs, Brn2 and Sox2 partner on a non-canonical SoxOct composite element to drive the expression of the Nestin gene [172]. However, genome wide studies did not support a broad application of this dimer configuration. The model of a switch of Sox2 from Oct4 to the neural class III POU factors is particularly appealing as Sox2 functions prominently in both pluripotent as well as neural lineages. Indeed, a switch from Oct4 to Brn1/2 on identical enhancer locations was suggested to maintain Sox2 expression in pluripotent and neural cells [173]. The partnership of Sox2 and class III POU TFs was further explored in genome-wide studies. Initially, Brn2 and Sox2 were reported to co-bind the canonical SoxOct element with Sox2 in NPCs reminiscent to the Sox2/Oct4 heterodimer in ESCs [174]. The differentiation of ESCs to NPCs led to the co-recruitment of Sox2/Brn2 to NPC-specific enhancers. Conversely, overexpression of Oct4 in NPCs led to generation of iPSCs presumably by re-directing Sox2 from neural to pluripotency enhancers [88, 89]. Whilst these studies indicated that the DNA sequence signature facilitating SoxOct heterodimerisation in ESCs versus NPC/NSCs is indistinguishable with the canonical SoxOct motif at its core, the set of bound genes is very different. Consistently, Sox2/Oct4 and Sox2/POUIII dimers (Oct6, Brn2 and Brn4) showed an indistinguishable cooperativity pattern when 324 composite DNA sequences were profiled in a recent cooperativity-by-sequencing (Coop-seq) study [175]. Similarly, EMSA measurements showed that the cooperativity constant for Sox2/Oct4 dimerisation was only about twofold higher than for Sox2/Oct6 dimerization on canonical SoxOct DNA [87]. This poses the question as to the molecular basis for the selection of unique gene sets by Sox2/Oct4 versus Sox2/POUIII complexes. A variation to the SoxOct element in the POUHD bound portion found in the Utf1 (ATGCTAGA) sequence had been attributed to this difference as only Sox2/Oct4 but not Sox2/Oct6 could effectively dimerize on this sequence [176]. However, genome-wide analysis could so far not detect obvious difference at this position. Mistri et al. also profiled the binding of Oct6, Brn1 and Brn2 in mouse NPCs [135]. Contrary to Lodato et al., here the homodimerisation promoting MORE was identified as the preferred motif for all three class III POU factors. In support of this finding, motif scanning with the MORE using data from Lodato et al. could detect the MORE in a large fraction of Brn2 binding sites [87, 135]. Analogously, re-analysis of Brn2 ChIP-seq data during the reprogramming of MEFs to neurons showed a strong enrichment of the MORE [85, 87]. Collectively, a picture emerges that differential binding to MORE sequences appears to contribute more strongly to the selection of unique sets of target genes by Oct4 and class III POUs whereas the capacity to dimerise with Sox2 on canonical SoxOct is shared. In another study, novel configurations of SoxOct elements were searched for in the enhancers of ESCs and a variation of the canonical SoxOct was discovered to show a modest level of enrichment [161]. Here, a single nucleotide separating Sox and Oct half-sites is eliminated and this sequence was hence designated ‘compressed’ SoxOct element (CATTGTATGCAAAT). Surprisingly, Sox2 and Oct4 are unable to co-bind this sequence whilst Sox17 and Oct4 dimerize very efficiently on it [161, 175, 177]. ChIP-seq verified the functional relevance of the compressed SoxOct motif after the overexpression of Sox17 in ESCs as well as after the retinoic acid induced endodermal differentiation of embryonic carcinoma cells [178]. By structural analysis, the molecular basis for the differential propensities of Sox2/Oct4 and Sox17/Oct4 dimers to differentially select canonical or compressed DNA elements could be elucidated. Residue 57 of helix 3 of the HMG box encodes a basic lysine in Sox2 but an acidic glutamate in Sox17 [141]. Grafting this residue into Sox17 to generate the engineered Sox17EK produced a protein that cooperates more strongly than Sox2 with Oct4 on the canonical SoxOct element. By contrast, Sox17EK loses the capacity to dimerize with Oct4 on the compressed element in vitro and in the context of chromatin [161, 178, 179]. The functional relevance of this finding was further tested in pluripotency reprogramming. Only Sox1, Sox3 and Sox15 can replace Sox2 in this experiment but Sox17 and other Sox family members cannot [86, 180]. Strikingly, Sox17EK and the analogous Sox7EK now acquire the capacity to generate iPSCs in mouse and human cells and substantially outperform wild-type Sox2 [161, 180]. Consistently, Sox17EK is able to support the self-renewal of ESCs depleted of Sox2 whilst Sox2KE loses this capacity [181]. This provides a compelling example how the subtle features of single amino acids in trans and of single nucleotides in cis define gene regulatory networks during embryogenesis. Moreover, this insight provides a molecular mechanism for the context dependent switch of the regulatory activity of Oct4. Relatedly, Sox17 has recently been found to be a key regulator of the differentiation of primordial germ cells (PGCs) from naïve pluripotent cells in human, but not mouse [182]. As Oct4 is expressed in pluripotent cells as well as PGC it will be of interest to explore whether an analogous Sox/Oct partner switch that relies on canonical and compressed motifs guides the redistribution from pluripotency to PGC enhancers.
Collectively, Sox factors evolved molecular interfaces allowing their selective association with POU TFs in the context of alternative composite DNA sequences, two of which (canonical and compressed SoxOct DNA elements) are widespread in developmental enhancers. By contrast, whether or not POU TFs evolved similarly selective interfaces is presently unclear.
Binding to methylated DNA
DNA methylation changes profoundly during the conversion of MEFs to iPSCs and the DNA-methylation level was initially reported to be negatively correlated with Oct4 binding [163]. Another study reported that Oct4 binding is independent of the DNA-methylation status at early stages of reprogramming, whereas at late stages Oct4 binding coincides with unmethylated DNA at pluripotency enhancers [183]. To directly examine the impact of cytosine methylation on DNA by TFs the high-throughput methylation-sensitive SELEX was developed [140]. It is generally thought that DNA methylation serves as a barrier to TF DNA interactions and that the remodelling of CpG methylation is essential for cell fate transition. Indeed, a large fraction of the profiled TFs showed sensitivity to enzymatically introduced methyl moieties at CpG dinucleotides. However, for a subset of TFs, DNA methylation facilitates rather than blocks binding. In particular, many homeodomain containing TFs show a preference for methylated DNA including Oct4. This study identified a tertiary Oct4 motif comprised of a palindrome consisting of two POUS half-sites ATGCGCAT. SELEX enrichment of this element is increased upon methylation suggesting that Oct4 preferentially targets it in the methylated state. The preference of Oct4 to bind this element in its methylated form was validated using ChIP-seq combined with whole-genome bisulfite sequencing in mouse embryonic stem cells where the DNA de-methylating enzymes Tet1-2 were knocked-out leading to an accumulation of methylated DNA. Interestingly, as the ability to bind methylated CpG DNA is largely restricted to TFs involved in embryonic development such as homeodomain TFs, the potency to bind methylated DNA may underlie their ability for epigenetic remodeling leading to cell fate switching.
The role of the linker
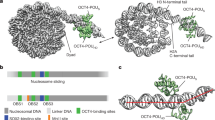
The length of the linker varies for different POU TFs (Fig. 3c). Structures of higher order POU/DNA complexes demonstrated a striking flexibility to accommodate alternative DNA sequences and to adopt very different conformations induced by the DNA (Fig. 3d, e). This versatility is enabled by the variable and structurally flexible linker that allows for the spatial reorganization of two domains endowing POU TFs with the ability to adopt diverse quaternary structures. The linker was not visible in earlier structures [115, 128, 130, 131, 142, 184]. However, the linker could be modeled for Brn5-POU bound to a non-octamer DNA sequence derived from the corticotrophin-releasing hormone promoter [185]. The relative orientation of the POUS and POUHD is flipped in Brn2 in comparison to the arrangement seen for Oct1 on the octamer element. Here, the linker extends the helix 4 of the POUS by 7.5 Å. The extended helix is followed by a sharp turn and an additional short helix. Similarly, a large part of the linker was visible in Oct4POU structure on PORE DNA element whilst it was invisible in the Oct1/PORE complex [186]. The linker of Oct4 forms an additional alpha-helix with amino acids unique to Oct4 but conserved amongst its orthologues. This finding led to the conclusion that Oct4link contributes to the distinct role of Oct4 in pluripotency reprogramming presumably by recruiting specific co-activators [186]. Consistently mutations to the linker were found to affect the induction of pluripotency [186,187,188]. Similarly, when the Oct4 linker is replaced by the Oct6 linker the resulting Oct4linkOct6 loses the competency to generate iPSCs [186]. However, given the poor sequence conservation the definition of the linker is ambiguous. More recently the boundaries of the linker were redefined taking structural information into account. The new definition removed an additional charge from the chimeric Oct4linkOct6 protein where the linker borders the POUHD (Oct4linkOct6: QGKRKKR, Oct6 derived residues underlined) and enabled iPSCs generation [87] whilst a version containing this additional charge was impaired (Oct4linkOct6: QGRKRKKR) [186]. Collectively, diversity in DNA recognition by POU TFs is largely achieved by modifications to the linker and to residues mediating the intra and intermolecular cooperativity. It is these regions that set POU protein apart and allow them to assemble into versatile dimeric configurations to bind alternative DNA elements to regulate non-redundant sets of genes leading to different regulatory outcomes.
Pioneering activity: a unique competency of Oct4?
During development the chromatin states and associated gene expression programs are successively changed until terminally differentiated cells are formed. Reprogramming experiments by forced TF expression have shown that a limited set of factors is capable of inducing the remodeling of chromatin even of terminally differentiated cells leading to changed gene expression programs and cell state conversions. It has been proposed that only a selected set of TFs that could access closed chromatin compacted by nucleosomes and higher order chromosomal assemblies is capable of this feat. These molecules were termed ‘pioneer TFs’ because they are required for the initial engagement of closed chromatin leading to the subsequent recruitment of non-pioneering TFs that would not be able to access these sites themselves [189]. The pioneer TF concept was proposed initially from studies on FoxA1 during the differentiation of mouse endodermal tissue to mature hepatocytes [190]. FoxA1 initially engages closed chromatin followed by the recruitment of companion TFs and gene activation. FoxA1 was also found to be able to bookmark highly compact chromatin during mitosis [191]. The DNA binding domain of FoxA1 and other forkhead TFs is of the winged–helix type [192]. This fold closely resembles the structure of the linker histone H1 [193, 194]. H1 has a major role in the compaction of histone octamers and the formation of higher order nucleosomal structures such as the 30 nm fiber [195, 196]. This similarity is attributed to the ability of FoxA1 to open nucleosomal arrays compacted by H1 as it can directly compete with H1 [197]. However, more recently additional TFs have been reported to possess pioneering activity including Oct4. The integration of micrococcal nuclease sequencing (MNAse-seq) and ChIP-seq data showed that Oct4 in combination with Klf4 and Sox2 binds DNA that was initially covered by nucleosomes at the onset of pluripotency reprogramming of human cells which is thought to be causative for rapid opening (Fig. 5a) [198]. Data were later supported by binding assays with in vitro reconstituted nucleosomes showing that Oct4, Sox2 and Klf4 bind nucleosome associated DNA with similar affinity as free DNA [199]. Motif discovery suggested that rather than binding full motifs, Oct4 only binds shortened degenerate versions of its classical binding sequence corresponding to POUS and POUHD half-sites. Soufi et al. reasoned this is because in the context of the full octamer Oct4 binds DNA with its sub-domains arranged on opposite faces (Fig. 3a). As this would lead to steric interference with histone proteins, only individual half-sites can be exposed and are accessible on the nucleosome surface. It will be interesting to test this model structurally or using binding assays with nucleosome associated DNA having different motif compositions. Regardless, structural similarity to the linker histone does not appear to be a defining feature of pioneer TFs. An extensive analysis by the Plath laboratory of the epigenetic changes during reprogramming of mouse embryonic fibroblasts to induced pluripotent stem cells came to a different conclusion as to the pioneering activity of Oct4 [164]. Here, is was reported that Oct4 and its companion reprogramming TFs predominantly bind pre-opened chromatin of somatic enhancers and promoters promiscuously at early reprogramming stages leading to the silencing of somatic genes. By contrast, pluripotency enhancers are bound and opened dynamically in a step-wise fashion in cooperation with somatic TFs rather than by a directed pioneering process starting immediately at the onset of reprogramming. Further, the starting chromatin state was suggested not to be predictive for the pluripotency enhancers the TFs will eventually target. In this view, reprogramming TFs do not actively trigger chromatin opening but rather passively follow and bind pluripotency enhancers after they were opened by an unexplained mechanism. It was concluded that there could be species-specific differences in the reprogramming process in mouse and human. A related study by the laboratory of Jacob Hanna performed a high-resolution assessment of the epigenetic dynamics of pluripotency reprogramming in mouse [200]. The cells reprogram near-deterministically within 8 days because of the depletion of the reprogramming barriers Mbd3 and Gatad2a of the NuRD complex. In this study, 74% of enhancers bound by OSK at day 1 are closed in MEFs. However, full DNA motifs rather than half motifs were discovered in these presumably nucleosome covered regions. Likewise, two additional studies conducted using mouse cells, emphasized that Sox2 and Oct4 have the ability to target compacted chromatin rather than preferring locations pre-opened in MEFs. First, a study by the Jose Polo’s laboratory used FACS (Fluorescence-activated cell sorting) to separate cells successfully progressing to pluripotency from cells that fail. The authors find that 75% of Oct4, Sox2 binding occurs at sites that are closed in MEFs but are opened at day 3 of reprogramming implying an active role of Sox2/Oct4 in the opening process [183]. Likewise, using a highly efficient chemically defined reprogramming system, Li and colleagues used time course ATAC-seq to show that the majority of somatic enhancers open in MEF are devoid of Sox and POU binding motifs whereas a large proportion of sites that undergo a closed-to-open transition contain matches to these motifs [201]. Collectively, these studies support a model where Oct4, Sox2 and Klf4 actively induce epigenetic switching of pluripotency enhancers and directly induce chromatin opening consistent with the pioneering model. Nevertheless, the discrepancies in the interpretations from these complementarity studies should be resolved, which could be due to the different reprogramming systems or because of alternative data analysis strategies. Likewise, the cis-regulatory codes dictating the engagement of closed versus open chromatin remain unclear and require further investigations.

Molecular basis for context-dependent functions. a According to current models Oct4 is be able to bind nucleosomal DNA and induce opening whilst Brn2 can only bind chromatin pre-opened by factors such as Ascl1 [85, 198, 199]. The colored rectangles denote cognate binding sites. b Pit1 bound as homodimer to a compact binding site in the Prolactin (Prl) results in transcriptional activation whilst the introduction of a TT spacer (marked in red) in the growth hormone (GH) promoter results in the recruitment of the NCoR co-repressor [132]. c Binding of Oct1 as homodimer to the PORE element recruits OBF1 to activate transcription whilst the homodimeric configuration on MORE impairs OBF1 recruitment and reduces the transcriptional response [129, 130]. d–g Single amino acid exchanges can switch functions of closely related paralogs. d Interchanging position 22 of the POUHD between Oct1 and Oct2 equips Oct2 with the capacity to recruit VP-16 and regulate transcription reminiscent to Oct1 [227, 228]. e An analogous exchange between Brn3a and Brn3b was reported to switch them from transcriptional activators to repressors on certain response elements [229]. f Oct4 co-binds with Sox2 to canonical SoxOct elements to control the transcription of pluripotency related genes but in the presence of Sox17, Oct4 is re-distributed to enhancers earmarked by compressed SoxOct elements to regulated extra-embryonic endoderm (XEN) genes [178]. Point mutations at the Oct4 interaction interface of Sox2 or Sox17 change binding and function of the resulting DNA dependent heterodimers. g Oct4 prefers heterodimerisation with Sox2 on canonical SoxOct elements whereas Oct6 prefers to form a homodimers on MORE elements. A single amino acid swap between Oct4 and Oct6 changes the binding preferences of the two proteins [87]. Solid green arrows represent context-dependent activities of wild type proteins; asterisks mark mutations and dashed arrows represent newly acquired functions
The pioneering question was also addressed for Brn2 (class III POU) during the highly efficient conversion of fibroblasts to mature neurons as part of the Brn2, Ascl1, Myt1l (BAM) cocktail [85] (Fig. 2c, d). Genomic binding analysis suggested that the HLH TF Ascl1 binds neural enhancers immediately after its forcible introduction into MEFs and was hence termed ‘on-target’ pioneer [85]. In contrast, Brn2 shows little chromatin engagement at the beginning of reprogramming and associates with relevant binding sites only after Ascl1 opened them. Thus, intriguingly, despite profound structural similarity, Oct4 is believed to function as pioneer but Brn2 is not (Fig. 5a). A careful analysis of the nucleosome binding and opening of Oct4 versus Brn2 should be carried out to test this model and to search for the molecular features endowing Oct4, but not Brn2, with pioneering capacity. It is possible that modified preference to DNA target sequence influence the ability to bind and remodel nucleosomes even of closely related factors. Alternatively, there may not be a clear-cut separation between pioneer and non-pioneer TFs. Further, whether ATP-dependent remodeling complexes are required for the opening process is currently debated [202, 203]. In conclusion, the molecular mechanism as to how POU TFs engage and remodel chromatin to direct changes of cell states awaits further mechanistic interrogation.
Different DNA binding modes direct alternative regulatory outcomes
Rather than forming rigid and structurally inert ‘enhanceosomes’ [204], POU factors are more likely to engage in flexible and dynamically changing partnerships in the context of different cis-regulatory regions. This versatility probably underlies context-dependent regulatory outcomes entailing the activation or repression of nearby genes but also include more complex epigenetic processes that do not lead to obvious effects on gene expression such as the maintenance of a ‘bivalent’ chromatin state. A consensus view is that most TFs regulate their target genes by triggering the formation of a loop between enhancers and promoters mediated by large molecular machineries such as the mediator facilitated by cohesion [205, 206]. Bivalent chromatin refers to the co-occurrence of ‘repressive’ H3K27me3 and ‘active’ H3K4me3 chromatin modifications which are particularly prominent at gene promoters of developmental genes in pluripotent cells [159, 207,208,209]. Proteomic studies indicated that Oct4 interacts with the machineries depositing the activation mark H3K4me3 as well as the depletion of repressive mark H3K27me3 probably assisted by WDR5 (H3K4me3 reader) and UTX (H3K27 demethylase) [210,211,212]. Working out how DNA sequences allosterically affect the recruitment of these various complexes is crucial to reveal the molecular underpinnings for the context dependent functions of POU TFs. Alternative binding modes occur on the level of binary and ternary POU/DNA complexes encompassing homotypic and heterotypic interactions. Switched binding configurations were demonstrated by structures of Oct1 [130], Oct6 [131] and Pit1 [128, 132] on PORE or MORE DNA leading to strikingly different homodimeric assemblies. In contrast to binary POU/DNA complexes on octamer DNA that lack protein–protein interactions, such contacts exist on the ternary PORE and MORE. This raised the question as to whether different DNA binding modes lead to different regulatory outcomes.
During the development of the pituitary gland Pit1 regulates both a lactotrop and a somatotrop programs from a common primordium by activating the growth hormone or prolactin genes, respectively [132]. The prolactin (Prl-1P) element resembles a degenerate MORE without spacer whilst the growth hormone element (GH-1) possesses a version with 2 base pair spacer. When the spacer is deleted from a reporter, expression is detected in lactotropes rather than somatotropes reminiscent to wild-type Prl-1P [132]. The context specific activation of the growth hormone in somatotrope cells but not in lactotropes was suggested to be mediated by differential recruitment of N-CoR (nuclear corepressor complex), which is sensitive to half-site spacing (Fig. 5b). Analogously, Oct1 dimers on PORE were shown to interact with the coactivator OBF-1 but this interaction does not occur when Oct1 is bound to the MORE because Oct1 residues required for OBF-1 recruitment are unavailable on the MORE as they shape the homodimer interface (Fig. 5c) [129]. Collectively, the assembly of subdomains on composite DNA elements as well as the spacing between half-sites critically influences cofactor recruitment and the transcriptional consequences of a binding event.
An interesting concept is that the preference for DNA sequences, target gene selection and the regulatory outcome of POU TFs can be influenced by post-translational modifications. This could allow a single POU TF to execute different regulatory programs in the same cell without epigenetic changes or the need for new co-factors but simply in response to intracellular signaling. POU factors are subjected to a variety of post-translational modifications including phosphorylation [213,214,215,216,217,218], O-GlcNAcylation [219,220,221], SUMOylation [222, 223] and ubiquitinylation [220, 224, 225]. Phosphorylation of the Oct4-POUS was suggested to more severely affect transcription from genes controlled by the PORE compared to MORE and SoxOct dependent targets [226]. In another study, cellular stress induced Oct1 phosphorylation at Ser385 of the POUHD was found to be deleterious for monomeric binding to the octamer but favors the association with promoter proximal MOREs [214]. Additionally, mutations of several potential phosphorylation sites including the sites corresponding to Oct1/Ser385 in Oct4 resulted in the complete abolishment of its reprogramming activity [187].
Outlook
In this review, we attempted to provide an in depth summary of our present understanding, how the ability of POU factors to bind DNA and chromatin in different configurations and with different partner factors relates to their capacity to reprogram cell states. We particularly emphasised the differences between paralogous POU genes and the unique ability of Oct4 to direct pluripotency reprogramming. The POU family is a particularly captivating group of TFs for their astonishing plasticity to bind a large set of composite DNA sequences either as homodimers or heterodimers. This plasticity likely dictates the selection of specific gene sets, the nature of the regulatory outcomes following binding, and changes to chromatin dynamics. This in turn determines context specific roles in embryonic development and during cellular reprogramming. Classical sequence comparison and mutagenesis studies revealed that even very subtle changes to the POU domain could have a tremendous functional impact. For example, residue 22 at helix 1 of the POUHD was found to be the sole determinant for differential gene regulation by Oct1 versus Oct2 and Brn3b versus Brn3a in some systems (Fig. 5d, e). The mere swap of this residue between Oct2 and Oct1 bestows Oct2 with the ability to recruit the VP16 co-activator to positively regulate transcription [227, 228]. Analogously, an isoleucine at this position is essential for the repressor function of Brn3b whilst a valine as found in Brn3a leads to transactivation [229]. Interchanging this residue swaps the regulatory activity of Brn3a and Brn3b. Residue 22 is solvent exposed and thus does not affect DNA binding and the dimeric configuration. Likewise, single amino acid swaps between Sox2 and Sox17 as well as between Oct4 and Oct6 can rebalance the association with heterotypic or homotypic partner proteins, thus switching their activities in pluripotency reprogramming (Fig. 5f, g) [87, 161]. These insights reinforce the notion that the structural properties that functionally discriminate POU TFs are predominantly provided by their DNA binding domains. Yet, overall sequence function relationships, both on the level of the trans acting POU factors and the cis DNA elements they are targeting, remain only poorly understood. Deep mutational scanning [230], with a focus on sites that were shown to set POU paralogs functionally and biochemically apart, provides a powerful approach to systematically define the key amino acids that led to the biochemical and functional diversification of POU TFs. Moreover, the side-by-side profiling of the epigenetic changes accompanying pluripotency and lineage reprogramming driven by paralogous POU factors provide a means to study the functional consequences of sequence diversifications. The POU domain not only mediates DNA recognition but also the DNA dependent partnership with other TFs as well as the selective recruitment of co-activators and repressors, which we are only beginning to work out. A number of proteomics studies have begun to address this problem with a focus on Oct4 in embryonic stem cells [231,232,233] and started to reveal the underpinnings of the cross-talk of Oct4 with epigenetic modifiers and the transcriptional machinery. However, these various studies revealed a surprisingly limited overlap for the identified sets of interaction partners in embryonic stem cells [231,232,233]. Future studies should contrast the interactomes of paralogous POU factors and define how the context of DNA binding sites changes the recruitment of co-activators. Here, studying the reliance of partner recruitment on the various DNA elements targeted by POU TFs (SoxOct, MORE, PORE) could reveal important insights as to the context specific regulatory programs directed by POU TFs. Ideally, such experiments should be carried out in the context of native chromatin. Future work should also address whether different binding modalities can switch regulatory responses. For example, delayed versus immediate transcriptional responses could be reliant on the DNA-bound configuration of POU TFs. Likewise, transient and sustained transcriptional responses could be modulated in this manner. The utilization of high throughput enhancer reporter assay such as STARR-seq (self-transcribing active regulatory region sequencing) [234] provides a method to study these questions systematically.
A molecular interpretation of genomic, epigenetic and functional data is impeded because the available structural information of POU TFs is currently limited to their DNA binding domains bound to a small set of DNA binding sites as monomers, homodimers or heterodimers. Structures of full-length POU proteins or higher order regulatory complexes are not available. Obtaining such structures may remain challenging in the years to come because of the large extent of structurally disordered portions outside of the POU domain and the highly dynamic and DNA context dependent assembly of TF complexes. Such complexes can likely be assembled only in vitro, once the DNA sequence requirements and the protein–protein interaction surfaces have been further refined. Biochemical and genomic assays demonstrated that POU factors could bind nucleosomes with high affinity [199]. The cis-regulatory context that facilitates binding of POU factors to nucleosome bound DNA, which is considered to be the first step eventually leading to an increase in the accessibility of such genomic regions, is not resolved so far. Structures of TF nucleosome complex are not available at present, thus precluding a molecular understanding of the pioneering process. However, advances to study higher order nucleosomal complexes by electron microscopy could put the goal of revealing the structural basis for TF: nucleosome recognition and pioneering activity within reach [195, 196, 235]. As a result of a more detailed dissection of the sequence-function relationships for POU factors and the structural basis for DNA, co-factor and chromatin recognition; efforts to switch or enhance reprogramming activities of POU factors could be invigorated. As a consequence, we expect structurally informed protein engineering to further advance reprogramming technologies by the design of artificial TFs based on the versatile scaffold of POU TFs [236].
References
Parslow TG, Blair DL, Murphy WJ, Granner DK (1984) Structure of the 5′ ends of immunoglobulin genes: a novel conserved sequence. Proc Natl Acad Sci USA 81(9):2650–2654
Carbon P, Murgo S, Ebel JP, Krol A, Tebb G, Mattaj LW (1987) A common octamer motif binding protein is involved in the transcription of U6 snRNA by RNA polymerase III and U2 snRNA by RNA polymerase II. Cell 51(1):71–79
LaBella F, Sive HL, Roeder RG, Heintz N (1988) Cell-cycle regulation of a human histone H2b gene is mediated by the H2b subtype-specific consensus element. Genes Dev 2(1):32–39
Pruijn GJ, van Driel W, van der Vliet PC (1986) Nuclear factor III, a novel sequence-specific DNA-binding protein from HeLa cells stimulating adenovirus DNA replication. Nature 322(6080):656–659. https://doi.org/10.1038/322656a0
Sturm R, Baumruker T, Franza BR Jr, Herr W (1987) A 100-kD HeLa cell octamer binding protein (OBP100) interacts differently with two separate octamer-related sequences within the SV40 enhancer. Genes Dev 1(10):1147–1160
Staudt LM, Singh H, Sen R, Wirth T, Sharp PA, Baltimore D (1986) A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature 323(6089):640–643. https://doi.org/10.1038/323640a0
Fletcher C, Heintz N, Roeder RG (1987) Purification and characterization of OTF-1, a transcription factor regulating cell cycle expression of a human histone H2b gene. Cell 51(5):773–781
Hanke JH, Landolfi NF, Tucker PW, Capra JD (1988) Identification of murine nuclear proteins that bind to the conserved octamer sequence of the immunoglobulin promoter region. Proc Natl Acad Sci USA 85(10):3560–3564
Scheidereit C, Heguy A, Roeder RG (1987) Identification and purification of a human lymphoid-specific octamer-binding protein (OTF-2) that activates transcription of an immunoglobulin promoter in vitro. Cell 51(5):783–793
O’Neill EA, Kelly TJ (1988) Purification and characterization of nuclear factor III (origin recognition protein C), a sequence-specific DNA binding protein required for efficient initiation of adenovirus DNA replication. J Biol Chem 263(2):931–937
Clerc RG, Corcoran LM, LeBowitz JH, Baltimore D, Sharp PA (1988) The B-cell-specific Oct-2 protein contains POU box- and homeo box-type domains. Genes Dev 2(12A):1570–1581
Ko HS, Fast P, McBride W, Staudt LM (1988) A human protein specific for the immunoglobulin octamer DNA motif contains a functional homeobox domain. Cell 55(1):135–144
Muller MM, Ruppert S, Schaffner W, Matthias P (1988) A cloned octamer transcription factor stimulates transcription from lymphoid-specific promoters in non-B cells. Nature 336(6199):544–551. https://doi.org/10.1038/336544a0
Scheidereit C, Cromlish JA, Gerster T, Kawakami K, Balmaceda CG, Currie RA, Roeder RG (1988) A human lymphoid-specific transcription factor that activates immunoglobulin genes is a homoeobox protein. Nature 336(6199):551–557. https://doi.org/10.1038/336551a0
Bodner M, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M (1988) The pituitary-specific transcription factor GHF-1 is a homeobox-containing protein. Cell 55(3):505–518
Ingraham HA, Chen RP, Mangalam HJ, Elsholtz HP, Flynn SE, Lin CR, Simmons DM, Swanson L, Rosenfeld MG (1988) A tissue-specific transcription factor containing a homeodomain specifies a pituitary phenotype. Cell 55(3):519–529
Finney M, Ruvkun G, Horvitz HR (1988) The C. elegans cell lineage and differentiation gene unc-86 encodes a protein with a homeodomain and extended similarity to transcription factors. Cell 55(5):757–769
Herr W, Sturm RA, Clerc RG, Corcoran LM, Baltimore D, Sharp PA, Ingraham HA, Rosenfeld MG, Finney M, Ruvkun G et al (1988) The POU domain: a large conserved region in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. Genes Dev 2(12A):1513–1516
McGinnis W, Garber RL, Wirz J, Kuroiwa A, Gehring WJ (1984) A homologous protein-coding sequence in Drosophila homeotic genes and its conservation in other metazoans. Cell 37(2):403–408
Scott MP, Weiner AJ (1984) Structural relationships among genes that control development: sequence homology between the Antennapedia, Ultrabithorax, and fushi tarazu loci of Drosophila. Proc Natl Acad Sci USA 81(13):4115–4119
Robertson M (1988) Homoeo boxes, POU proteins and the limits to promiscuity. Nature 336(6199):522–524. https://doi.org/10.1038/336522a0
Herr W, Cleary MA (1995) The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev 9(14):1679–1693
Jerabek S, Merino F, Scholer HR, Cojocaru V (2014) OCT4: dynamic DNA binding pioneers stem cell pluripotency. Biochim Biophys Acta 1839(3):138–154. https://doi.org/10.1016/j.bbagrm.2013.10.001
Phillips K, Luisi B (2000) The virtuoso of versatility: POU proteins that flex to fit. J Mol Biol 302(5):1023–1039. https://doi.org/10.1006/jmbi.2000.4107
Remenyi A, Tomilin A, Scholer HR, Wilmanns M (2002) Differential activity by DNA-induced quarternary structures of POU transcription factors. Biochem Pharmacol 64(5–6):979–984
Ryan AK, Rosenfeld MG (1997) POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev 11(10):1207–1225
Tantin D (2013) Oct transcription factors in development and stem cells: insights and mechanisms. Development 140(14):2857–2866. https://doi.org/10.1242/dev.095927
Verrijzer CP, Van der Vliet PC (1993) POU domain transcription factors. Biochim Biophys Acta 1173(1):1–21
Wegner M, Drolet DW, Rosenfeld MG (1993) POU-domain proteins: structure and function of developmental regulators. Curr Opin Cell Biol 5(3):488–498
Gold DA, Gates RD, Jacobs DK (2014) The early expansion and evolutionary dynamics of POU class genes. Mol Biol Evol 31(12):3136–3147. https://doi.org/10.1093/molbev/msu243
Rosenfeld MG (1991) POU-domain transcription factors: pou-er-ful developmental regulators. Genes Dev 5(6):897–907
Holland PW, Booth HA, Bruford EA (2007) Classification and nomenclature of all human homeobox genes. BMC Biol 5:47. https://doi.org/10.1186/1741-7007-5-47
Scholer HR (1991) Octamania: the POU factors in murine development. Trends Genet 7(10):323–329
Hutchins AP, Yang Z, Li Y, He F, Fu X, Wang X, Li D, Liu K, He J, Wang Y, Chen J, Esteban MA, Pei D (2017) Models of global gene expression define major domains of cell type and tissue identity. Nucleic Acids Res 45(5):2354–2367. https://doi.org/10.1093/nar/gkx054
Sturm RA, Das G, Herr W (1988) The ubiquitous octamer-binding protein Oct-1 contains a POU domain with a homeo box subdomain. Genes Dev 2(12A):1582–1599
Landolfi NF, Capra JD, Tucker PW (1986) Interaction of cell-type-specific nuclear proteins with immunoglobulin VH promoter region sequences. Nature 323(6088):548–551. https://doi.org/10.1038/323548a0
Staudt LM, Clerc RG, Singh H, LeBowitz JH, Sharp PA, Baltimore D (1988) Cloning of a lymphoid-specific cDNA encoding a protein binding the regulatory octamer DNA motif. Science 241(4865):577–580
Andersen B, Weinberg WC, Rennekampff O, McEvilly RJ, Bermingham JR Jr, Hooshmand F, Vasilyev V, Hansbrough JF, Pittelkow MR, Yuspa SH, Rosenfeld MG (1997) Functions of the POU domain genes Skn-1a/i and Tst-1/Oct-6/SCIP in epidermal differentiation. Genes Dev 11(14):1873–1884
Matsumoto I, Ohmoto M, Narukawa M, Yoshihara Y, Abe K (2011) Skn-1a (Pou2f3) specifies taste receptor cell lineage. Nat Neurosci 14(6):685–687. https://doi.org/10.1038/nn.2820
Andersen B, Schonemann MD, Pearse RV 2nd, Jenne K, Sugarman J, Rosenfeld MG (1993) Brn-5 is a divergent POU domain factor highly expressed in layer IV of the neocortex. J Biol Chem 268(31):23390–23398
He X, Treacy MN, Simmons DM, Ingraham HA, Swanson LW, Rosenfeld MG (1989) Expression of a large family of POU-domain regulatory genes in mammalian brain development. Nature 340(6228):35–41. https://doi.org/10.1038/340035a0
Mathis JM, Simmons DM, He X, Swanson LW, Rosenfeld MG (1992) Brain 4: a novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J 11(7):2551–2561
Wey E, Lyons GE, Schafer BW (1994) A human POU domain gene, mPOU, is expressed in developing brain and specific adult tissues. Eur J Biochem 220(3):753–762
Monuki ES, Weinmaster G, Kuhn R, Lemke G (1989) SCIP: a glial POU domain gene regulated by cyclic AMP. Neuron 3(6):783–793
Suzuki N, Rohdewohld H, Neuman T, Gruss P, Scholer HR (1990) Oct-6: a POU transcription factor expressed in embryonal stem cells and in the developing brain. EMBO J 9(11):3723–3732
Nakai S, Kawano H, Yudate T, Nishi M, Kuno J, Nagata A, Jishage K, Hamada H, Fujii H, Kawamura K et al (1995) The POU domain transcription factor Brn-2 is required for the determination of specific neuronal lineages in the hypothalamus of the mouse. Genes Dev 9(24):3109–3121
Schonemann MD, Ryan AK, McEvilly RJ, O’Connell SM, Arias CA, Kalla KA, Li P, Sawchenko PE, Rosenfeld MG (1995) Development and survival of the endocrine hypothalamus and posterior pituitary gland requires the neuronal POU domain factor Brn-2. Genes Dev 9(24):3122–3135
Scholer HR, Hatzopoulos AK, Balling R, Suzuki N, Gruss P (1989) A family of octamer-specific proteins present during mouse embryogenesis: evidence for germline-specific expression of an Oct factor. EMBO J 8(9):2543–2550
Schreiber E, Harshman K, Kemler I, Malipiero U, Schaffner W, Fontana A (1990) Astrocytes and glioblastoma cells express novel octamer-DNA binding proteins distinct from the ubiquitous Oct-1 and B cell type Oct-2 proteins. Nucleic Acids Res 18(18):5495–5503
Gerrero MR, McEvilly RJ, Turner E, Lin CR, O’Connell S, Jenne KJ, Hobbs MV, Rosenfeld MG (1993) Brn-3.0: a POU-domain protein expressed in the sensory, immune, and endocrine systems that functions on elements distinct from known octamer motifs. Proc Natl Acad Sci USA 90(22):10841–10845
Lillycrop KA, Budrahan VS, Lakin ND, Terrenghi G, Wood JN, Polak JM, Latchman DS (1992) A novel POU family transcription factor is closely related to Brn-3 but has a distinct expression pattern in neuronal cells. Nucleic Acids Res 20(19):5093–5096
Turner EE, Jenne KJ, Rosenfeld MG (1994) Brn-3.2: a Brn-3-related transcription factor with distinctive central nervous system expression and regulation by retinoic acid. Neuron 12(1):205–218
Xiang M, Zhou L, Peng YW, Eddy RL, Shows TB, Nathans J (1993) Brn-3b: a POU domain gene expressed in a subset of retinal ganglion cells. Neuron 11(4):689–701
Ninkina NN, Stevens GE, Wood JN, Richardson WD (1993) A novel Brn3-like POU transcription factor expressed in subsets of rat sensory and spinal cord neurons. Nucleic Acids Res 21(14):3175–3182
Zhou H, Yoshioka T, Nathans J (1996) Retina-derived POU-domain factor-1: a complex POU-domain gene implicated in the development of retinal ganglion and amacrine cells. J Neurosci 16(7):2261–2274
Lenardo MJ, Staudt L, Robbins P, Kuang A, Mulligan RC, Baltimore D (1989) Repression of the IgH enhancer in teratocarcinoma cells associated with a novel octamer factor. Science 243(4890):544–546
Okamoto KOH, Okuda A, Sakai M, Muramatsu M, Hamada H (1990) A novel octamer binding transcription factor is differentially expressed in mouse embryonic cells. Cell 60(3):461–472. https://doi.org/10.1016/0092-8674(90)90597-8
Scholer HR, Ruppert S, Suzuki N, Chowdhury K, Gruss P (1990) New type of POU domain in germ line-specific protein Oct-4. Nature 344(6265):435–439. https://doi.org/10.1038/344435a0
Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A (1998) Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95(3):379–391
Niwa H, Miyazaki J, Smith AG (2000) Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 24(4):372–376. https://doi.org/10.1038/74199
Yuan H, Corbi N, Basilico C, Dailey L (1995) Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev 9(21):2635–2645
Takeda J, Seino S, Bell GI (1992) Human Oct3 gene family: cDNA sequences, alternative splicing, gene organization, chromosomal location, and expression at low levels in adult tissues. Nucleic Acids Res 20(17):4613–4620
Mizuno N, Kosaka M (2008) Novel variants of Oct-3/4 gene expressed in mouse somatic cells. J Biol Chem 283(45):30997–31004. https://doi.org/10.1074/jbc.M802992200
Lee J, Kim HK, Rho JY, Han YM, Kim J (2006) The human OCT-4 isoforms differ in their ability to confer self-renewal. J Biol Chem 281(44):33554–33565. https://doi.org/10.1074/jbc.M603937200
Atlasi Y, Mowla SJ, Ziaee SA, Gokhale PJ, Andrews PW (2008) OCT4 spliced variants are differentially expressed in human pluripotent and nonpluripotent cells. Stem Cells 26(12):3068–3074. https://doi.org/10.1634/stemcells.2008-0530
Andersen B, Pearse RV 2nd, Schlegel PN, Cichon Z, Schonemann MD, Bardin CW, Rosenfeld MG (1993) Sperm 1: a POU-domain gene transiently expressed immediately before meiosis I in the male germ cell. Proc Natl Acad Sci USA 90(23):11084–11088
Pearse RV 2nd, Drolet DW, Kalla KA, Hooshmand F, Bermingham JR Jr, Rosenfeld MG (1997) Reduced fertility in mice deficient for the POU protein sperm-1. Proc Natl Acad Sci USA 94(14):7555–7560
Frankenberg SR, Frank D, Harland R, Johnson AD, Nichols J, Niwa H, Scholer HR, Tanaka E, Wylie C, Brickman JM (2014) The POU-er of gene nomenclature. Development 141(15):2921–2923. https://doi.org/10.1242/dev.108407
Takeda H, Matsuzaki T, Oki T, Miyagawa T, Amanuma H (1994) A novel POU domain gene, zebrafish pou2: expression and roles of two alternatively spliced twin products in early development. Genes Dev 8(1):45–59
Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD (1989) Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci USA 86(14):5434–5438
Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B cells into macrophages. Cell 117(5):663–676
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861–872. https://doi.org/10.1016/j.cell.2007.11.019
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. https://doi.org/10.1016/j.cell.2006.07.024
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318(5858):1917–1920. https://doi.org/10.1126/science.1151526
Bar-Nur O, Verheul C, Sommer AG, Brumbaugh J, Schwarz BA, Lipchina I, Huebner AJ, Mostoslavsky G, Hochedlinger K (2015) Lineage conversion induced by pluripotency factors involves transient passage through an iPSC stage. Nat Biotechnol 33(7):761–768. https://doi.org/10.1038/nbt.3247
Han DW, Tapia N, Hermann A, Hemmer K, Hoing S, Arauzo-Bravo MJ, Zaehres H, Wu G, Frank S, Moritz S, Greber B, Yang JH, Lee HT, Schwamborn JC, Storch A, Scholer HR (2012) Direct reprogramming of fibroblasts into neural stem cells by defined factors. Cell Stem Cell 10(4):465–472. https://doi.org/10.1016/j.stem.2012.02.021
Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, Lipton SA, Zhang K, Ding S (2011) Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci USA 108(19):7838–7843. https://doi.org/10.1073/pnas.1103113108
Lujan E, Chanda S, Ahlenius H, Sudhof TC, Wernig M (2012) Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc Natl Acad Sci USA 109(7):2527–2532. https://doi.org/10.1073/pnas.1121003109
Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T, Seiferling D, Quandel T, Hoffmann P, Nothen MM, Brustle O, Edenhofer F (2012) Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell 10(4):473–479. https://doi.org/10.1016/j.stem.2012.03.003
Zhu S, Ambasudhan R, Sun W, Kim HJ, Talantova M, Wang X, Zhang M, Zhang Y, Laurent T, Parker J, Kim HS, Zaremba JD, Saleem S, Sanz-Blasco S, Masliah E, McKercher SR, Cho YS, Lipton SA, Kim J, Ding S (2014) Small molecules enable OCT4-mediated direct reprogramming into expandable human neural stem cells. Cell Res 24(1):126–129. https://doi.org/10.1038/cr.2013.156
Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA, Ding S (2011) Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9(2):113–118. https://doi.org/10.1016/j.stem.2011.07.002
Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M (2011) Induction of human neuronal cells by defined transcription factors. Nature 476(7359):220–223. https://doi.org/10.1038/nature10202
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463(7284):1035–1041. https://doi.org/10.1038/nature08797
Wang H, Cao N, Spencer CI, Nie B, Ma T, Xu T, Zhang Y, Wang X, Srivastava D, Ding S (2014) Small molecules enable cardiac reprogramming of mouse fibroblasts with a single factor, Oct4. Cell Rep 6(5):951–960. https://doi.org/10.1016/j.celrep.2014.01.038
Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, Drechsel D, Martynoga B, Castro DS, Webb AE, Sudhof TC, Brunet A, Guillemot F, Chang HY, Wernig M (2013) Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155(3):621–635. https://doi.org/10.1016/j.cell.2013.09.028
Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26(1):101–106. https://doi.org/10.1038/nbt1374
Jerabek S, Ng CK, Wu G, Arauzo-Bravo MJ, Kim KP, Esch D, Malik V, Chen Y, Velychko S, MacCarthy CM, Yang X, Cojocaru V, Scholer HR, Jauch R (2017) Changing POU dimerization preferences converts Oct6 into a pluripotency inducer. EMBO Rep 18(2):319–333. https://doi.org/10.15252/embr.201642958
Kim JB, Greber B, Arauzo-Bravo MJ, Meyer J, Park KI, Zaehres H, Scholer HR (2009) Direct reprogramming of human neural stem cells by OCT4. Nature 461(7264):649–653. https://doi.org/10.1038/nature08436
Kim JB, Sebastiano V, Wu G, Arauzo-Bravo MJ, Sasse P, Gentile L, Ko K, Ruau D, Ehrich M, van den Boom D, Meyer J, Hubner K, Bernemann C, Ortmeier C, Zenke M, Fleischmann BK, Zaehres H, Scholer HR (2009) Oct4-induced pluripotency in adult neural stem cells. Cell 136(3):411–419. https://doi.org/10.1016/j.cell.2009.01.023
Ebert AD, Yu J, Rose FF Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457(7227):277–280. https://doi.org/10.1038/nature07677
Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461(7262):402–406. https://doi.org/10.1038/nature08320
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321(5893):1218–1221. https://doi.org/10.1126/science.1158799
Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, Isacson O, Jaenisch R (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136(5):964–977. https://doi.org/10.1016/j.cell.2009.02.013
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ (2008) Disease-specific induced pluripotent stem cells. Cell 134(5):877–886. https://doi.org/10.1016/j.cell.2008.07.041
Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, Bao X, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA (2012) Generation of human induced pluripotent stem cells from urine samples. Nat Protoc 7(12):2080–2089. https://doi.org/10.1038/nprot.2012.115
Lujan E, Wernig M (2012) The many roads to Rome: induction of neural precursor cells from fibroblasts. Curr Opin Genet Dev 22(5):517–522. https://doi.org/10.1016/j.gde.2012.07.002
Lin C, Yu C, Ding S (2013) Toward directed reprogramming through exogenous factors. Curr Opin Genet Dev 23(5):519–525. https://doi.org/10.1016/j.gde.2013.06.002
Yu C, Liu K, Tang S, Ding S (2014) Chemical approaches to cell reprogramming. Curr Opin Genet Dev 28:50–56. https://doi.org/10.1016/j.gde.2014.09.006
Li K, Zhu S, Russ HA, Xu S, Xu T, Zhang Y, Ma T, Hebrok M, Ding S (2014) Small molecules facilitate the reprogramming of mouse fibroblasts into pancreatic lineages. Cell Stem Cell 14(2):228–236. https://doi.org/10.1016/j.stem.2014.01.006
Zhu S, Rezvani M, Harbell J, Mattis AN, Wolfe AR, Benet LZ, Willenbring H, Ding S (2014) Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature 508(7494):93–97. https://doi.org/10.1038/nature13020
Efe JA, Hilcove S, Kim J, Zhou H, Ouyang K, Wang G, Chen J, Ding S (2011) Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat Cell Biol 13(3):215–222. https://doi.org/10.1038/ncb2164
Li J, Huang NF, Zou J, Laurent TJ, Lee JC, Okogbaa J, Cooke JP, Ding S (2013) Conversion of human fibroblasts to functional endothelial cells by defined factors. Arterioscler Thromb Vasc Biol 33(6):1366–1375. https://doi.org/10.1161/ATVBAHA.112.301167
Szabo E, Rampalli S, Risueno RM, Schnerch A, Mitchell R, Fiebig-Comyn A, Levadoux-Martin M, Bhatia M (2010) Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468(7323):521–526. https://doi.org/10.1038/nature09591
Maza I, Caspi I, Zviran A, Chomsky E, Rais Y, Viukov S, Geula S, Buenrostro JD, Weinberger L, Krupalnik V, Hanna S, Zerbib M, Dutton JR, Greenleaf WJ, Massarwa R, Novershtern N, Hanna JH (2015) Transient acquisition of pluripotency during somatic cell transdifferentiation with iPSC reprogramming factors. Nat Biotechnol 33(7):769–774. https://doi.org/10.1038/nbt.3270
Marro S, Pang ZP, Yang N, Tsai MC, Qu K, Chang HY, Sudhof TC, Wernig M (2011) Direct lineage conversion of terminally differentiated hepatocytes to functional neurons. Cell Stem Cell 9(4):374–382. https://doi.org/10.1016/j.stem.2011.09.002
Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L, Aubry L, Vanti WB, Moreno H, Abeliovich A (2011) Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell 146(3):359–371. https://doi.org/10.1016/j.cell.2011.07.007
Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR (2011) MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476(7359):228–231. https://doi.org/10.1038/nature10323
Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A, Bjorklund A, Lindvall O, Jakobsson J, Parmar M (2011) Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci USA 108(25):10343–10348. https://doi.org/10.1073/pnas.1105135108
Verrijzer CP, Alkema MJ, van Weperen WW, Van Leeuwen HC, Strating MJ, van der Vliet PC (1992) The DNA binding specificity of the bipartite POU domain and its subdomains. EMBO J 11(13):4993–5003
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38(4):576–589. https://doi.org/10.1016/j.molcel.2010.05.004
Assa-Munt N, Mortishire-Smith RJ, Aurora R, Herr W, Wright PE (1993) The solution structure of the Oct-1 POU-specific domain reveals a striking similarity to the bacteriophage lambda repressor DNA-binding domain. Cell 73(1):193–205
Dekker N, Cox M, Boelens R, Verrijzer CP, van der Vliet PC, Kaptein R (1993) Solution structure of the POU-specific DNA-binding domain of Oct-1. Nature 362(6423):852–855. https://doi.org/10.1038/362852a0
Cox M, van Tilborg PJ, de Laat W, Boelens R, van Leeuwen HC, van der Vliet PC, Kaptein R (1995) Solution structure of the Oct-1 POU homeodomain determined by NMR and restrained molecular dynamics. J Biomol NMR 6(1):23–32
Morita EH, Shirakawa M, Hayashi F, Imagawa M, Kyogoku Y (1995) Structure of the Oct-3 POU-homeodomain in solution, as determined by triple resonance heteronuclear multidimensional NMR spectroscopy. Protein Sci 4(4):729–739. https://doi.org/10.1002/pro.5560040412
Klemm JD, Rould MA, Aurora R, Herr W, Pabo CO (1994) Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell 77(1):21–32
Ferraris L, Stewart AP, Kang J, DeSimone AM, Gemberling M, Tantin D, Fairbrother WG (2011) Combinatorial binding of transcription factors in the pluripotency control regions of the genome. Genome Res 21(7):1055–1064. https://doi.org/10.1101/gr.115824.110
Baburajendran N, Jauch R, Tan CY, Narasimhan K, Kolatkar PR (2011) Structural basis for the cooperative DNA recognition by Smad4 MH1 dimers. Nucleic Acids Res 39(18):8213–8222. https://doi.org/10.1093/nar/gkr500
Kim S, Brostromer E, Xing D, Jin J, Chong S, Ge H, Wang S, Gu C, Yang L, Gao YQ, Su XD, Sun Y, Xie XS (2013) Probing allostery through DNA. Science 339(6121):816–819. https://doi.org/10.1126/science.1229223
Narasimhan K, Pillay S, Huang YH, Jayabal S, Udayasuryan B, Veerapandian V, Kolatkar P, Cojocaru V, Pervushin K, Jauch R (2015) DNA-mediated cooperativity facilitates the co-selection of cryptic enhancer sequences by SOX2 and PAX6 transcription factors. Nucleic Acids Res 43(3):1513–1528. https://doi.org/10.1093/nar/gku1390
Merino F, Bouvier B, Cojocaru V (2015) Cooperative DNA recognition modulated by an interplay between protein–protein interactions and DNA-mediated allostery. PLoS Comput Biol 11(6):e1004287. https://doi.org/10.1371/journal.pcbi.1004287
Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, Kuznetsov H, Wang CF, Coburn D, Newburger DE, Morris Q, Hughes TR, Bulyk ML (2009) Diversity and complexity in DNA recognition by transcription factors. Science 324(5935):1720–1723. https://doi.org/10.1126/science.1162327
Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, Zheng H, Goity A, van Bakel H, Lozano JC, Galli M, Lewsey MG, Huang E, Mukherjee T, Chen X, Reece-Hoyes JS, Govindarajan S, Shaulsky G, Walhout AJM, Bouget FY, Ratsch G, Larrondo LF, Ecker JR, Hughes TR (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158(6):1431–1443. https://doi.org/10.1016/j.cell.2014.08.009
Takayama Y, Clore GM (2011) Intra- and intermolecular translocation of the bi-domain transcription factor Oct1 characterized by liquid crystal and paramagnetic NMR. Proc Natl Acad Sci USA 108(22):E169–176. https://doi.org/10.1073/pnas.1100050108
Kemler I, Schreiber E, Muller MM, Matthias P, Schaffner W (1989) Octamer transcription factors bind to two different sequence motifs of the immunoglobulin heavy chain promoter. EMBO J 8(7):2001–2008
LeBowitz JH, Clerc RG, Brenowitz M, Sharp PA (1989) The Oct-2 protein binds cooperatively to adjacent octamer sites. Genes Dev 3(10):1625–1638
Poellinger L, Roeder RG (1989) Octamer transcription factors 1 and 2 each bind to two different functional elements in the immunoglobulin heavy-chain promoter. Mol Cell Biol 9(2):747–756
Rhee JM, Gruber CA, Brodie TB, Trieu M, Turner EE (1998) Highly cooperative homodimerization is a conserved property of neural POU proteins. J Biol Chem 273(51):34196–34205
Jacobson EM, Li P, Leon-del-Rio A, Rosenfeld MG, Aggarwal AK (1997) Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev 11(2):198–212
Tomilin A, Remenyi A, Lins K, Bak H, Leidel S, Vriend G, Wilmanns M, Scholer HR (2000) Synergism with the coactivator OBF-1 (OCA-B, BOB-1) is mediated by a specific POU dimer configuration. Cell 103(6):853–864
Remenyi A, Tomilin A, Pohl E, Lins K, Philippsen A, Reinbold R, Scholer HR, Wilmanns M (2001) Differential dimer activities of the transcription factor Oct-1 by DNA-induced interface swapping. Mol Cell 8(3):569–580
Jauch R, Choo SH, Ng CK, Kolatkar PR (2011) Crystal structure of the dimeric Oct6 (POU3f1) POU domain bound to palindromic MORE DNA. Proteins 79(2):674–677. https://doi.org/10.1002/prot.22916
Scully KM, Jacobson EM, Jepsen K, Lunyak V, Viadiu H, Carriere C, Rose DW, Hooshmand F, Aggarwal AK, Rosenfeld MG (2000) Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science 290(5494):1127–1131
Botquin V, Hess H, Fuhrmann G, Anastassiadis C, Gross MK, Vriend G, Scholer HR (1998) New POU dimer configuration mediates antagonistic control of an osteopontin preimplantation enhancer by Oct-4 and Sox-2. Genes Dev 12(13):2073–2090
Skowronska-Krawczyk D, Ma Q, Schwartz M, Scully K, Li W, Liu Z, Taylor H, Tollkuhn J, Ohgi KA, Notani D, Kohwi Y, Kohwi-Shigematsu T, Rosenfeld MG (2014) Required enhancer-matrin-3 network interactions for a homeodomain transcription program. Nature 514(7521):257–261. https://doi.org/10.1038/nature13573
Mistri TK, Devasia AG, Chu LT, Ng WP, Halbritter F, Colby D, Martynoga B, Tomlinson SR, Chambers I, Robson P, Wohland T (2015) Selective influence of Sox2 on POU transcription factor binding in embryonic and neural stem cells. EMBO Rep 16(9):1177–1191. https://doi.org/10.15252/embr.201540467
Sharov AA, Ko MS (2009) Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res 16(5):261–273. https://doi.org/10.1093/dnares/dsp014
Lins K, Remenyi A, Tomilin A, Massa S, Wilmanns M, Matthias P, Scholer HR (2003) OBF1 enhances transcriptional potential of Oct1. EMBO J 22(9):2188–2198. https://doi.org/10.1093/emboj/cdg199
BabuRajendran N, Palasingam P, Narasimhan K, Sun W, Prabhakar S, Jauch R, Kolatkar PR (2010) Structure of Smad1 MH1/DNA complex reveals distinctive rearrangements of BMP and TGF-beta effectors. Nucleic Acids Res 38(10):3477–3488. https://doi.org/10.1093/nar/gkq046
Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, Palin K, Vaquerizas JM, Vincentelli R, Luscombe NM, Hughes TR, Lemaire P, Ukkonen E, Kivioja T, Taipale J (2013) DNA-binding specificities of human transcription factors. Cell 152(1–2):327–339. https://doi.org/10.1016/j.cell.2012.12.009
Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, Nitta KR, Taipale M, Popov A, Ginno PA, Domcke S, Yan J, Schubeler D, Vinson C, Taipale J (2017) Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356(6337):eaaj2239. https://doi.org/10.1126/science.aaj2239
Palasingam P, Jauch R, Ng CK, Kolatkar PR (2009) The structure of Sox17 bound to DNA reveals a conserved bending topology but selective protein interaction platforms. J Mol Biol 388(3):619–630. https://doi.org/10.1016/j.jmb.2009.03.055
Remenyi A, Lins K, Nissen LJ, Reinbold R, Scholer HR, Wilmanns M (2003) Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev 17(16):2048–2059. https://doi.org/10.1101/gad.269303
Jauch R, Ng CK, Narasimhan K, Kolatkar PR (2012) Crystal structure of the Sox4 HMG/DNA complex suggests a mechanism for the positional interdependence in DNA recognition. Biochem J 443(1):39–47. https://doi.org/10.1042/BJ20111768
Klaus M, Prokoph N, Girbig M, Wang X, Huang YH, Srivastava Y, Hou L, Narasimhan K, Kolatkar PR, Francois M, Jauch R (2016) Structure and decoy-mediated inhibition of the SOX18/Prox1–DNA interaction. Nucleic Acids Res 44(8):3922–3935. https://doi.org/10.1093/nar/gkw130
Werner MH, Huth JR, Gronenborn AM, Clore GM (1995) Molecular basis of human 46X, Y sex reversal revealed from the three-dimensional solution structure of the human SRY-DNA complex. Cell 81(5):705–714
Hou L, Srivastava Y, Jauch R (2016) Molecular basis for the genome engagement by Sox proteins. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2016.08.005
Ambrosetti DC, Basilico C, Dailey L (1997) Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein–protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol Cell Biol 17(11):6321–6329
Dailey L, Yuan H, Basilico C (1994) Interaction between a novel F9-specific factor and octamer-binding proteins is required for cell-type-restricted activity of the fibroblast growth factor 4 enhancer. Mol Cell Biol 14(12):7758–7769
Nishimoto M, Fukushima A, Okuda A, Muramatsu M (1999) The gene for the embryonic stem cell coactivator UTF1 carries a regulatory element which selectively interacts with a complex composed of Oct-3/4 and Sox-2. Mol Cell Biol 19(8):5453–5465
Nakatake Y, Fukui N, Iwamatsu Y, Masui S, Takahashi K, Yagi R, Yagi K, Miyazaki J, Matoba R, Ko MS, Niwa H (2006) Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol 26(20):7772–7782. https://doi.org/10.1128/MCB.00468-06
Tokuzawa Y, Kaiho E, Maruyama M, Takahashi K, Mitsui K, Maeda M, Niwa H, Yamanaka S (2003) Fbx15 is a novel target of Oct3/4 but is dispensable for embryonic stem cell self-renewal and mouse development. Mol Cell Biol 23(8):2699–2708
Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, Robson P (2005) Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem 280(26):24731–24737. https://doi.org/10.1074/jbc.M502573200
Kuroda T, Tada M, Kubota H, Kimura H, Hatano SY, Suemori H, Nakatsuji N, Tada T (2005) Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol 25(6):2475–2485. https://doi.org/10.1128/MCB.25.6.2475-2485.2005
Tomioka M, Nishimoto M, Miyagi S, Katayanagi T, Fukui N, Niwa H, Muramatsu M, Okuda A (2002) Identification of Sox-2 regulatory region which is under the control of Oct-3/4-Sox-2 complex. Nucleic Acids Res 30(14):3202–3213
Chew JL, Loh YH, Zhang W, Chen X, Tam WL, Yeap LS, Li P, Ang YS, Lim B, Robson P, Ng HH (2005) Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol 25(14):6031–6046. https://doi.org/10.1128/MCB.25.14.6031-6046.2005
Okumura-Nakanishi S, Saito M, Niwa H, Ishikawa F (2005) Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J Biol Chem 280(7):5307–5317. https://doi.org/10.1074/jbc.M410015200
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122(6):947–956. https://doi.org/10.1016/j.cell.2005.08.020
Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH (2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38(4):431–440. https://doi.org/10.1038/ng1760
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133(6):1106–1117. https://doi.org/10.1016/j.cell.2008.04.043
Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, Ng HH, Bourque G (2010) Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42(7):631–634. https://doi.org/10.1038/ng.600
Jauch R, Aksoy I, Hutchins AP, Ng CK, Tian XF, Chen J, Palasingam P, Robson P, Stanton LW, Kolatkar PR (2011) Conversion of Sox17 into a pluripotency reprogramming factor by reengineering its association with Oct4 on DNA. Stem Cells 29(6):940–951. https://doi.org/10.1002/stem.639
Tapia N, MacCarthy C, Esch D, Gabriele Marthaler A, Tiemann U, Arauzo-Bravo MJ, Jauch R, Cojocaru V, Scholer HR (2015) Dissecting the role of distinct OCT4-SOX2 heterodimer configurations in pluripotency. Sci Rep 5:13533. https://doi.org/10.1038/srep13533
Chen J, Chen X, Li M, Liu X, Gao Y, Kou X, Zhao Y, Zheng W, Zhang X, Huo Y, Chen C, Wu Y, Wang H, Jiang C, Gao S (2016) Hierarchical Oct4 binding in concert with primed epigenetic rearrangements during somatic cell reprogramming. Cell Rep 14(6):1540–1554. https://doi.org/10.1016/j.celrep.2016.01.013
Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K (2017) Cooperative binding of transcription factors orchestrates reprogramming. Cell 168(3):442–459 e420. https://doi.org/10.1016/j.cell.2016.12.016
Chen J, Zhang Z, Li L, Chen BC, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, Tjian R, Liu Z (2014) Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156(6):1274–1285. https://doi.org/10.1016/j.cell.2014.01.062
Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, Taipale J (2015) DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527(7578):384–388. https://doi.org/10.1038/nature15518
Dailey L, Basilico C (2001) Coevolution of HMG domains and homeodomains and the generation of transcriptional regulation by Sox/POU complexes. J Cell Physiol 186(3):315–328
Kamachi Y, Uchikawa M, Kondoh H (2000) Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 16(4):182–187
Wilson M, Koopman P (2002) Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12(4):441–446
Kuhlbrodt K, Herbarth B, Sock E, Enderich J, Hermans-Borgmeyer I, Wegner M (1998) Cooperative function of POU proteins and SOX proteins in glial cells. J Biol Chem 273(26):16050–16057
Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M (1998) Sox10, a novel transcriptional modulator in glial cells. J Neurosci 18(1):237–250
Tanaka S, Kamachi Y, Tanouchi A, Hamada H, Jing N, Kondoh H (2004) Interplay of SOX and POU factors in regulation of the Nestin gene in neural primordial cells. Mol Cell Biol 24(20):8834–8846. https://doi.org/10.1128/MCB.24.20.8834-8846.2004
Catena R, Tiveron C, Ronchi A, Porta S, Ferri A, Tatangelo L, Cavallaro M, Favaro R, Ottolenghi S, Reinbold R, Scholer H, Nicolis SK (2004) Conserved POU binding DNA sites in the Sox2 upstream enhancer regulate gene expression in embryonic and neural stem cells. J Biol Chem 279(40):41846–41857. https://doi.org/10.1074/jbc.M405514200
Lodato MA, Ng CW, Wamstad JA, Cheng AW, Thai KK, Fraenkel E, Jaenisch R, Boyer LA (2013) SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet 9(2):e1003288. https://doi.org/10.1371/journal.pgen.1003288
Chang YK, Srivastava Y, Hu C, Joyce A, Yang X, Zuo Z, Havranek JJ, Stormo GD, Jauch R (2017) Quantitative profiling of selective Sox/POU pairing on hundreds of sequences in parallel by Coop-seq. Nucleic Acids Res 45(2):832–845. https://doi.org/10.1093/nar/gkw1198
Nishimoto M, Miyagi S, Yamagishi T, Sakaguchi T, Niwa H, Muramatsu M, Okuda A (2005) Oct-3/4 maintains the proliferative embryonic stem cell state via specific binding to a variant octamer sequence in the regulatory region of the UTF1 locus. Mol Cell Biol 25(12):5084–5094. https://doi.org/10.1128/MCB.25.12.5084-5094.2005
Ng CK, Li NX, Chee S, Prabhakar S, Kolatkar PR, Jauch R (2012) Deciphering the Sox–Oct partner code by quantitative cooperativity measurements. Nucleic Acids Res 40(11):4933–4941. https://doi.org/10.1093/nar/gks153
Aksoy I, Jauch R, Chen J, Dyla M, Divakar U, Bogu GK, Teo R, Leng Ng CK, Herath W, Lili S, Hutchins AP, Robson P, Kolatkar PR, Stanton LW (2013) Oct4 switches partnering from Sox2 to Sox17 to reinterpret the enhancer code and specify endoderm. EMBO J 32(7):938–953. https://doi.org/10.1038/emboj.2013.31
Merino F, Ng CK, Veerapandian V, Scholer HR, Jauch R, Cojocaru V (2014) Structural basis for the SOX-dependent genomic redistribution of OCT4 in stem cell differentiation. Structure 22(9):1274–1286. https://doi.org/10.1016/j.str.2014.06.014
Aksoy I, Jauch R, Eras V, Chng WB, Chen J, Divakar U, Ng CK, Kolatkar PR, Stanton LW (2013) Sox transcription factors require selective interactions with Oct4 and specific transactivation functions to mediate reprogramming. Stem Cells 31(12):2632–2646. https://doi.org/10.1002/stem.1522
Niwa H, Nakamura A, Urata M, Shirae-Kurabayashi M, Kuraku S, Russell S, Ohtsuka S (2016) The evolutionally-conserved function of group B1 Sox family members confers the unique role of Sox2 in mouse ES cells. BMC Evol Biol 16:173. https://doi.org/10.1186/s12862-016-0755-4
Irie N, Weinberger L, Tang WW, Kobayashi T, Viukov S, Manor YS, Dietmann S, Hanna JH, Surani MA (2015) SOX17 is a critical specifier of human primordial germ cell fate. Cell 160(1–2):253–268. https://doi.org/10.1016/j.cell.2014.12.013
Knaupp AS, Buckberry S, Pflueger J, Lim SM, Ford E, Larcombe MR, Rossello FJ, de Mendoza A, Alaei S, Firas J, Holmes ML, Nair SS, Clark SJ, Nefzger CM, Lister R, Polo JM (2017) Transient and permanent reconfiguration of chromatin and transcription factor occupancy drive reprogramming. Cell Stem Cell 21(6):834–845 e836. https://doi.org/10.1016/j.stem.2017.11.007
Williams DC Jr, Cai M, Clore GM (2004) Molecular basis for synergistic transcriptional activation by Oct1 and Sox2 revealed from the solution structure of the 42-kDa Oct1.Sox2.Hoxb1-DNA ternary transcription factor complex. J Biol Chem 279(2):1449–1457. https://doi.org/10.1074/jbc.M309790200
Pereira JH, Kim SH (2009) Structure of human Brn-5 transcription factor in complex with CRH gene promoter. J Struct Biol 167(2):159–165. https://doi.org/10.1016/j.jsb.2009.05.003
Esch D, Vahokoski J, Groves MR, Pogenberg V, Cojocaru V, Vom Bruch H, Han D, Drexler HC, Arauzo-Bravo MJ, Ng CK, Jauch R, Wilmanns M, Scholer HR (2013) A unique Oct4 interface is crucial for reprogramming to pluripotency. Nat Cell Biol 15(3):295–301. https://doi.org/10.1038/ncb2680
Jin W, Wang L, Zhu F, Tan W, Lin W, Chen D, Sun Q, Xia Z (2016) Critical POU domain residues confer Oct4 uniqueness in somatic cell reprogramming. Sci Rep 6:20818. https://doi.org/10.1038/srep20818
Kong X, Liu J, Li L, Yue L, Zhang L, Jiang H, Xie X, Luo C (2015) Functional interplay between the RK motif and linker segment dictates Oct4-DNA recognition. Nucleic Acids Res 43(9):4381–4392. https://doi.org/10.1093/nar/gkv323
Zaret KS, Mango SE (2016) Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev 37:76–81. https://doi.org/10.1016/j.gde.2015.12.003
Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS (1996) Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev 10(13):1670–1682
Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS (2013) Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev 27(3):251–260. https://doi.org/10.1101/gad.206458.112
Clark KL, Halay ED, Lai E, Burley SK (1993) Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 364(6436):412–420. https://doi.org/10.1038/364412a0
Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM (1993) Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 362(6417):219–223. https://doi.org/10.1038/362219a0
Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, Clark KL, Burley SK, Zaret KS (1998) Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J 17(1):244–254. https://doi.org/10.1093/emboj/17.1.244
Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, Menoni H, Papin C, Skoufias DA, Kurumizaka H, Lavery R, Hamiche A, Hayes JJ, Schultz P, Angelov D, Petosa C, Dimitrov S (2017) Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol Cell 66(3):384–397 e388. https://doi.org/10.1016/j.molcel.2017.04.012
Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G (2014) Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344(6182):376–380. https://doi.org/10.1126/science.1251413
Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS (2002) Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9(2):279–289
Soufi A, Donahue G, Zaret KS (2012) Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151(5):994–1004. https://doi.org/10.1016/j.cell.2012.09.045
Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161(3):555–568. https://doi.org/10.1016/j.cell.2015.03.017
Zviran A, Mor N, Rais Y, Gingold H, Peles S, Chomsky E, Viukov S, Buenrostro JD, Weinberger L, Manor YS, Krupalnik V, Zerbib M, Hezroni H, Jaitin DA, Larastiaso D, Gilad S, Benjamin S, Mousa A, Ayyash M, Sheban D, Bayerl J, Castrejon AA, Massarwa R, Maza I, Hanna S, Amit I, Stelzer Y, Ulitsky I, Greenleaf WJ, Pilpel Y, Novershtern N, Hanna JH (2017) High-resolution dissection of conducive reprogramming trajectory to ground state pluripotency. bioRxiv. https://doi.org/10.1101/184135
Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, Liu H, Wang X, Wu F, Kuang J, Hutchins AP, Chen J, Pei D (2017) Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell 21(6):819–833 e816. https://doi.org/10.1016/j.stem.2017.10.012
Swinstead EE, Paakinaho V, Presman DM, Hager GL (2016) Pioneer factors and ATP-dependent chromatin remodeling factors interact dynamically: a new perspective: Multiple transcription factors can effect chromatin pioneer functions through dynamic interactions with ATP-dependent chromatin remodeling factors. BioEssays 38(11):1150–1157. https://doi.org/10.1002/bies.201600137
Zaret KS, Lerner J, Iwafuchi-Doi M (2016) Chromatin scanning by dynamic binding of pioneer factors. Mol Cell 62(5):665–667. https://doi.org/10.1016/j.molcel.2016.05.024
Panne D, Maniatis T, Harrison SC (2007) An atomic model of the interferon-beta enhanceosome. Cell 129(6):1111–1123. https://doi.org/10.1016/j.cell.2007.05.019
Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467(7314):430–435. https://doi.org/10.1038/nature09380
Nozawa K, Schneider TR, Cramer P (2017) Core mediator structure at 3.4 A extends model of transcription initiation complex. Nature 545(7653):248–251. https://doi.org/10.1038/nature22328
Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, Fisher AG (2006) Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8(5):532–538. https://doi.org/10.1038/ncb1403
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125(2):315–326. https://doi.org/10.1016/j.cell.2006.02.041
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448(7153):553–560. https://doi.org/10.1038/nature06008
Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A (2011) Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell 8(1):96–105. https://doi.org/10.1016/j.stem.2010.12.001
Ang YS, Tsai SY, Lee DF, Monk J, Su J, Ratnakumar K, Ding J, Ge Y, Darr H, Chang B, Wang J, Rendl M, Bernstein E, Schaniel C, Lemischka IR (2011) Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145(2):183–197. https://doi.org/10.1016/j.cell.2011.03.003
Mansour AA, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, Krupalnik V, Zerbib M, Amann-Zalcenstein D, Maza I, Geula S, Viukov S, Holtzman L, Pribluda A, Canaani E, Horn-Saban S, Amit I, Novershtern N, Hanna JH (2012) The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488(7411):409–413. https://doi.org/10.1038/nature11272
Brumbaugh J, Hou Z, Russell JD, Howden SE, Yu P, Ledvina AR, Coon JJ, Thomson JA (2012) Phosphorylation regulates human OCT4. Proc Natl Acad Sci USA 109(19):7162–7168. https://doi.org/10.1073/pnas.1203874109
Kang J, Gemberling M, Nakamura M, Whitby FG, Handa H, Fairbrother WG, Tantin D (2009) A general mechanism for transcription regulation by Oct1 and Oct4 in response to genotoxic and oxidative stress. Genes Dev 23(2):208–222. https://doi.org/10.1101/gad.1750709
Lin Y, Yang Y, Li W, Chen Q, Li J, Pan X, Zhou L, Liu C, Chen C, He J, Cao H, Yao H, Zheng L, Xu X, Xia Z, Ren J, Xiao L, Li L, Shen B, Zhou H, Wang YJ (2012) Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol Cell 48(4):627–640. https://doi.org/10.1016/j.molcel.2012.08.030
Nieto L, Joseph G, Stella A, Henri P, Burlet-Schiltz O, Monsarrat B, Clottes E, Erard M (2007) Differential effects of phosphorylation on DNA binding properties of N Oct-3 are dictated by protein/DNA complex structures. J Mol Biol 370(4):687–700. https://doi.org/10.1016/j.jmb.2007.04.072
Schild-Poulter C, Shih A, Tantin D, Yarymowich NC, Soubeyrand S, Sharp PA, Hache RJ (2007) DNA-PK phosphorylation sites on Oct-1 promote cell survival following DNA damage. Oncogene 26(27):3980–3988. https://doi.org/10.1038/sj.onc.1210165
Segil N, Roberts SB, Heintz N (1991) Mitotic phosphorylation of the Oct-1 homeodomain and regulation of Oct-1 DNA binding activity. Science 254(5039):1814–1816
Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, Kwon YW, Cho EJ, Youn HD (2012) O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 11(1):62–74. https://doi.org/10.1016/j.stem.2012.03.001
Kang J, Shen Z, Lim JM, Handa H, Wells L, Tantin D (2013) Regulation of Oct1/Pou2f1 transcription activity by O-GlcNAcylation. FASEB J 27(7):2807–2817. https://doi.org/10.1096/fj.12-220897
Webster DM, Teo CF, Sun Y, Wloga D, Gay S, Klonowski KD, Wells L, Dougan ST (2009) O-GlcNAc modifications regulate cell survival and epiboly during zebrafish development. BMC Dev Biol 9:28. https://doi.org/10.1186/1471-213X-9-28
Wei F, Scholer HR, Atchison ML (2007) Sumoylation of Oct4 enhances its stability, DNA binding, and transactivation. J Biol Chem 282(29):21551–21560. https://doi.org/10.1074/jbc.M611041200
Zhang Z, Liao B, Xu M, Jin Y (2007) Post-translational modification of POU domain transcription factor Oct-4 by SUMO-1. FASEB J 21(12):3042–3051. https://doi.org/10.1096/fj.06-6914com
Kang J, Goodman B, Zheng Y, Tantin D (2011) Dynamic regulation of Oct1 during mitosis by phosphorylation and ubiquitination. PLoS One 6(8):e23872. https://doi.org/10.1371/journal.pone.0023872
Xu HM, Liao B, Zhang QJ, Wang BB, Li H, Zhong XM, Sheng HZ, Zhao YX, Zhao YM, Jin Y (2004) Wwp2, an E3 ubiquitin ligase that targets transcription factor Oct-4 for ubiquitination. J Biol Chem 279(22):23495–23503. https://doi.org/10.1074/jbc.M400516200
Saxe JP, Tomilin A, Scholer HR, Plath K, Huang J (2009) Post-translational regulation of Oct4 transcriptional activity. PLoS One 4(2):e4467. https://doi.org/10.1371/journal.pone.0004467
Lai JS, Cleary MA, Herr W (1992) A single amino acid exchange transfers VP16-induced positive control from the Oct-1 to the Oct-2 homeo domain. Genes Dev 6(11):2058–2065
Pomerantz JL, Kristie TM, Sharp PA (1992) Recognition of the surface of a homeo domain protein. Genes Dev 6(11):2047–2057
Dawson SJ, Palmer RD, Morris PJ, Latchman DS (1998) Functional role of position 22 in the homeodomain of Brn-3 transcription factors. NeuroReport 9(10):2305–2309
Fowler DM, Fields S (2014) Deep mutational scanning: a new style of protein science. Nat Methods 11(8):801–807. https://doi.org/10.1038/nmeth.3027
Ding J, Xu H, Faiola F, Ma’ayan A, Wang J (2012) Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res 22(1):155–167. https://doi.org/10.1038/cr.2011.179
Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, Choudhary J (2010) An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell 6(4):382–395. https://doi.org/10.1016/j.stem.2010.03.004
van den Berg DL, Snoek T, Mullin NP, Yates A, Bezstarosti K, Demmers J, Chambers I, Poot RA (2010) An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6(4):369–381. https://doi.org/10.1016/j.stem.2010.02.014
Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A (2013) Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339(6123):1074–1077. https://doi.org/10.1126/science.1232542
Farnung L, Vos SM, Wigge C, Cramer P (2017) Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 550(7677):539–542. https://doi.org/10.1038/nature24046
Yang X, Malik V, Jauch R (2015) Reprogramming cells with synthetic proteins. Asian J Androl 17(3):394–402. https://doi.org/10.4103/1008-682X.145433
Tsubooka N, Ichisaka T, Okita K, Takahashi K, Nakagawa M, Yamanaka S (2009) Roles of Sall4 in the generation of pluripotent stem cells from blastocysts and fibroblasts. Genes Cells 14(6):683–694. https://doi.org/10.1111/j.1365-2443.2009.01301.x
Maekawa M, Yamaguchi K, Nakamura T, Shibukawa R, Kodanaka I, Ichisaka T, Kawamura Y, Mochizuki H, Goshima N, Yamanaka S (2011) Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 474(7350):225–229. https://doi.org/10.1038/nature10106
Wernig M, Meissner A, Cassady JP, Jaenisch R (2008) c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2(1):10–12. https://doi.org/10.1016/j.stem.2007.12.001
Feng B, Jiang J, Kraus P, Ng JH, Heng JC, Chan YS, Yaw LP, Zhang W, Loh YH, Han J, Vega VB, Cacheux-Rataboul V, Lim B, Lufkin T, Ng HH (2009) Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nat Cell Biol 11(2):197–203. https://doi.org/10.1038/ncb1827
Maherali N, Hochedlinger K (2009) Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr Biol 19(20):1718–1723. https://doi.org/10.1016/j.cub.2009.08.025
Lyssiotis CA, Foreman RK, Staerk J, Garcia M, Mathur D, Markoulaki S, Hanna J, Lairson LL, Charette BD, Bouchez LC, Bollong M, Kunick C, Brinker A, Cho CY, Schultz PG, Jaenisch R (2009) Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA 106(22):8912–8917. https://doi.org/10.1073/pnas.0903860106
Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisua Belmonte JC (2009) Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460(7259):1140–1144. https://doi.org/10.1038/nature08311
Chen J, Liu J, Yang J, Chen Y, Chen J, Ni S, Song H, Zeng L, Ding K, Pei D (2011) BMPs functionally replace Klf4 and support efficient reprogramming of mouse fibroblasts by Oct4 alone. Cell Res 21(1):205–212. https://doi.org/10.1038/cr.2010.172
Moon JH, Heo JS, Kim JS, Jun EK, Lee JH, Kim A, Kim J, Whang KY, Kang YK, Yeo S, Lim HJ, Han DW, Kim DW, Oh S, Yoon BS, Scholer HR, You S (2011) Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res 21(9):1305–1315. https://doi.org/10.1038/cr.2011.107
Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, Ding S (2008) Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 3(5):568–574. https://doi.org/10.1016/j.stem.2008.10.004
Li Y, Zhang Q, Yin X, Yang W, Du Y, Hou P, Ge J, Liu C, Zhang W, Zhang X, Wu Y, Li H, Liu K, Wu C, Song Z, Zhao Y, Shi Y, Deng H (2011) Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res 21(1):196–204. https://doi.org/10.1038/cr.2010.142
Tsai SY, Bouwman BA, Ang YS, Kim SJ, Lee DF, Lemischka IR, Rendl M (2011) Single transcription factor reprogramming of hair follicle dermal papilla cells to induced pluripotent stem cells. Stem Cells 29(6):964–971. https://doi.org/10.1002/stem.649
Tsai SY, Clavel C, Kim S, Ang YS, Grisanti L, Lee DF, Kelley K, Rendl M (2010) Oct4 and klf4 reprogram dermal papilla cells into induced pluripotent stem cells. Stem Cells 28(2):221–228. https://doi.org/10.1002/stem.281
Eminli S, Foudi A, Stadtfeld M, Maherali N, Ahfeldt T, Mostoslavsky G, Hock H, Hochedlinger K (2009) Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat Genet 41(9):968–976. https://doi.org/10.1038/ng.428
Sugii S, Kida Y, Kawamura T, Suzuki J, Vassena R, Yin YQ, Lutz MK, Berggren WT, Izpisua Belmonte JC, Evans RM (2010) Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc Natl Acad Sci USA 107(8):3558–3563. https://doi.org/10.1073/pnas.0910172106
Wu T, Wang H, He J, Kang L, Jiang Y, Liu J, Zhang Y, Kou Z, Liu L, Zhang X, Gao S (2011) Reprogramming of trophoblast stem cells into pluripotent stem cells by Oct4. Stem Cells 29(5):755–763. https://doi.org/10.1002/stem.617
Shi Y, Do JT, Desponts C, Hahm HS, Scholer HR, Ding S (2008) A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell 2(6):525–528. https://doi.org/10.1016/j.stem.2008.05.011
Silva J, Barrandon O, Nichols J, Kawaguchi J, Theunissen TW, Smith A (2008) Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol 6(10):e253. https://doi.org/10.1371/journal.pbio.0060253
Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, Arauzo-Bravo MJ, Ruau D, Han DW, Zenke M, Scholer HR (2008) Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 454(7204):646–650. https://doi.org/10.1038/nature07061
Utikal J, Maherali N, Kulalert W, Hochedlinger K (2009) Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci 122(Pt 19):3502–3510. https://doi.org/10.1242/jcs.054783
Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S (2008) Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 321(5889):699–702. https://doi.org/10.1126/science.1154884
Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M, Creyghton MP, Steine EJ, Cassady JP, Foreman R, Lengner CJ, Dausman JA, Jaenisch R (2008) Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133(2):250–264. https://doi.org/10.1016/j.cell.2008.03.028
Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K (2008) Induced pluripotent stem cells generated without viral integration. Science 322(5903):945–949. https://doi.org/10.1126/science.1162494
Tan KY, Eminli S, Hettmer S, Hochedlinger K, Wagers AJ (2011) Efficient generation of iPS cells from skeletal muscle stem cells. PLoS One 6(10):e26406. https://doi.org/10.1371/journal.pone.0026406
Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, Clark AT, Plath K (2008) Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA 105(8):2883–2888. https://doi.org/10.1073/pnas.0711983105
Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451(7175):141–146. https://doi.org/10.1038/nature06534
Zhao Y, Yin X, Qin H, Zhu F, Liu H, Yang W, Zhang Q, Xiang C, Hou P, Song Z, Liu Y, Yong J, Zhang P, Cai J, Liu M, Li H, Li Y, Qu X, Cui K, Zhang W, Xiang T, Wu Y, Zhao Y, Liu C, Yu C, Yuan K, Lou J, Ding M, Deng H (2008) Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 3(5):475–479. https://doi.org/10.1016/j.stem.2008.10.002
Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA (2008) Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 26(11):1269–1275. https://doi.org/10.1038/nbt.1502
Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, Edel M, Boue S, Izpisua Belmonte JC (2008) Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26(11):1276–1284. https://doi.org/10.1038/nbt.1503
Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K (2008) A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3(3):340–345. https://doi.org/10.1016/j.stem.2008.08.003
Yan X, Qin H, Qu C, Tuan RS, Shi S, Huang GT (2010) iPS cells reprogrammed from human mesenchymal-like stem/progenitor cells of dental tissue origin. Stem Cells Dev 19(4):469–480. https://doi.org/10.1089/scd.2009.0314
Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, Blasco MA, Serrano M (2009) The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460(7259):1136–1139. https://doi.org/10.1038/nature08290
Zhao HX, Li Y, Jin HF, Xie L, Liu C, Jiang F, Luo YN, Yin GW, Li Y, Wang J, Li LS, Yao YQ, Wang XH (2010) Rapid and efficient reprogramming of human amnion-derived cells into pluripotency by three factors OCT4/SOX2/NANOG. Differentiation 80(2–3):123–129. https://doi.org/10.1016/j.diff.2010.03.002
Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C, Zweigerdt R, Gruh I, Meyer J, Wagner S, Maier LS, Han DW, Glage S, Miller K, Fischer P, Scholer HR, Martin U (2009) Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 5(4):434–441. https://doi.org/10.1016/j.stem.2009.08.021
Giorgetti A, Montserrat N, Aasen T, Gonzalez F, Rodriguez-Piza I, Vassena R, Raya A, Boue S, Barrero MJ, Corbella BA, Torrabadella M, Veiga A, Izpisua Belmonte JC (2009) Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell 5(4):353–357. https://doi.org/10.1016/j.stem.2009.09.008
Liu H, Ye Z, Kim Y, Sharkis S, Jang YY (2010) Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology 51(5):1810–1819. https://doi.org/10.1002/hep.23626
Aoki T, Ohnishi H, Oda Y, Tadokoro M, Sasao M, Kato H, Hattori K, Ohgushi H (2010) Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC. Tissue Eng Part A 16(7):2197–2206. https://doi.org/10.1089/ten.TEA.2009.0747
Bar-Nur O, Russ HA, Efrat S, Benvenisty N (2011) Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 9(1):17–23. https://doi.org/10.1016/j.stem.2011.06.007
Acknowledgements
We thank Andrew Hutchins, Sergiy Velychko for discussions and Yogesh Srivastava for help with structural models. R.J. is supported by the Ministry of Science and Technology of China (2013DFE33080, 2016YFA0100700, 2017YFA0105103) by the National Natural Science Foundation of China (Grant No. 31471238), a 100 talent award of the Chinese Academy of Sciences and by a Science and Technology Planning Projects of Guangdong Province, China (2017B030314056 and 2016A050503038). V.M. thanks the CAS-TWAS (Chinese Academy of Sciences–The World Academy of Sciences) President’s Fellowship and UCAS (University of Chinese Academy of Science) for financial and infrastructure support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Malik, V., Zimmer, D. & Jauch, R. Diversity among POU transcription factors in chromatin recognition and cell fate reprogramming. Cell. Mol. Life Sci. 75, 1587–1612 (2018). https://doi.org/10.1007/s00018-018-2748-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2748-5