
Abstract
In the central nervous system, most excitatory post-synapses are small subcellular structures called dendritic spines. Their structure and morphological remodeling are tightly coupled to changes in synaptic transmission. The F-actin cytoskeleton is the main driving force of dendritic spine remodeling and sustains synaptic plasticity. It is therefore essential to understand how changes in synaptic transmission can regulate the organization and dynamics of actin binding proteins (ABPs). In this review, we will provide a detailed description of the organization and dynamics of F-actin and ABPs in dendritic spines and will discuss the current models explaining how the actin cytoskeleton sustains both structural and functional synaptic plasticity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 1888, Ramón y Cajal described for the first time that the surface of a neuron “appears bristling with thorns or short spines” [1, 2]. In subsequent work, he speculated that dendritic spines received axonal contacts and that their morphological changes were associated with neuronal function and learning processes [3–5]. Cajal’s hypothesis that spines connect axons and dendrites was confirmed in 1959 by the first visualization of pre- and post-synaptic contacts by electron microscopy (EM) [6–8]. Although it was widely believed that long-lasting changes in synaptic function are the cellular basis of learning and memory, [9–11] it was only recently that direct links between synaptic activity, dendritic spine morphology and the formation of a memory trace in vivo were demonstrated [12, 13].
Dendritic spines are generally composed of a spine head (200 nm to 1 µm in diameter) that is connected to dendrites by a thin spine neck (100–200 nm thick) [14–17]. Spine heads contain the post-synaptic density (PSD), a macro-molecular structure essential for synaptic transmission which mediates adhesion to the pre-synapse and anchoring of post-synaptic glutamate receptors. Despite their architectural role, dendritic spines are highly dynamic structures that undergo distinct morphological changes on timescales of seconds to minutes, hours and years, both in vitro and in vivo [18–22]. The morphological remodeling of spines is correlated with changes in synaptic transmission. Synaptic plasticity-induced stimuli such as long-term potentiation (LTP) or long-term depression (LTD) cause not only the formation or removal of new spines [23–27] but also the enlargement or shrinkage of pre-existing spines [27–31]. However, the in vivo longitudinal measurement of a single spine experiencing morphological remodeling during LTP and LTD has not yet been observed [32–34]. In this review, this type of spine morphological remodeling will be referred to as structural plasticity, while functional plasticity is defined as increased or decreased synaptic transmission.
While the function of dendritic spines had been the major scope of pioneer studies, current studies are thoroughly focusing on their molecular composition. Biochemical and proteomic studies have shown that dendritic spines are composed of a plethora of proteins covering a wide range of functions including membrane receptors and channels, scaffolding proteins, adhesion proteins, molecular motors, GTPases, kinases/phosphatases and cytoskeleton proteins [35]. Actin and actin binding proteins (ABPs) are particularly enriched in PSD and accumulate in dendritic spine heads [35–39], with F-actin dynamics being the driving force of spine morphological remodeling [19, 40, 41]. Indeed, the actin cytoskeleton sustains the formation of dendritic spines during neuron development and their enlargement and shrinkage upon increase and decrease synaptic activity, respectively [28, 29, 42–44].
Filamentous actin (F-actin) consists of two-stranded helical polymers arising from the polymerization of globular actin (G-actin) (Fig. 1a) [45, 46]. Actin assembles in a head-to-tail manner giving rise to the structural polarity of F-actin. Therefore, polymerization and depolymerization occur preferentially at the barbed end and the pointed end, respectively. This creates a net flow of actin monomers within the filament called actin treadmilling. Fluorescence microscopy revealed that the nature of ABPs but also the spatiotemporal coordination of their activity determine the organization and dynamics of F-actin in motile structures such as the lamellipodium and filopodium (Fig. 1a) [45, 47]. In the past decade, seminal studies have established the central role of F-actin dynamics and ABPs in spine morphogenesis and in synaptic plasticity-induced morphological remodeling [43, 44, 48, 49]. The spatial resolution of conventional fluorescence microscopy is, however, limited by the diffraction of light (around 250 nm in the lateral direction), whereas many sub-synaptic structures are typically below this diffraction limit (synaptic cleft: 20 nm; PSD thickness: 25–50 nm; spine neck width: 100–200 nm). To date, EM provided most of our knowledge about synapse ultrastructure, including the synaptic cleft size, the PSD and F-actin organization in dendritic spines [15, 39, 50, 51]. In 2014, Eric Betzig, William Moerner and Stefan Hell were awarded the Nobel Prize in Chemistry for the recent development of single-molecule and super-resolution light microscopy. These techniques can circumvent the diffraction limit of light and have provided new insights into protein organization and dynamics in different cellular systems including neurons [52, 53]. We are therefore just starting to reach a deep understanding of the sequence of molecular events leading to dendritic spines functional and structural plasticity, such as neurotransmitter receptor dynamics and organization, PSD remodeling, and spine neck morphology [54–56]. In this review, we will focus on the organization and dynamics of the F-actin cytoskeleton in dendritic spines, the signaling cascades driving F-actin remodeling upon changes in synaptic activity, and the interplay between F-actin remodeling and spine structural and functional plasticity. We will also highlight how super-resolution microscopy and single-molecule tracking approaches provided new understanding on the architecture and the dynamics of actin and actin regulatory proteins in dendritic spines.

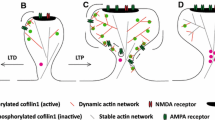
Comparison of F-actin and ABPs organization in the lamellipodium and in dendritic spines. a In lamellipodia of motile cells, nucleation and elongation of F-actin are co-localized at the tip of membrane protrusion triggering a fast and concerted rearward flow of the branched F-actin network. Activation of the Arp2/3 complex is mediated by the synchronized convergence of prenylated Rac-GTP, PIP3, IRSp53 and the WAVE complex, while elongation of F-actin filaments are powered by VASP and FMNL2, all these proteins being localized at the lamellipodium tip. Capping proteins are also enriched at the lamellipodium tip, while cortactin associates with the entire F-actin network. ADF/cofilin associates with the entire F-actin network and induces F-actin severing. b Dendritic spine heads are characterized by slow and non-polarized motions of the branched F-actin pool. This distinct F-actin dynamics in spines could be sustained by spatial segregation between stationary nucleation zones and delocalizing elongation zones. Indeed, since the PSD is a persistent confinement zone for the WAVE complex and IRSp53, branched F-actin nucleation occurs with a higher probability at the PSD vicinity, while elongation of F-actin filaments by VASP and FMNL2 occur at the tip of finger-like protrusions. Dendritic spines are also composed of a stable F-actin pool localizing at the base of the spine head and stabilized by the crosslinking activity of myosin II. Dendritic spine necks are composed of both branched and non-branched long F-actin filaments that exhibit slow and non-polarized motions
Actin dynamics in dendritic spines
Actin dynamics in dendritic filopodia
During neuronal development, highly motile structures called dendritic filopodia will emerge from dendrites [18]. Enlargement of dendritic filopodia occurs soon after their contact with axons leading to the formation of a dendritic spine [39, 57–60]. As opposed to the tight linear F-actin bundles present in filopodia from motile cells, dendritic filopodia are composed of unbundled F-actin of mixed orientations [39, 61]. However, dendritic filopodia display a fast retrograde actin flow powered by continuous polymerization of actin against the tip membrane protrusion, as observed for the lamellipodium and filopodium of motile cells and neuronal growth cones [62–67]. The molecular motor myosin II, accumulates at the base of dendritic filopodia and contributes to the retrograde F-actin flow [39, 66]. The initial contact between dendritic filopodia and the axon is driven by N-cadherin adhesion proteins and drastically reduces the retrograde F-actin flow [60]. This interplay between cell adhesion and actin dynamics is characteristic of a model used to explain cell migration called the ‘molecular clutch’ [68–72]. The molecular clutch consists of an assembly of ABPs that transmit forces generated by the F-actin flow to adhesive structures allowing cells to exert grip on the surrounding substrate or cells and to generate membrane protrusions [70]. According to this model, dendritic filopodia that are not contacting axons are in the slipping mode and display fast rearward F-actin motion driven by F-actin polymerization and myosin II activity. Upon filopodia/axon contact, clutch engagement between N-cadherin and F-actin, possibly mediated by α-catenin, will immobilize actin filaments counteracting myosin II activity and leading to a reduction of the F-actin flow, which causes F-actin growth to push against the plasma membrane. The resulting F-actin accumulation, decreased motility and dendritic filopodia enlargement might therefore be required for the initiation of dendritic spines [60].
Actin dynamics in mature dendritic spines
Motility of dendritic spines is characterized by the formation and retraction of membrane protrusions [22, 73, 74]. However, the precise role of those spine protrusions is still unknown but might be needed to continuously adjust the position of the post-synaptic structure to the pre-synaptic one. Dendritic spines are F-actin-rich protrusions with a dynamic equilibrium between G-actin and F-actin [75]. As for dendritic filopodia, F-actin dynamics was demonstrated to be the driving force of dendritic spine motility. Indeed, pharmacological treatments preventing F-actin polymerization (cytochalasin-D) or promoting F-actin depolymerization (latrunculin-A) stop spine dynamics [19, 41]. Fluorescence recovery after photobleaching (FRAP) and photoactivation experiments provided more insights into actin dynamics in dendritic spines [76, 77]. These studies revealed that F-actin was composed of a large dynamic pool (80–95 %; 40 s turnover time constant) and a small stable pool (5–20 %; 17 min turnover time constant) (Fig. 1b); the size of the stable pool being positively correlated with spine volume. While the stable pool remains stationary at the base of the spine heads, the dynamic pool exhibits a slow retrograde flow (5–20 nm/s) from the tip to the base of the spine head [77, 78]. The recent development of single-particle tracking photoactivation localization microscopy (sptPALM) [79, 80] has enabled F-actin tracking in dendritic spines [67, 78, 81]. Those studies did not reveal a concerted flow of F-actin from the tip to the base of the spine head but rather an overall non-polarized motion. A large fraction of actin molecules had no detectable motion, while the remaining ones displayed retrograde and anterograde motions with heterogeneous speeds (8–40 nm/s). These dynamic properties are different from those found in other protrusive structures such as the lamellipodium of motile cells, axonal growth cones or dendritic filopodia where F-actin movements are faster, highly polarized rearward and driven by F-actin growth against the tip of membrane protrusions [66, 67, 82, 83]. Therefore, slow actin movements found in dendritic spines might result from a complex pattern of force generation, involving polymerization of new filaments pushing older filaments, F-actin severing and recycling, but also protrusion ruffling [22, 77, 81, 84–86]. Interestingly, F-actin motions in dendritic spine necks are also slow and non-polarized [39, 66]. This complex F-actin dynamics could also arise from the distinct spatial distribution and dynamics characterizing F-actin pools and from a specific spatiotemporal organization of ABPs in spines.
Actin organization in dendritic spines
Formation and organization of the branched F-actin network in dendritic spines
Essential F-actin regulators are located in spines, and have been found to critically control spinogenesis, spine morphology and synaptic plasticity [44]. Despite having different F-actin dynamics as compared to the lamellipodium, dendritic spine heads are also composed of a dense branched F-actin network, while the spine neck is composed of both branched and non-branched long F-actin filaments [39, 61, 87, 88]. The actin-related protein 2/3 (Arp2/3) complex is the only known actin nucleator forming side branches on a “mother” actin filament [89–92]. Knockout of a single Arp2/3 subunit decreases F-actin turnover and spine head size, induces a progressive spine loss, alters spine structural plasticity and leads to behavioral abnormalities [93]. However, the Arp2/3 complex possesses low intrinsic activity and requires nucleation promoting factors (NPFs) for an efficient activation [92]. NPFs such as the WAVE complex and N-WASP can activate Arp2/3 to induce branched actin nucleation; while cortactin stabilizes Arp2/3 during and after nucleation [92, 94, 95]. The Arp2/3 complex can also be inhibited by PICK1 [96]. Importantly, these proteins are all present in dendritic spines, interact directly or indirectly with PSD components and are involved in the regulation of spine density, morphology and plasticity (WAVE [97–101]; cortactin [102, 103]; N-WASP [104, 105]; PICK1 [96, 106]).
Filament length is also an important factor determining F-actin network dynamics [107]. Profilin binds with a one-to-one ratio to G-actin favoring F-actin polymerization at the barbed end [108, 109]. Profilin-II is enriched in spines upon increased synaptic activity and preventing its binding to G-actin destabilizes dendritic spines [110]. Vasodilator-stimulated phosphoprotein (VASP) and formins promote barbed end F-actin polymerization while capping proteins (CP or CapZ) and EGFR substrate pathway #8 (Eps8) cap the barbed end to prevent F-actin polymerization [111–115]. Interestingly, knockout of Eps8 and knockdown of CP lead to the formation of thin long spines [116, 117], whereas decreasing the activity of the formin mouse diaphanous 2 (mDia2) increases the proportion of large spines [61]. Actin filaments decoration by sub-stoichiometric densities of actin depolymerization factor/cofilin (ADF/cofilin), in coordination with coronin and actin interacting protein 1 (AIP1) recruitment, will induce severing and stochastic disassembly of F-actin [118–121]. Numerous studies have shown that ADF/cofilin and its upstream regulators are tightly regulating dendritic spine shape [29, 61, 122, 123].
Electron microscopy provided the first insights into F-actin organization in dendritic spines [36, 39, 51]. By performing single-molecule localization microscopy (SMLM) and single-particle tracking, our recent study has now revealed the specific nanoscale organization of ABPs in dendritic spines [67] (Fig. 1b). Incorporation of Arp2/3 complexes into the F-actin network occurred in close vicinity to the PSD, and the WAVE complex was found to form a single stable domain overlapping with PSD-95 [67]. The insulin receptor substrate p53 (IRSp53), an I-BAR protein binding and regulating the WAVE complex, was also shown to form a stable domain overlapping with PSD-95 [67, 124, 125]. PSD-95, IRSp53 and the WAVE complex share similar diffusion properties suggesting that branched F-actin nucleation occurs at the membrane apposed to the PSD [67]. Consistently, IRSp53 and subunits of the WAVE and Arp2/3 complexes can directly and/or indirectly bind to components of the PSD, including PSD-95, Shank1, Shank3 and CaMKII [101, 126–129]. In addition, the post-synaptic cell adhesion protein neuroligin-1 bears an interacting sequence binding directly to the WAVE complex [130]. Thus, sequestration of the WAVE complex and IRSp53 at the PSD might provide an efficient mechanism for spatiotemporal control of the branched F-actin network during changes in synaptic transmission. SMLM has also shown that VASP and the formin-like proteins2 (FMNL2), but not the WAVE complex, accumulate at the tip of finger-like protrusions growing away from the PSD [67]. Thus, in contrast to the lamellipodium, zones of branched F-actin nucleation and elongation do not colocalize at protrusion tips [47, 82, 83, 131] (Fig. 1). This demonstrates that it is the respective nanoscale organization of F-actin regulators and not their nature that determines the shape and dynamics of protrusive structures. Those finger-like protrusions might arise from the branched F-actin network but are unlikely to be formed by elongation of tight F-actin bundles, as demonstrated for filopodia emerging from the lamellipodium [39, 132]. The densely packed PSD could represent a physical barrier forcing F-actin barbed ends to grow away and generate finger-like protrusions in dendritic spines. Consistent with this hypothesis, in an in vitro reconstituted system, branched F-actin networks can elongate their barbed ends away for a nucleating surface [84]. Because of the inverse relationship between the rate of F-actin flow and membrane protrusion, the nanoscale segregation between stationary nucleation zones and the delocalizing elongation zones could also explain the slow and non-polarized motion of branched F-actin networks in dendritic spines [67, 133] (Fig. 1b). Despite these recent findings, the current models cannot yet assign a specific F-actin nanoscale organization to the previously described stable and dynamic F-actin pools.
Heterogeneity of F-actin crosslinker organization in dendritic spines
F-actin organization is also controlled by crosslinking proteins. Dendritic spines are for instance enriched in F-actin crosslinkers including α-actinin, neurabins and the neuron-specific F-actin parallel bundler drebrin (α-actinin [134–136]; neurabins [137–141]; drebrin [142–145]). Fascin, however, which stabilizes parallel F-actin bundles in conventional filopodia, appears absent from dendritic filopodia and spines [39, 146, 147]. While α-actinin knockdown delays spine maturation [136] and neurabin knockdown decreases surface glutamate receptor surface expression [148], drebrin knockdown was suggested to promote spine maturation [149]. This discrepancy might arise from specific binding capabilities to different fractions of the F-actin networks. On the one hand, α-actinin was shown to be enriched at the PSD where it could bind subunits of the GluN receptor (NMDA receptor), and therefore link and stabilize GluN receptors with the branched F-actin network [150]. On the other hand, drebrin is located at the spine center where it can associate with the stable F-actin pool and prevent the F-actin remodeling required for spine maturation [51, 151, 152].
The Ca2+/calmodulin-dependent protein kinase II (CaMKII), an essential protein involved in post-synaptic plasticity [153–155], can directly bundle and stabilize F-actin in its inactive state via its CaMKIIβ subunit [156, 157]. Under basal conditions, the binding of CaMKII to F-actin prevents other ABPs, such as the Arp2/3 complex and ADF/cofilin, to interact with F-actin, but upon GluN receptor activation, autophosphorylation of CaMKII prompts its release from F-actin allowing these ABPs to bind and regulate F-actin organization and dynamics [158]. Accordingly, recent sptPALM experiments showed that CaMKIIβ shares similar dynamics to F-actin and that depolymerization of the F-actin network (latrunculin-A) or dissociation of the CaMKIIβ subunit from F-actin increase CaMKII diffusion [158, 159]. The formation of F-actin bundles by CaMKIIβ could suggest an association with the stable F-actin pool [156]. However, CaMKIIβ displays inward and forward movements in dendritic spines similar to actin and Arp2/3, rather suggesting an association with the dynamic F-actin pool [158]. In the following sections, we will see how the activation status of CaMKII during synaptic transmission can act as a gating mechanism to trigger the nanoscale reorganization of the F-actin network [49, 128, 158].
The molecular motor myosin II can bind and generate forces on F-actin, leading to F-actin networks’ contraction and/or disassembly. Myosin II is therefore a crucial regulator of F-actin dynamics and network architecture in the lamellipodium and neuronal growth cones [64, 65, 133, 160–164]. EM and SMLM studies have shown that myosin II is found in both dendritic spine heads and necks [39, 66] and that knockdown or pharmacological inhibition of myosin II induces filopodium-like morphologies [165–168]. Myosin II function in dendritic spines might be more complex than just enhancing the retrograde F-actin flow [60, 169, 170]. A recent study suggests that myosin IIb can stabilize the stable F-actin pool through actin crosslinking, while its contractile activity increases the turnover rate of the dynamic F-actin pool [170]. This dual function might arise from differences in the structural organization of actin filaments in the stable versus dynamic pool. A very elegant in vitro study has shown that myosin II, as well as myosin VI, can disassemble antiparallel but not parallel F-actin bundles leading to a selective actomyosin contractility and disassembly [163]. Taken together with the localization of the parallel bundler drebrin, this suggests that the stable F-actin pool is mainly composed of parallel F-actin bundles stabilized by drebrin and myosin II.
Other ABPs, such as actin binding protein 1 (ABP1), synaptopodin and MAP1B, can be enriched in dendritic spine and regulate spine morphology [171–176]. However, the mechanism of F-actin regulation by those ABPs remains largely unclear, and precludes any speculation about their role in F-actin network organization and dynamics.
Revealing the nanoscale organization and dynamics of a subset of ABPs led to a deeper understanding of the mechanisms linking F-actin generated forces to dendritic spine motility during basal synaptic transmission. In the following sections, we will focus on how changes in synaptic activity can lead to transient and long-lasting reorganization of the F-actin network.
The interplay between the actin cytoskeleton and synaptic plasticity
The strength of synaptic transmission can be either increased or decreased on a timescale of seconds to months. These processes, called long-term potentiation (LTP) and long-term depression (LTD), correlate with the enlargement and shrinkage of dendritic spines, respectively [27–29, 31, 43]. Therefore, the organization and dynamics of the F-actin network requires to be adjusted accordingly to the strength of synaptic transmission [177]. In this section, we will describe the signaling pathways that can regulate F-actin networks during synaptic plasticity, the sequence of events leading to the morphological remodeling of dendritic spines and how the actin cytoskeleton sustains both structural and functional synaptic plasticity.
Signaling pathways that control activity-induced reorganization of the actin cytoskeleton
Neuron depolarization leads to the pre-synaptic release of glutamate and to the activation of post-synaptic receptors. During synaptic plasticity, the opening of post-synaptic GluN receptors (NMDA receptors) and voltage-gated calcium channels induces Ca2+ influx into dendritic spines and activation of calmodulin (CaM) sensitive proteins [153, 178]. CaM binding to CaMKII releases its auto-inhibition allowing autophosphorylation and leading to transient kinase activity [153, 155] (Fig. 2a). As mentioned previously, CaMKII autophosphorylation induces its dissociation from F-actin. Once activated, CaMKII can in turn phosphorylate several synaptic proteins, including regulators of small Rho-familly GTPases. RhoGTPases can switch from an active GTP-bound state to an inactive GDP-bound state by intrinsic GTPase activity. Guanine nucleotide exchange factors (GEFs) activate RhoGTPases by stimulating the release of bound GDP and allowing the binding of GTP. Conversely, GTP-activating proteins (GAPs) can inactivate RhoGTPases by increasing GTPase activity. Rac1, Cdc42 and RhoA are among the best characterized small RhoGTPases and have been extensively described as F-actin regulators in non-neuronal cells [179–182]. Rac1 is responsible for lamellipodium formation, while Cdc42 is more involved in the formation of conventional filopodia, and RhoA plays a role in the assembly of stress fibers and adhesion sites. CaMKII can directly phosphorylate and activate Rac1 GEFs such as Tiam1 and Kalirin7 but can also directly phosphorylate and inactivate Rac1 GAP, p250GAP, both signaling cascades leading to enhanced Rac1 activity [183–186]. CaMKII phosphorylation of neurabin-II leads to the recruitment of a RhoA GEF, Lfc (“Lbc’s first cousin”) [187, 188]. CaMKII activity is also partially involved in Cdc42 activation, although the signaling pathway has not been described in dendritic spines [189]. An increase in intracellular Ca2+ can activate other CaM sensitive proteins involved in the regulation of F-actin networks. CaMKK and CaMKI form a multiprotein complex with another Rac1 GEF, βPIX (“Pak-interacting exchange factor”), resulting in Rac1 activation [190, 191].

Activation of ABPs and reorganization of the F-actin cytoskeleton during LTP. a Selected signaling cascades driving F-actin remodeling upon Ca2+ influx via GluN Receptors. CAMKII is a key triggering protein both involved in functional and structural LTP. CAMKII can activate small RhoGTPases (Rac1, Cdc42, RhoA) by phosphorylating GEFs (Tiam1, Karilin7) and GAPs (p250GAAP). This will control the spatiotemporal activation of several ABPs (Arp2/3, myosin II, cofilin). Arrows indicate positive regulation. T-shaped bars indicate negative regulation. GluN NMDA receptor, CaM calmodulin, CaMK calcium-calmodulin dependent protein kinase, GEF guanine nucleotide exchange factor, GAP GTPase activating protein, βPIX β PAK interacting exchange factor, Tiam1 T cell lymphoma invasion and metastasis-inducing protein 1, Rac1 Ras-related C3 botulinum toxin substrate 1, Cdc42 cell division cycle 42, RhoA Ras homologous member A, ROCK Rho-associated, coiled-coil containing protein kinase, PAK p21-activated kinase, LIMK LIM-kinase, N-WASP neuronal Wiskott–Aldrich syndrome protein, WAVE WASP-family verprolin homologous protein, MLC myosin light chain, Arp2/3 actin-related-protein 2/3. b A model for F-actin reorganization during LTP. LTP induces a transient and long-lasting increase of the spine head size, a shortening and widening of spine neck and a concomitant ABPs and CaMKII spine recruitment. Activated CAMKII will dissociate from F-actin and phosphorylate multiple proteins leading to a fast F-actin reorganization and a transient spine head enlargement. This transient reorganization is characterized by the formation of an enlargement F-actin pool, an increase in the F-actin/G-actin ratio and increased concentration of cofilin and Arp2/3. Note that inactivation of cofilin is detected only 2 min after LTP induction, suggesting that cofilin might be only transiently active before it gets inactivated to prevent further depolymerization. This might be important to only transiently sever F-actin filaments in order to generate new F-actin barbed ends, and to increase F-actin polymerization and branching. During the long-lasting spine head enlargement, most ABPs return to their basal concentration, suggesting the formation of a larger dynamic and stable F-actin pool. Those larger dendritic spines most likely provides a “tag” for the capture of newly synthetized synaptic proteins in order to sustain late LTP
Many targets of RhoGTPases are present in dendritic spines and are critical regulators of the actin cytoskeleton (Fig. 2a). The WAVE complex and N-WASP, NPFs of the Arp2/3 complex, are two main downstream targets of Rac1 and Cdc42, respectively [192–194]. p21-activated kinase (PAK) is a downstream target of Rac1 and Cdc42, while Rho-associated, coiled-coil-containing protein kinase (ROCK), is a downstream target of RhoA [180, 195–197]. Both PAK and ROCK can activate LIM-kinase (LIMK) that can in turn phosphorylate ADF/cofilin preventing its binding and severing activities. This inhibition of ADF/cofilin can be counterbalanced by other signaling pathways. CaM activates protein phosphatase 2B (PP2B or calcineurin) triggering slingshot activation, which dephosphorylates and activates ADF/cofilin leading to enhanced F-actin severing [198, 199]. PAK and ROCK can also phosphorylate the myosin light chain (MLC) leading to increased myosin II activity [177, 195]. Finally, the Arp2/3 complex inhibitor PICK1 can also be regulated by synaptic activity since it is a low-affinity Ca2+ sensor, which binds to Rac1 and Cdc42 and is regulated by the GTPase ADP-ribosylation factor 1 (Arf1) [200–202]. Although many studies revealed the complex signaling pathways leading to F-actin network reorganization, their spatiotemporal integration during synaptic plasticity is far from being understood [181, 189, 203–205].
Morphological remodeling of dendritic spines and F-actin reorganization during LTP and LTD
Early EM studies already reported that LTP induction would increase dendritic spine head and PSD size [23, 206–208], increase the number of dendritic spine protrusions [25] and induce wider and shorter spine necks [207]. However, EM studies cannot provide longitudinal measurements for single spines during LTP induction. Kasai and co-workers performed two-photon glutamate uncaging to release glutamate at single dendritic spines [28, 209]. This stimulation induced spine head enlargement, GluA1 recruitment and an increase in GluA-mediated current [28, 210]. More recently, time-lapse STED microscopy revealed that shortening and widening of the spine neck occurred in a concerted fashion with spine head enlargement [31]. These changes in spine head volume display two distinct phases, a transient large volume increase (<5 min; Δvolume 200–400 % increase) followed by long-lasting smaller volume increase for small dendritic spines (60 min; Δvolume 50–150 % increase), whereas spine neck changes seem to display only a long-lasting change (Δ with 30 % increase; Δlength 30 % decrease; [31]). Consistent with previous studies demonstrating that F-actin dynamics supports spine motility, pharmacological treatments that increased F-actin depolymerization (latrunculin-A) prevented transient and long-lasting changes of dendritic spine head [28].
In vivo, LTP induction was shown to correlate with increased F-actin content in dendritic spines visualized by EM [211]. Fluorescence resonance energy transfer (FRET) also revealed an increased F-actin/G-actin ratio after pre-synaptic fiber tetanic stimulation, suggesting that LTP promotes F-actin polymerization [75]. Subsequently, a very elegant study explored F-actin reorganization by probing photoactivable actin dynamics after two-photon glutamate uncaging [77]. Single spine activation, which triggered spine enlargement, induced the formation of a transient enlargement F-actin pool distributed throughout the spine head, displaying a distinct turnover compared to the stable and dynamic F-actin pools (Fig. 2b). The localization and dynamics of this enlargement pool often synchronized with spine membrane protrusions, as if F-actin polymerization drove the enlargement. However, increased volumes triggered by LTP last longer than the life-time of the enlargement pool, suggesting that the long-lasting spine enlargement might be sustained by transfer of F-actin from the enlargement into the stable pool.
GluN receptor-dependent Ca2+ influx during LTP, which leads to activation of the intracellular signaling pathways, could increase F-actin polymerization and reorganization [153, 212]. CaMKII is necessary and sufficient for LTP induction; it is therefore not surprising that pharmacological inhibition of CaMKII or expression of a CaMKIIα kinase dead mutant in knock-in mouse disrupt the long-lasting volume increase of dendritic spines [28, 205, 213]. Combining FRET with two-photon glutamate uncaging, Yasuda and co-workers demonstrated that CaMKII activity was only transiently increased (~2 min) during spine head enlargement [205]. As mentioned in “Actin organization in dendritic spines” and “The interplay between the actin cytoskeleton and synaptic plasticity”, active CaMKIIβ will dissociate from F-actin and phosphorylate multiple proteins. Recently, Okamoto and colleagues showed that CaMKIIβ dissociation from F-actin is required for structural and functional plasticity of spines, and that this dissociation is not sufficient to induce structural plasticity but permits ADF/cofilin binding to F-actin [158]. The small RhoGTPases Cdc42 and RhoA showed increases in transient (~2 min) but also long-lasting (~40 min) activity [189]. These increased activities are dependent on CaMKII and GluN receptor. Although this latter study did not study Rac1 activity pattern during LTP, a previous study reported that kalirin-7 phosphorylation by CaMKII and subsequent Rac1 activation would lead to dendritic spine enlargement [186]. Overall, those studies suggest that the sustained activities of Rac1, Cdc42 and RhoA might be the main mechanism to relay the transient CaMKII activity for efficient F-actin remodeling.
The main downstream targets of those proteins will regulate F-actin branched nucleation by Arp2/3, ADF/cofilin severing activity and myosin II function (Fig. 2a). Studying the spatiotemporal coordination of various proteins, Hayashi and co-workers have provided new insights into F-actin cytoskeleton reorganization during two-photon glutamate uncaging-induced LTP [49] (Fig. 2b). Concomitantly with spine enlargement, ABPs and CaMKII were translocated into spines. However, while the concentration of cofilin, AIP1 and Arp2/3 in spines increased during the transient spine enlargement phase, the concentration of the crosslinking proteins CaMKIIβ, α-actinin and drebrin A decreased (relative to the spine volume) [49]. This qualitative switch in ABPs concentration in spines suggests that the F-actin network undergoes a fast reorganization that might be important for the formation of the F-actin enlargement pool. Phosphorylation and inactivation of cofilin is detected only 2 min after LTP induction [214], suggesting that cofilin might be only transiently active before it gets inactivated by LIMK to prevent further F-actin severing [49]. Activation of Rac1 downstream pathways might be the key for this short time window activation of ADF/cofilin. Indeed, activation of Rac1 will lead to increased F-actin branching via activation of WAVE and Arp2/3 complexes but also decreased F-actin severing via phosphorylation of ADF/cofilin by PAK/LIMK pathways. A delayed activation of the PAK/LIMK pathway as compared to the fast increase of AIP1 and ADF/cofilin concentrations might provide this 2-min time window for efficient F-actin severing [120]. Interestingly, this transient activation fits with the CaMKII time window of activity and its dissociation from the F-actin network [158, 189]. This model is in agreement with the role that ADF/cofilin might play to trigger membrane protrusions [83, 215]. In that model, transient F-actin severing generates new F-actin barbeds ends, increasing F-actin branching and polymerization without totally depolymerizing the F-actin network. Thus, during these 2-min time windows, the actin cytoskeleton in spines could experience drastic remodeling. Our recent findings provided some insights into the links between spine enlargement and the nanoscale reorganization of ABPs [67]. SMLM experiments demonstrated that Rac1 activation correlates with its immobilization in spines, spine enlargement and delocalization of the WAVE complex from the PSD. In addition, we found that Shank3 overexpression, which causes spine enlargement and neuropsychiatric disorders [129, 216], also induces delocalization of the WAVE complex from the PSD [67]. Thus, alteration of spine morphology could rely on the long-lasting or transient nanoscale relocalization of ABPs, e.g., the WAVE complex, leading to remodeling of the entire dendritic spine structure.
During the long-lasting spine volume increase, most ABPs returned to their basal concentration, whereas cofilin showed a persistent accumulation at the base of the spine and increased F-actin binding [49]. Actin filaments fully decorated by ADF/cofilin are stable, promoting F-actin bundle stabilization in the absence of AIP1 [120, 121, 217]. The long-lasting ADF/cofilin pool might therefore be associated with the stable F-actin pool. Accordingly, drebrin-A is also slightly enriched at potentiated spines during the long-lasting spine head enlargement [49, 218]. Maintaining high-concentration levels of ADF/cofilin during a precise time window could be an interesting mechanism to efficiently depotentiate newly potentiated synapses [49, 219].
Although GluN receptor antagonist and F-actin depolymerization (latrunculin-A) totally disrupt transient and long-lasting spine remodeling, inhibition of CaMKII, Cdc42, ADF/cofilin or Arp2/3 blocks only the long-lasting volume changes [28, 49, 93, 189, 205]. Thus, Ca2+ influx might remodel the F-actin network via distinct signaling pathways such as activation of CaMKI or release of internal calcium stores [174, 220]. Activation of GluN receptor can also recruit and stabilize β-catenin in dendritic spines [221]. According to the molecular clutch hypothesis, connection of N-cadherin/β-catenin to the acto-myosin network could transmit forces that will lead to spine enlargement [60, 70, 222, 223]. Supporting the requirement of a clutch mechanism for dendritic spine enlargement during LTP, inhibition of RhoA and ROCK, needed for myosin II activity, also disrupted transient and long-lasting volume changes [60, 189]. All these studies highlight the need for a spatiotemporal coordination of F-actin polymerization, branching, severing and bundling for transient and long-lasting remodeling of the different F-actin pools in order to sustain structural LTP.
Induction of synaptic LTD by electrical or chemical stimulation can lead to dendritic spine shrinkage [29, 30, 75, 224, 225]. Adapting the two-photon glutamate uncaging to induce LTP, Zito and co-workers established a low frequency uncaging (LFU) protocol to induce LTD at individual spines [30]. Although LFU does not allow the probing of structural remodeling at early time scales, the authors were able to induce a long-lasting and saturable volume shrinkage of small and large dendritic spines (Δvolume 25 % decrease). Combining two-photon uncaging of glutamate and GABA to induce a distinct form of LTD, Kasai and coworkers could induce larger volume changes (ΔVolume 40 % decrease) and even spine elimination [226]. Spine shrinkage was first shown to correlate with a decreased F-actin/G-actin ratio after pre-synaptic fiber tetanic stimulation, suggesting F-actin depolymerization [75], which, during GluN receptor-dependent LTD, would be powered by activation of ADF/cofilin by calcineurin via slingshot [29, 123, 198, 224]. Inhibition of GluN receptors and calcineurin and also a phospho-cofilin peptide can therefore abolish spine shrinkage [224, 226]. However, shrinkage of large spines requires additional signaling from mGluR and internal calcium stores [30]. Dendritic spine shrinkage is also blocked after knockdown of the Arp2/3 inhibitor PICK1 or expression of a PICK1 mutant unable to bind Arp2/3 [106]. Therefore, as for LTP, regulation of F-actin organization and dynamics might be critical mechanisms for structural LTD.
How does the actin cytoskeleton sustain functional LTP and LTD?
Dendritic spine structural plasticity and functional plasticity share similar triggering molecules, such as CaMKII for LTP and calcineurin for LTD. However, structural plasticity and functional plasticity can be dissociated as they do not strictly rely on the same intracellular signaling pathways [43, 199, 224, 227–230]. Although regulation of F-actin network assembly and disassembly is not sufficient to induce synaptic plasticity, it is required for its formation in vitro and in vivo [211, 231–233]. Likewise, knocking out proteins involved in F-actin network organization and dynamics, such as Rac1, IRSp53, subunits of WAVE or Arp2/3 complexes, PAK1, PAK3, LIMK, ADF/cofilin and Eps8 displayed impaired LTP and/or LTD and also spatial and working memory deficits [93, 98, 100, 117, 122, 234–239]. Neurological disorders associated with abnormal spine morphologies such as autism spectrum disorders (ASD) and schizophrenia can also be triggered by genetic deregulation of proteins such as IRSp53, Shank3 and FMRP (fragile X mental retardation protein) that directly interact with subunits of the WAVE and Arp2/3 complexes [129, 216, 239–242]. What could therefore be the mechanisms behind F-actin cytoskeleton sustainment of synaptic plasticity?
GluN and GluA receptors play a major role in the induction and maintenance of synaptic plasticity, respectively [199, 243, 244]. Trafficking of GluN and GluA in and out of dendritic spines, but also their anchorage and stabilization via interaction with PSD scaffold proteins, are crucial mechanisms for LTP and LTD [245–248]). PSD dynamic architecture and receptor trafficking are supported and regulated by the F-actin cytoskeleton [43, 86, 249]. F-actin can regulate in many ways the dynamics of key PSD scaffold proteins such as PSD-95, GKAP, Shank and Homer [35]. First, actin filaments are in close proximity with the PSD [36]. Second, the Arp2/3 and WAVE complexes and cortactin can directly bind Shank3 [101, 129]. Third, proteins involved in the activation of the WAVE complex, such as IRSp53 and the Rac1 GEF βPIX, can interact with Shank family proteins and PSD95 [126, 127, 250–252]. Acute pharmacological treatment inducing F-actin depolymerization (latrunculin-A) disrupts PSD-95 nanoscale organization and dynamics but only slightly affects its content in dendritic spines, probably because of its direct association with the plasma membrane [253–255]. A similar treatment decreases GKAP, Homer1C and Shank2 content in immature but not mature spines [253, 254]. Thus, PSD scaffold matrix remodeling relies on F-actin dynamics, but the intermolecular assembly between PSD proteins might be strengthened during dendritic spine maturation to become independent from the F-actin network [256]. Interestingly, as for PSD95 proteins, the WAVE complex and IRSp53 are still present in dendritic spines after F-actin depolymerization, highlighting the tight functional coupling between the PSD and F-actin regulators [67]. Pharmacological treatments which induce F-actin depolymerization also disrupt GluA dynamic architecture, increasing their surface diffusion, decreasing their spine content and preventing their spine insertion during LTP [230, 238, 254, 257]. Furthermore, inhibiting F-actin dynamics without disrupting the F-actin network (low concentrations of latrunculin-A) does not decrease GluA content but still prevents insertion of new GluA receptors upon synaptic stimulation [228, 230, 232]. Altogether, these findings suggest, first, that PSD reorganization by F-actin controls the anchorage of a synaptic pool of GluA receptors; second that PSD remodeling provides constant accessibility of new binding sites for GluA; and finally that maintenance and insertion of GluA receptors relies on the F-actin cytoskeleton [230, 238, 254, 258–260].
Disrupting F-actin dynamics could also lead to impaired exocytosis and endocytosis therefore preventing insertion and removal of GluA receptors during LTP and LTD, respectively [249, 261–265]. The plasma membrane t-SNARE syntaxin 4 binds to actin and is involved in exocytosis in dendritic spines and LTP [261, 266]. However, F-actin depolymerization does not change the frequency and proportion of exocytotic events in the somatodendritic compartment [267]. Spine-localized endocytosis of GluA receptors is, on the other hand, well established and relies on the actin cytoskeleton [249]. The Arp2/3 inhibitor PICK1 binds to GluA2/3 subunits and plays a critical role in their surface expression. Preventing PICK1 binding to Arp2/3 disrupts GluA internalization during LTD induction therefore occluding LTD [96, 106]. Interestingly, PICK1 is a Ca2+ sensor, and the PICK1-mediated inhibition of Arp2/3 is enhanced by GluA2-PICK1 interaction thus providing spatiotemporal control of GluA endocytosis during LTD [96, 200]. Since PICK1 knockdown also prevents LTP, branched F-actin nucleation and its regulation by PICK1 could be more generally involved in the recycling endosomal pathway and in the constant supply of GluA receptors [249, 268, 269].
Intracellular transport of recycling endosomes (RE) into and out of dendritic spines relies on myosin motors moving on actin filaments: myosin V and myosin VI walking towards the barbed- and pointed-end, respectively. Spine necks are composed of both branched and non-branched long F-actin filaments of mixed orientation, thus providing tracks for myosin V and VI to enter and exit dendritic spines [39, 66]. As mentioned previously, LTP induction induces shortening and widening of the spine neck and could therefore facilitate in and out trafficking mediated by myosin motors [31]. Myosin VI was first shown to be involved in stimulation-induced GluA1 endocytosis [270]. Myosin Vb was also shown to be recruited to RE upon Ca2+ influx and LTP induction, triggering translocation of RE into spines and GluA1 surface insertion [271]. Furthermore, acute blockade of Myosin Vb ATP activity, needed for its movements along F-actin, attenuates LTP maintenance [271].
While the mechanisms described above were shown to be essential to sustain the early phase of LTP and LTD (1–4 h), protein synthesis is required to sustain the later phases (≥4 h) [49, 199, 272–277]. Late LTP (L-LTP) requires, for instance, the supply of newly synthetized PSD proteins to increase the number of available binding sites for GluA receptors in order to prevent synaptic saturation [278]. Interestingly, inhibiting CaMKII activity only during LTP induction or inhibiting F-actin dynamics during LTP induction up to 30 min after induction are sufficient to occlude L-LTP [233, 279]. Therefore, activation of the CaMKII signaling cascade, reorganization of the F-actin cytoskeleton and dendritic spine morphological remodeling could all represent a synaptic “tag” for efficient delivery and capture of newly synthetized proteins in order to sustain long-term synaptic plasticity and memory storage [49, 280].
Conclusion/outlook
In this review, we have highlighted that, as for many other motile sub-cellular compartments, dendritic spine motility and remodeling are sustained by F-actin cytoskeleton dynamics. The unique F-actin dynamics of dendritic spines results from the nanoscale organization of various ABPs within different sub-spine compartments [67]. However, to completely understand how the nanoscale organization of ABPs can lead to the coexistence of several F-actin pools with distinct dynamic properties will require further studies. Correlative live cell and super-resolution microscopy could, for instance, enable correlation between F-actin dynamics and ABP nanoscale organization [281]. Because of the heterogeneity of dendritic spine morphologies, multiplex SMLM could represent a powerful tool to visualize membrane, ABPs, PSD proteins and post-synaptic receptors within an individual spine. Multiplex SMLM can be achieved by linking DNA-PAINT (DNA-Point Accumulation for Imaging in Nanoscale Topography) docking strands to antibodies [282, 283]; by using virtual grids and sequential immunostaining and imaging [284]; or more recently by spectrally resolving single molecules from different dyes [285]. This last approach might, however, be challenging regarding the density of proteins in dendritic spines.
Regulation of the spatiotemporal activity of ABPs is a fundamental mechanism to induce spine structural plasticity during LTP and LTD. Recent work has provided a detailed picture of this spatiotemporal coordination [49, 189, 286]. However, those studies could not provide the nanoscale organization of ABPs at sub-spine resolution. So far, SMLM has reached the highest resolution for fluorescence microscopy on both live and fixed biological samples in 2D and 3D [53, 80, 287, 288]. Deep tissue and in vivo imaging of dendritic spines is, however, still a challenge [289, 290]. Super-resolution microscopy techniques, such as simulated emission depletion microscopy (STED), can perform live recordings of dendritic spines in brain slices and in vivo [22, 31, 291]. However, high-power lasers used for STED induce high photobleaching and requires high labeling density or replenishment of a cytosolic fluorophore. Reversible saturable optical fluorescence transitions microscopy can overcome those drawbacks by exploiting reversibly photoswitchable fluorophores [292, 293]. Structured illumination microscopy is also well suited for three-dimensional multi-color live imaging, and recent improvements of its spatial resolution in combination with light sheet microscopy could represent a valuable tool to study the spatiotemporal reorganization of synaptic proteins in brain slices and in vivo during synaptic plasticity [294–299].
Spine structural plasticity and the underlying reorganization of F-actin networks could represent a synaptic “tag” required for the maintenance of functional plasticity [75, 153, 233, 280]. Recently, Kasai and co-workers have demonstrated that motor task learning could be disrupted by optical shrinkage of the potentiated spines, therefore establishing a direct link between dendritic spine morphology and the formation of a memory trace in vivo [13]. The exact mechanism by which the F-actin cytoskeleton sustains functional plasticity is, however, still unclear. Most likely, it involves efficient delivery and capture of newly synthetized proteins as well as the regulation of PSD proteins and post-synaptic receptors dynamics and stability. The coordinated structural remodeling of pre- and post-synaptic structures could also be a key mechanism for maintenance of functional plasticity and might require tight coordination between the F-actin cytoskeleton and trans-synaptic adhesion proteins [286, 300]. Further development in optogenetics will allow the manipulation of synaptic protein interactions in vitro and in vivo and could tackle those hypotheses to refine the model behind the sustainment of functional synaptic plasticity by F-actin cytoskeleton reorganization [301, 302]. However, how this knowledge, acquired at a single spine and neuron level, can be implemented in neural networks models that can generate behavior, cognition and mental disease remains a main challenge in neuroscience [303, 304].
Abbreviations
- ABP:
-
Actin binding proteins
- ASD:
-
Autism spectrum disorders
- EM:
-
Electron microscopy
- FRAP:
-
Fluorescence recovery after photobleaching
- FRET:
-
Fluorescence resonance energy transfer
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- NPFs:
-
Nucleation promoting factors
- PSD:
-
Post-synaptic density
- SMLM:
-
Single molecule localization microscopy
- sptPALM:
-
Single particle tracking photoactivation localization microscopy
- STED:
-
Simulated emission depletion microscopy
References
Ramón y Cajal S S (1888) Estructura de los centros nerviosos de las aves. Rev Trim Histol Norm Pat 1:1–10
Yuste R (2015) The discovery of dendritic spines by Cajal. Front Neuroanat 9:1–6. doi:10.3389/fnana.2015.00018
Ramón y Cajal S (1891) Significación fisiológica de las expansiones protoplásmicas y nerviosas de la sustancia gris. Rev Cienc Med Barcelona 22:23
Ramón y Cajal S (1893) Neue darstellung vom histologischen bau des centralnervensystem. Arch Anat Entwick 319–428
Ramón y Cajal S (1894) La fine structure des centres nerveux. The croonian lecture. Proc R Soc Lond B 55:443–468
Gray EG (1959) Electron microscopy of synaptic contacts on dendrite spines of the cerebral cortex. Nature 183:1592–1593. doi:10.1038/1831592a0
Gray EG (1959) Axo-somatic and axo-dendritic synapses of the cerebral cortex. J Anat 93:420–433. doi:10.1038/1831592a0
Guillery RW (2000) Early electron microscopic observations of synaptic structures in the cerebral cortex: a view of the contributions made by George Gray (1924–1999). Trends Neurosci 23:594–598. doi:10.1016/S0166-2236(00)01635-0
Hebb DO (1949) The organization of behaviour: a neuropsychological theory. Wiley, New York
Kandel ER (2009) The biology of memory: a forty-year perspective. J Neurosci 29:12748–12756. doi:10.1523/JNEUROSCI.3958-09.2009
Squire LR (2009) Memory and brain systems: 1969–2009. J Neurosci 29:12711–12716. doi:10.1523/JNEUROSCI.3575-09.2009
Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R (2014) Engineering a memory with LTD and LTP. Nature 511:348–352. doi:10.1038/nature13294
Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H (2015) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525:333–338. doi:10.1038/nature15257
Harris KM, Jensen FE, Tsao B (1992) Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci 12:2685–2705
Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31:47–67. doi:10.1146/annurev.neuro.31.060407.125646
Nägerl UV, Willig KI, Hein B, Hell SW, Bonhoeffer T (2008) Live-cell imaging of dendritic spines by STED microscopy. Proc Natl Acad Sci USA 105:18982–18987. doi:10.1073/pnas.0810028105
Izeddin I, Specht CG, Lelek M, Darzacq X, Triller A, Zimmer C, Dahan M (2011) Super-resolution dynamic imaging of dendritic spines using a low-affinity photoconvertible actin probe. PLoS ONE 6:e15611. doi:10.1371/journal.pone.0015611
Dailey E, Smith S (1996) The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16:2983–2994
Fischer M, Kaech S, Knutti D, Matus A (1998) Rapid actin-based plasticity in dendritic spines. Neuron 20:847–854
Lendvai B, Stern EA, Chen B, Svoboda K (2000) Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404:876–881. doi:10.1038/35009107
Grutzendler J, Kasthuri N, Gan W-B (2002) Long-term dendritic spine stability in the adult cortex. Nature 420:812–816. doi:10.1038/nature01276
Berning S, Willig KI, Steffens H, Dibaj P, Hell SW (2012) Nanoscopy in a living mouse brain. Science 335:551. doi:10.1126/science.1215369
Buchs PA, Muller D (1996) Induction of long-term potentiation is associated with major ultrastructural changes of activated synapses. Proc Natl Acad Sci USA 93:8040–8045
Engert F, Bonhoeffer T (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399:66–70. doi:10.1038/19978
Toni N, Buchs P, Nikonenko I, Bron C, Muller D (1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402:421–425. doi:10.1038/46574
Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T (2004) Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44:759–767. doi:10.1016/j.neuron.2004.11.016
Nishiyama J, Yasuda R (2015) Biochemical computation for spine structural plasticity. Neuron 87:63–75. doi:10.1016/j.neuron.2015.05.043
Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. doi:10.1038/nature02617
Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44:749–757. doi:10.1016/j.neuron.2004.11.011
Oh WC, Hill TC, Zito K (2013) Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc Natl Acad Sci USA 110:E305–E312. doi:10.1073/pnas.1214705110
Tønnesen J, Katona G, Rózsa B, Nägerl UV (2014) Spine neck plasticity regulates compartmentalization of synapses. Nat Neurosci 17:678–685. doi:10.1038/nn.3682
Noguchi J, Nagaoka A, Watanabe S, Ellis-Davies GCR, Kitamura K, Kano M, Matsuzaki M, Kasai H (2011) In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J Physiol 589:2447–2457. doi:10.1113/jphysiol.2011.207100
Loewenstein Y, Kuras A, Rumpel S (2011) Multiplicative dynamics underlie the emergence of the log-normal distribution of spine sizes in the neocortex in vivo. J Neurosci 31:9481–9488. doi:10.1523/JNEUROSCI.6130-10.2011
Zhang Y, Cudmore RH, Lin D-T, Linden DJ, Huganir RL (2015) Visualization of NMDA receptor-dependent AMPA receptor synaptic plasticity in vivo. Nat Neurosci 18:402–407. doi:10.1038/nn.3936
Sheng M, Hoogenraad CC (2007) The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76:823–847. doi:10.1146/annurev.biochem.76.060805.160029
Fifkova E, Delay RJ (1982) Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol 95:345–350
Kaech S, Fischer M, Doll T, Matus A (1997) Isoform specificity in the relationship of actin to dendritic spines. J Neurosci 17:9565–9572
Cheng D, Hoogenraad C, Rush J, Schlager M, Duong D, Xu P, Wijayawardana S, Hanfelt J, Nakagawa T, Sheng M, Peng J (2006) Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics 5:1158–1170. doi:10.1074/mcp.D500009-MCP200
Korobova F, Svitkina T (2010) Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell 21:165–176. doi:10.1091/mbc.E09-07-0596
Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R (1999) Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA 96:13438–13443
Korkotian E, Segal M (2001) Regulation of dendritic spine motility in cultured hippocampal neurons. J Neurosci 21:6115–6124
Portera-Cailliau C, Pan DT, Yuste R (2003) Activity-regulated dynamic behavior of early dendritic protrusions: evidence for different types of dendritic filopodia. J Neurosci 23:7129–7142
Cingolani LA, Goda Y (2008) Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9:344–356. doi:10.1038/nrn2373
Hotulainen P, Hoogenraad CC (2010) Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189:619–629. doi:10.1083/jcb.201003008
Pollard TD, Cooper JA (2009) Actin, a central player in cell shape and movement. Science 326:1208–1212. doi:10.1126/science.1175862
Carlier M-F, Pernier J, Montaville P, Shekhar S, Kühn S (2015) Control of polarized assembly of actin filaments in cell motility. Cell Mol Life Sci 72:3051–3067. doi:10.1007/s00018-015-1914-2
Pilo Boyl P, Witke W (2014) Small, smaller. dendritic spine. EMBO J 33:2737–2739. doi:10.15252/embj.201490137
Lin W-H, Webb DJ (2009) Actin and actin-binding proteins: masters of dendritic spine formation, morphology, and function. Open Neurosci J 3:54–66. doi:10.2174/1874082000903020054
Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82:444–459. doi:10.1016/j.neuron.2014.03.021
Chen X, Winters C, Azzam R, Li X, Galbraith JA, Leapman RD, Reese TS (2008) Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci USA 105:4453–4458. doi:10.1073/pnas.0800897105
Rácz B, Weinberg RJ (2013) Microdomains in forebrain spines: an ultrastructural perspective. Mol Neurobiol 47:77–89. doi:10.1007/s12035-012-8345-y
Huang B, Bates M, Zhuang X (2009) Super-resolution fluorescence microscopy. Annu Rev Biochem 78:993–1016. doi:10.1146/annurev.biochem.77.061906.092014
Maglione M, Sigrist SJ (2013) Seeing the forest tree by tree: super-resolution light microscopy meets the neurosciences. Nat Neurosci 16:790–797. doi:10.1038/nn.3403
Choquet D, Triller A (2013) The dynamic synapse. Neuron 80:691–703. doi:10.1016/j.neuron.2013.10.013
Adrian M, Kusters R, Wierenga CJ, Storm C, Hoogenraad CC, Kapitein LC (2014) Barriers in the brain: resolving dendritic spine morphology and compartmentalization. Front Neuroanat 8:1–12. doi:10.3389/fnana.2014.00142
MacGillavry HD, Hoogenraad CC (2015) The internal architecture of dendritic spines revealed by super-resolution imaging: what did we learn so far? Exp Cell Res 335:180–186. doi:10.1016/j.yexcr.2015.02.024
Ziv NE, Smith SJ (1996) Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17:91–102
Yuste R, Bonhoeffer T (2004) Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci 5:24–34. doi:10.1038/nrn1300
Zuo Y, Lin A, Chang P, Gan W-B (2005) Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46:181–189. doi:10.1016/j.neuron.2005.04.001
Chazeau A, Garcia M, Czondor K, Perrais D, Tessier B, Giannone G, Thoumine O (2015) Mechanical coupling between transsynaptic N-cadherin adhesions and actin flow stabilizes dendritic spines. Mol Biol Cell 26:859–873. doi:10.1091/mbc.E14-06-1086
Hotulainen P, Llano O, Smirnov S, Tanhuanpää K, Faix J, Rivera C, Lappalainen P (2009) Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol 185:323–339. doi:10.1083/jcb.200809046
Mallavarapu A, Mitchison T (1999) Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J Cell Biol 146:1097–1106. doi:10.1083/jcb.146.5.1097
Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G (2004) Two distinct actin networks drive the protrusion of migrating cells. Science 305:1782–1786. doi:10.1126/science.1100533
Medeiros NA, Burnette DT, Forscher P (2006) Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat Cell Biol 8:215–226. doi:10.1038/ncb1367
Giannone G, Dubin-Thaler BJ, Rossier O, Cai Y, Chaga O, Jiang G, Beaver W, Döbereiner H-G, Freund Y, Borisy G, Sheetz MP (2007) Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128:561–575. doi:10.1016/j.cell.2006.12.039
Tatavarty V, Das S, Yu J (2012) Polarization of actin cytoskeleton is reduced in dendritic protrusions during early spine development in hippocampal neuron. Mol Biol Cell 23:3167–3177. doi:10.1091/mbc.E12-02-0165
Chazeau A, Mehidi A, Nair D, Gautier JJ, Leduc C, Chamma I, Kage F, Kechkar A, Thoumine O, Rottner K, Choquet D, Gautreau A, Sibarita J-B, Giannone G (2014) Nanoscale segregation of actin nucleation and elongation factors determines dendritic spine protrusion. EMBO J 33:2745–2764. doi:10.15252/embj.201488837
Mitchison T, Kirschner M (1988) Cytoskeletal dynamics and nerve growth. Neuron 1:761–772
Suter DM, Forscher P (2000) Substrate-cytoskeletal coupling as a mechanism for the regulation of growth cone motility and guidance. J Neurobiol 44:97–113. doi:10.1002/1097-4695(200008)44:2<97:AID-NEU2>3.3.CO;2-L
Giannone G, Mège R-M, Thoumine O (2009) Multi-level molecular clutches in motile cell processes. Trends Cell Biol 19:475–486. doi:10.1016/j.tcb.2009.07.001
Bard L, Boscher C, Lambert M, Mège R-M, Choquet D, Thoumine O (2008) A molecular clutch between the actin flow and N-cadherin adhesions drives growth cone migration. J Neurosci 28:5879–5890. doi:10.1523/JNEUROSCI.5331-07.2008
Garcia M, Leduc C, Lagardère M, Argento A, Sibarita J-B, Thoumine O (2015) Two-tiered coupling between flowing actin and immobilized N-cadherin/catenin complexes in neuronal growth cones. Proc Natl Acad Sci USA 112:201423455. doi:10.1073/pnas.1423455112
Spacek J, Harris KM (2004) Trans-endocytosis via spinules in adult rat hippocampus. J Neurosci 24:4233–4241. doi:10.1523/JNEUROSCI.0287-04.2004
Tao-Cheng J-H, Dosemeci A, Gallant PE, Miller S, Galbraith JA, Winters CA, Azzam R, Reese TS (2009) Rapid turnover of spinules at synaptic terminals. Neuroscience 160:42–50. doi:10.1016/j.neuroscience.2009.02.031
Okamoto K-I, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112. doi:10.1038/nn1311nn1311
Star EN, Kwiatkowski DJ, Murthy VN (2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5:239–246. doi:10.1038/nn811
Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GCR, Kasai H (2008) The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57:719–729. doi:10.1016/j.neuron.2008.01.013
Frost NA, Shroff H, Kong H, Betzig E, Blanpied TA (2010) Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron 67:86–99. doi:10.1016/j.neuron.2010.05.026
Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–1645. doi:10.1126/science.1127344
Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J (2008) High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods 5:155–157. doi:10.1038/nmeth.1176
Tatavarty V, Kim E-J, Rodionov V, Yu J (2009) Investigating sub-spine actin dynamics in rat hippocampal neurons with super-resolution optical imaging. PLoS ONE 4:e7724. doi:10.1371/journal.pone.0007724
Pollard TD (2003) The cytoskeleton, cellular motility and the reductionist agenda. Nature 422:741–745. doi:10.1038/nature01598
Lai FPL, Szczodrak M, Block J, Faix J, Breitsprecher D, Mannherz HG, Stradal TEB, Dunn GA, Small JV, Rottner K (2008) Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J 27:982–992. doi:10.1038/emboj.2008.34
Achard V, Martiel J-L, Michelot A, Guérin C, Reymann A-C, Blanchoin L, Boujemaa-Paterski R (2010) A “primer”-based mechanism underlies branched actin filament network formation and motility. Curr Biol 20:423–428. doi:10.1016/j.cub.2009.12.056
Sykes C, Plastino J (2010) Cell biology: actin filaments up against a wall. Nature 464:365–366. doi:10.1038/464365a
Frost NA, Kerr JM, Lu HE, Blanpied TA (2010) A network of networks: cytoskeletal control of compartmentalized function within dendritic spines. Curr Opin Neurobiol 20:578–587. doi:10.1016/j.conb.2010.06.009
Rácz B, Weinberg RJ (2008) Organization of the Arp2/3 complex in hippocampal spines. J Neurosci 28:5654–5659. doi:10.1523/JNEUROSCI.0756-08.2008
Burette AC, Lesperance T, Crum J, Martone M, Volkmann N, Ellisman MH, Weinberg RJ (2012) Electron tomographic analysis of synaptic ultrastructure. J Comp Neurol 520:2697–2711. doi:10.1002/cne.23067
Blanchoin L, Amann KJ, Higgs HN, Marchand J, Kaiser DA, Pollard TD (2000) Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature 171:1007–1011
Amann KJ, Pollard TD (2001) Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc Natl Acad Sci USA 98:15009–15013. doi:10.1073/pnas.211556398
Goley ED, Welch MD (2006) The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 7:713–726. doi:10.1038/nrm2026
Campellone KG, Welch MD (2010) A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 11:237–251. doi:10.1038/nrm2867
Kim IH, Racz B, Wang H, Burianek L, Weinberg R, Yasuda R, Wetsel WC, Soderling SH (2013) Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci 33:6081–6092. doi:10.1523/JNEUROSCI.0035-13.2013
Helgeson LA, Nolen BJ (2013) Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife 2:e00884. doi:10.7554/eLife.00884
Helgeson LA, Prendergast JG, Wagner AR, Rodnick-Smith M, Nolen BJ (2014) Interactions with actin monomers, actin filaments, and Arp2/3 complex define the roles of WASP family proteins and cortactin in coordinately regulating branched actin networks. J Biol Chem 289:28856–28869. doi:10.1074/jbc.M114.587527
Rocca DL, Martin S, Jenkins EL, Hanley JG (2008) Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol 10:259–271. doi:10.1038/ncb1688
Kim Y, Sung JY, Ceglia I, Lee K-W, Ahn J-H, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, Greengard P (2006) Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442:814–817. doi:10.1038/nature04976
Grove M, Demyanenko G, Echarri A, Zipfel PA, Quiroz ME, Rodriguiz RM, Playford M, Martensen SA, Robinson MR, Wetsel WC, Maness PF, Pendergast AM (2004) ABI2-deficient mice exhibit defective cell migration, aberrant dendritic spine morphogenesis, and deficits in learning and memory. Mol Cell Biol 24:10905–10922. doi:10.1128/MCB.24.24.10905
Pilpel Y, Segal M (2005) Rapid WAVE dynamics in dendritic spines of cultured hippocampal neurons is mediated by actin polymerization. J Neurochem 95:1401–1410. doi:10.1111/j.1471-4159.2005.03467.x
Soderling SH, Guire ES, Kaech S, White J, Zhang F, Schutz K, Langeberg LK, Banker G, Raber J, Scott JD (2007) A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J Neurosci 27:355–365. doi:10.1523/JNEUROSCI.3209-06.2006
Proepper C, Johannsen S, Liebau S, Dahl J, Vaida B, Bockmann J, Kreutz MR, Gundelfinger ED, Boeckers TM (2007) Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J 26:1397–1409. doi:10.1038/sj.emboj.7601569
Hering H, Sheng M (2003) Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23:11759–11769 (pii:23/37/11759)
Racz B, Weinberg RJ (2004) The subcellular organization of cortactin in hippocampus. J Neurosci 24:10310–10317. doi:10.1523/JNEUROSCI.2080-04.2004
Lee S, Lee K, Hwang S, Kim SH, Song WK, Park ZY, Chang S (2006) SPIN90/WISH interacts with PSD-95 and regulates dendritic spinogenesis via an N-WASP-independent mechanism. EMBO J 25:4983–4995. doi:10.1038/sj.emboj.7601349
Wegner AM, Nebhan CA, Hu L, Majumdar D, Meier KM, Weaver AM, Webb DJ (2008) N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J Biol Chem 283:15912–15920. doi:10.1074/jbc.M801555200
Nakamura Y, Wood CL, Patton AP, Jaafari N, Henley JM, Mellor JR, Hanley JG (2011) PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J 30:719–730. doi:10.1038/emboj.2010.357
Mejillano MR, Kojima S, Applewhite DA, Gertler FB, Svitkina TM, Borisy GG (2004) Lamellipodial versus filopodial mode of the actin nanomachinery. Cell 118:363–373. doi:10.1016/j.cell.2004.07.019
Goldschmidt-Clermont PJ, Machesky LM, Doberstein SK, Pollard TD (1991) Mechanism of the interaction of human platelet profilin with actin. J Cell Biol 113:1081–1089
Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J (2000) Molecular cell biology, 4th edn. Section 18.2: the dynamics of actin assembly. W. H. Freeman, New York
Ackermann M, Matus A (2003) Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci 6:1194–1200. doi:10.1038/nn1135nn1135
Chesarone MA, DuPage AG, Goode BL (2010) Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol 11:62–74. doi:10.1038/nrm2816
Breitsprecher D, Kiesewetter AK, Linkner J, Vinzenz M, Stradal TEB, Small JV, Curth U, Dickinson RB, Faix J (2011) Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J 30:456–467. doi:10.1038/emboj.2010.348
Breitsprecher D, Jaiswal R, Bombardier JP, Gould CJ, Gelles J, Goode BL (2012) Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science 336:1164–1168. doi:10.1126/science.1218062
Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, Cooper JA (2014) Capping protein regulators fine-tune actin assembly dynamics. Nat Rev Mol Cell Biol 15:677–689. doi:10.1038/nrm3869
Disanza A, Carlier M-F, Stradal TEB, Didry D, Frittoli E, Confalonieri S, Croce A, Wehland J, Di Fiore PP, Scita G (2004) Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nat Cell Biol 6:1180–1188. doi:10.1038/ncb1199
Fan Y, Tang X, Vitriol E, Chen G, Zheng JQ (2011) Actin capping protein is required for dendritic spine development and synapse formation. J Neurosci 31:10228–10233. doi:10.1523/JNEUROSCI.0115-11.2011
Menna E, Zambetti S, Morini R, Donzelli A, Disanza A, Calvigioni D, Braida D, Nicolini C, Orlando M, Fossati G, Cristina Regondi M, Pattini L, Frassoni C, Francolini M, Scita G, Sala M, Fahnestock M, Matteoli M (2013) Eps8 controls dendritic spine density and synaptic plasticity through its actin-capping activity. EMBO J 32:1730–1744. doi:10.1038/emboj.2013.107
Suarez C, Roland J, Boujemaa-Paterski R, Kang H, McCullough BR, Reymann A-C, Guérin C, Martiel J-L, De la Cruz EM, Blanchoin L (2011) Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr Biol 21:862–868. doi:10.1016/j.cub.2011.03.064
McCullough BR, Grintsevich EE, Chen CK, Kang H, Hutchison AL, Henn A, Cao W, Suarez C, Martiel J-L, Blanchoin L, Reisler E, De La Cruz EM (2011) Cofilin-linked changes in actin filament flexibility promote severing. Biophys J 101:151–159. doi:10.1016/j.bpj.2011.05.049
Jansen S, Collins A, Chin SM, Ydenberg CA, Gelles J, Goode BL (2015) Single-molecule imaging of a three-component ordered actin disassembly mechanism. Nat Commun 6:7202. doi:10.1038/ncomms8202
Gressin L, Guillotin A, Guérin C, Blanchoin L, Michelot A (2015) Architecture dependence of actin filament network disassembly. Curr Biol 25:1437–1447. doi:10.1016/j.cub.2015.04.011
Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z (2002) Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35:121–133. doi:10.1016/S0896-6273(02)00758-4
Zhou L, Jones EV, Murai KK (2012) EphA signaling promotes actin-based dendritic spine remodeling through slingshot phosphatase. J Biol Chem 287:9346–9359. doi:10.1074/jbc.M111.302802
Scita G, Confalonieri S, Lappalainen P, Suetsugu S (2008) IRSp53: crossing the road of membrane and actin dynamics in the formation of membrane protrusions. Trends Cell Biol 18:52–60. doi:10.1016/j.tcb.2007.12.002
Suetsugu S, Gautreau A (2012) Synergistic BAR-NPF interactions in actin-driven membrane remodeling. Trends Cell Biol 22:141–150. doi:10.1016/j.tcb.2012.01.001
Bockmann J, Kreutz MR, Gundelfinger ED, Böckers TM (2002) ProSAP/Shank postsynaptic density proteins interact with insulin receptor tyrosine kinase substrate IRSp53. J Neurochem 83:1013–1017
Choi J, Ko J, Racz B, Burette A, Lee J-R, Kim S, Na M, Lee HW, Kim K, Weinberg RJ, Kim E (2005) Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci 25:869–879. doi:10.1523/JNEUROSCI.3212-04.2005
Park E, Chi S, Park D (2012) Activity-dependent modulation of the interaction between CaMKIIα and Abi1 and its involvement in spine maturation. J Neurosci 32:13177–13188. doi:10.1523/JNEUROSCI.2257-12.2012
Han K, Holder JL, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, Yu P, Sun H, Breman AM, Patel A, Lu H-C, Zoghbi HY (2013) SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503:72–77. doi:10.1038/nature12630
Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, Rosen MK (2014) The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 156:195–207. doi:10.1016/j.cell.2013.11.048
Chen XJ, Squarr AJ, Stephan R, Chen B, Higgins TE, Barry DJ, Martin MC, Rosen MK, Bogdan S, Way M (2014) Ena/VASP proteins cooperate with the WAVE complex to regulate the actin cytoskeleton. Dev Cell 30:569–584. doi:10.1016/j.devcel.2014.08.001
Yang C, Svitkina T (2011) Filopodia initiation: focus on the Arp2/3 complex and formins. Cell Adh Migr 5(5):402–408. doi: 10.4161/cam.5.5.16971
Giannone G, Dubin-Thaler BJ, Döbereiner H-G, Kieffer N, Bresnick AR, Sheetz MP (2004) Periodic lamellipodial contractions correlate with rearward actin waves. Cell 116:431–443
Ylänne J, Scheffzek K, Young P, Saraste M (2001) Crystal structure of the alpha-actinin rod reveals an extensive torsional twist. Structure 9:597–604. doi:10.1016/S0969-2126(01)00619-0
Nakagawa T, Engler JA, Sheng M (2004) The dynamic turnover and functional roles of α-actinin in dendritic spines. Neuropharmacology 47:734–745. doi:10.1016/j.neuropharm.2004.07.022
Hodges JL, Vilchez SM, Asmussen H, Whitmore LA, Horwitz AR (2014) α-actinin-2 mediates spine morphology and assembly of the post-synaptic density in hippocampal neurons. PLoS ONE 9:e101770. doi:10.1371/journal.pone.0101770
Allen PB, Ouimet CC, Greengard P (1997) Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA 94:9956–9961. doi:10.1073/pnas.94.18.9956
Nakanishi H, Obaishi H, Satoh A, Wada M, Mandai K, Satoh K, Nishioka H, Matsuura Y, Mizoguchi A, Takai Y (1997) Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol 139:951–961
Satoh A, Nakanishi H, Obaishi H, Wada M, Takahashi K, Satoh K, Hirao K, Nishioka H, Hata Y, Al E (1998) Neurabin-II/spinophilin: an actin filament-binding protein with one PDZ domain localized at cadherin-based cell-cell adhesion sites. J Biol Chem 273:3470–3475. doi:10.1074/jbc.273.6.3470
Zito K, Knott G, Shepherd GMG, Shenolikar S, Svoboda K (2004) Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron 44:321–334. doi:10.1016/j.neuron.2004.09.022
Sarrouilhe D, di Tommaso A, Métayé T, Ladeveze V (2006) Spinophilin: from partners to functions. Biochimie 88:1099–1113. doi:10.1016/j.biochi.2006.04.010
Aoki C, Sekino Y, Hanamura K, Fujisawa S, Mahadomrongkul V, Ren Y, Shirao T (2005) Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J Comp Neurol 483:383–402. doi:10.1002/cne.20449
Mikati MA, Grintsevich EE, Reisler E (2013) Drebrin-induced stabilization of actin filaments. J Biol Chem 288:19926–19938. doi:10.1074/jbc.M113.472647
Worth DC, Daly CN, Geraldo S, Oozeer F, Gordon-Weeks PR (2013) Drebrin contains a cryptic F-actin-bundling activity regulated by Cdk5 phosphorylation. J Cell Biol 202:793–806. doi:10.1083/jcb.201303005
Kreis P, Hendricusdottir R, Kay L, Papageorgiou IE, van Diepen M, Mack T, Ryves J, Harwood A, Leslie NR, Kann O, Parsons M, Eickholt BJ (2013) Phosphorylation of the actin binding protein drebrin at S647 is regulated by neuronal activity and PTEN. PLoS ONE 8:1–12. doi:10.1371/journal.pone.0071957
Jayo A, Parsons M (2010) Fascin: a key regulator of cytoskeletal dynamics. Int J Biochem Cell Biol 42:1614–1617. doi:10.1016/j.biocel.2010.06.019
Breitsprecher D, Koestler SA, Chizhov I, Nemethova M, Mueller J, Goode BL, Small JV, Rottner K, Faix J (2011) Cofilin cooperates with fascin to disassemble filopodial actin filaments. J Cell Sci 124:3305–3318. doi:10.1242/jcs.086934
Terry-Lorenzo R, Roadcap D, Otsuka T, Blanpied T, Zamorano P, Garner C, Shenolikar S, Ehlers M (2005) Neurabin/protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol Biol Cell 16:2349–2362. doi:10.1091/mbc.E04-12-1054
Biou V, Brinkhaus H, Malenka RC, Matus A (2008) Interactions between drebrin and Ras regulate dendritic spine plasticity. Eur J Neurosci 27:2847–2859. doi:10.1111/j.1460-9568.2008.06269.x
Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M (1997) Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature 385:439–442. doi:10.1038/385439a0
Shirao T, González-Billault C (2013) Actin filaments and microtubules in dendritic spines. J Neurochem 126:155–164. doi:10.1111/jnc.12313
Mizui T, Sekino Y, Yamazaki H, Ishizuka Y, Takahashi H, Kojima N, Kojima M, Shirao T (2014) Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS ONE 9:e85367. doi:10.1371/journal.pone.0085367
Okamoto K, Bosch M, Hayashi Y (2009) The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag? Physiology (Bethesda) 24:357–366. doi:10.1152/physiol.00029.2009
Lisman J, Yasuda R, Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13:169–182. doi:10.1038/nrn3192
Hell JW (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81:249–265. doi:10.1016/j.neuron.2013.12.024
Okamoto K-I, Narayanan R, Lee SH, Murata K, Hayashi Y (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci USA 104:6418–6423. doi:10.1073/pnas.0701656104
Lin Y-C, Redmond L (2008) CaMKIIbeta binding to stable F-actin in vivo regulates F-actin filament stability. Proc Natl Acad Sci USA 105:15791–15796. doi:10.1073/pnas.0804399105
Kim K, Lakhanpal G, Lu HE, Khan M, Suzuki A, Kato Hayashi M, Narayanan R, Luyben TT, Matsuda T, Nagai T, Blanpied TA, Hayashi Y, Okamoto K (2015) A temporary gating of actin remodeling during synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87:813–826. doi:10.1016/j.neuron.2015.07.023
Lu HE, MacGillavry HD, Frost NA, Blanpied TA (2014) Multiple spatial and kinetic subpopulations of CaMKII in spines and dendrites as resolved by single-molecule tracking PALM. J Neurosci 34:7600–7610. doi:10.1523/JNEUROSCI.4364-13.2014
Wilson CA, Tsuchida MA, Allen GM, Barnhart EL, Applegate KT, Yam PT, Ji L, Keren K, Danuser G, Theriot JA (2010) Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature 465:373–377. doi:10.1038/nature08994
Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, Lippincott-Schwartz J (2011) A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol 13:371–381. doi:10.1038/ncb2205
Yang Q, Zhang X-F, Pollard TD, Forscher P (2012) Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J Cell Biol 197:939–956. doi:10.1083/jcb.201111052
Reymann A-C, Boujemaa-Paterski R, Martiel J-L, Guérin C, Cao W, Chin HF, De La Cruz EM, Théry M, Blanchoin L (2012) Actin network architecture can determine myosin motor activity. Science 336:1310–1314. doi:10.1126/science.1221708
Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, Betzig E, Lippincott-Schwartz J (2014) A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J Cell Biol 205:83–96. doi:10.1083/jcb.201311104
Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF (2005) A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci 25:3379–3388. doi:10.1523/JNEUROSCI.3553-04.2005
Ryu J, Liu L, Wong TP, Wu DC, Burette A, Weinberg R, Wang YT, Sheng M (2006) A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron 49:175–182. doi:10.1016/j.neuron.2005.12.017
Hodges JL, Newell-Litwa K, Asmussen H, Vicente-Manzanares M, Horwitz AR (2011) Myosin IIb activity and phosphorylation status determines dendritic spine and post-synaptic density morphology. PLoS ONE 6:e24149. doi:10.1371/journal.pone.0024149
Rubio MDM, Johnson R, Miller CA, Huganir RL, Rumbaugh G (2011) Regulation of synapse structure and function by distinct myosin II motors. J Neurosci 31:1448–1460. doi:10.1523/JNEUROSCI.3294-10.2011.Regulation
Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, Huganir RL, Muzyczka N, Gall CM, Miller CA, Lynch G, Rumbaugh G (2010) Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67:603–617. doi:10.1016/j.neuron.2010.07.016
Koskinen M, Bertling E, Hotulainen R, Tanhuanpää K, Hotulainen P (2014) Myosin IIb controls actin dynamics underlying the dendritic spine maturation. Mol Cell Neurosci 61:56–64. doi:10.1016/j.mcn.2014.05.008
Qualmann B, Boeckers TM, Jeromin M, Gundelfinger ED, Kessels MM (2004) Linkage of the actin cytoskeleton to the postsynaptic density via direct interactions of Abp1 with the ProSAP/Shank family. J Neurosci 24:2481–2495. doi:10.1523/JNEUROSCI.5479-03.2004
Haeckel A, Ahuja R, Gundelfinger ED, Qualmann B, Kessels MM (2008) The actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head and synapse formation. J Neurosci 28:10031–10044. doi:10.1523/JNEUROSCI.0336-08.2008
Vlachos A, Korkotian E, Schonfeld E, Copanaki E, Deller T, Segal M (2009) Synaptopodin regulates plasticity of dendritic spines in hippocampal neurons. J Neurosci 29:1017–1033. doi:10.1523/JNEUROSCI.5528-08.2009
Korkotian E, Frotscher M, Segal M (2014) Synaptopodin regulates spine plasticity: mediation by calcium stores. J Neurosci 34:11641–11651. doi:10.1523/JNEUROSCI.0381-14.2014
Cueille N, Blanc CT, Popa-Nita S, Kasas S, Catsicas S, Dietler G, Riederer BM (2007) Characterization of MAP1B heavy chain interaction with actin. Brain Res Bull 71:610–618. doi:10.1016/j.brainresbull.2006.12.003
Tortosa E, Montenegro-Venegas C, Benoist M, Hartel S, Gonzalez-Billault C, Esteban JA, Avila J (2011) Microtubule-associated protein 1B (MAP1B) is required for dendritic spine development and synaptic maturation. J Biol Chem 286:40638–40648. doi:10.1074/jbc.M111.271320
Saneyoshi T, Hayashi Y (2012) The Ca(2+) and Rho GTPase signaling pathways underlying activity-dependent actin remodeling at dendritic spines. Cytoskeleton (Hoboken) 69:545–554. doi:10.1002/cm.21037
Lisman JE, Raghavachari S, Tsien RW (2007) The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci 8:597–609. doi:10.1038/nrn2191
Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279(5350):509–514. doi:10.1126/science.279.5350.509
Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701. doi:10.1038/nrm2476
Guilluy C, Garcia-Mata R, Burridge K (2011) Rho protein crosstalk: another social network? Trends Cell Biol 21:718–726. doi:10.1016/j.tcb.2011.08.002
Stankiewicz TR, Linseman DA (2014) Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci 8:314. doi:10.3389/fncel.2014.00314
Fleming IN, Elliott CM, Buchanan FG, Downes CP, Exton JH (1999) Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem 274:12753–12758. doi:10.1074/jbc.274.18.12753
Okabe T, Nakamura T, Nishimura YN, Kohu K, Ohwada S, Morishita Y, Akiyama T (2003) RICS, a novel GTPase-activating protein for Cdc42 and Rac1, is involved in the beta-catenin-N-cadherin and N-methyl-d-aspartate receptor signaling. J Biol Chem 278:9920–9927. doi:10.1074/jbc.M208872200
Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME (2005) The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45:525–538. doi:10.1016/j.neuron.2005.01.024
Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P (2007) Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56:640–656. doi:10.1016/j.neuron.2007.10.005
Grossman SD, Futter M, Snyder GL, Allen PB, Nairn AC, Greengard P, Hsieh-Wilson LC (2004) Spinophilin is phosphorylated by Ca2+/calmodulin-dependent protein kinase II resulting in regulation of its binding to F-actin. J Neurochem 90:317–324. doi:10.1111/j.1471-4159.2004.02491.x
Ryan XP, Alldritt J, Svenningsson P, Allen PB, Wu G-Y, Nairn AC, Greengard P (2005) The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron 47:85–100. doi:10.1016/j.neuron.2005.05.013
Murakoshi H, Wang H, Yasuda R (2011) Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472:100–104. doi:10.1038/nature09823
Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR (2008) Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron 57:94–107. doi:10.1016/j.neuron.2007.11.016
Wayman GA, Lee Y-S, Tokumitsu H, Silva AJ, Silva A, Soderling TR (2008) Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron 59:914–931. doi:10.1016/j.neuron.2008.08.021
Takenawa T, Suetsugu S (2007) The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 8:37–48. doi:10.1038/nrm2069
Derivery E, Gautreau A (2010) Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. BioEssays 32:119–131. doi:10.1002/bies.200900123
Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK (2010) Structure and control of the actin regulatory WAVE complex. Nature 468:533–538. doi:10.1038/nature09623
Riento K, Ridley AJ (2003) Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4:446–456. doi:10.1038/nrm1128
Rane CK, Minden A (2014) P21 activated kinases: structure, regulation, and functions. Small GTPases 5:37–41. doi:10.4161/sgtp.28003
Julian L, Olson MF (2014) Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases 5:e29846. doi:10.4161/sgtp.29846
Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T (2002) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108:233–246
Collingridge GL, Peineau S, Howland JG, Wang YT (2010) Long-term depression in the CNS. Nat Rev Neurosci 11:459–473. doi:10.1038/nrn2867
Hanley JG, Henley JM (2005) PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J 24:3266–3278. doi:10.1038/sj.emboj.7600801
Rocca DL, Amici M, Antoniou A, Blanco Suarez E, Halemani N, Murk K, McGarvey J, Jaafari N, Mellor JR, Collingridge GL, Hanley JG (2013) The small GTPase Arf1 modulates Arp2/3-mediated actin polymerization via PICK1 to regulate synaptic plasticity. Neuron 79:293–307. doi:10.1016/j.neuron.2013.05.003
Rocca DL, Hanley JG (2015) PICK1 links AMPA receptor stimulation to Cdc42. Neurosci Lett 585:155–159. doi:10.1016/j.neulet.2014.11.046
Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G (2009) Coordination of Rho GTPase activities during cell protrusion. Nature 461:99–103. doi:10.1038/nature08242
Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM (2009) A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461:104–108. doi:10.1038/nature08241
Lee SR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R (2009) Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458:299–304. doi:10.1038/nature07842
Van Harreveld A, Fifkova E (1975) Swelling of dendritic spines in the fascia dentata after stimulation of the perforant fibers as a mechanism of post-tetanic potentiation. Exp Neurol 49:736–749. doi:10.1016/0014-4886(75)90055-2
Fifková E, Anderson CL (1981) Stimulation-induced changes in dimensions of stalks of dendritic spines in the dentate molecular layer. Exp Neurol 74:621–627. doi:10.1016/0014-4886(81)90197-7
Desmond N, Levy W (1986) Changes in the postsynaptic density with long-term potentiation in the dentate gyrus. J Comp Neurol 253:476–482. doi:10.1002/cne.902530405
Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4:1086–1092. doi:10.1038/nn736
Patterson MA, Szatmari EM, Yasuda R (2010) AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci USA 107:15951–15956. doi:10.1073/pnas.0913875107
Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003) Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38:447–460
Rochefort NL, Konnerth A (2012) Dendritic spines: from structure to in vivo function. EMBO Rep 13:699–708. doi:10.1038/embor.2012.102
Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, Obata K, Imoto K, Yanagawa Y, Manabe T, Okabe S (2009) Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci 29:7607–7618. doi:10.1523/JNEUROSCI.0707-09.2009
Chen LY, Rex CS, Casale MS, Gall CM, Lynch G (2007) Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci 27:5363–5372. doi:10.1523/JNEUROSCI.0164-07.2007
Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS (2004) Cofilin promotes actin polymerization and defines the direction of cell motility. Science 304:743–746. doi:10.1126/science.1094561
Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N (2012) SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 17:71–84. doi:10.1038/mp.2011.57
Andrianantoandro E, Pollard TD (2006) Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell 24:13–23. doi:10.1016/j.molcel.2006.08.006
Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW (2013) Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci 33:16471–16482. doi:10.1523/JNEUROSCI.0661-13.2013
Yang Y, Wang X-B, Frerking M, Zhou Q (2008) Spine expansion and stabilization associated with long-term potentiation. J Neurosci 28:5740–5751. doi:10.1523/JNEUROSCI.3998-07.2008
Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR (2010) Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci 30:11565–11575. doi:10.1523/JNEUROSCI.1746-10.2010
Tai C-Y, Mysore SP, Chiu C, Schuman EM (2007) Activity-regulated N-cadherin endocytosis. Neuron 54:771–785. doi:10.1016/j.neuron.2007.05.013
Kobielak A, Pasolli HA, Fuchs E (2004) Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol 6:21–30. doi:10.1038/ncb1075
Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng L-H, Colman DR, Miki N (2004) Cadherin activity is required for activity-induced spine remodeling. J Cell Biol 167:961–972. doi:10.1083/jcb.200406030
Wang X, Yang Y, Zhou Q (2007) Independent expression of synaptic and morphological plasticity associated with long-term depression. J Neurosci 27:12419–12429. doi:10.1523/JNEUROSCI.2015-07.2007
He K, Lee A, Song L, Kanold PO, Lee H-K (2011) AMPA receptor subunit GluR1 (GluA1) serine-845 site is involved in synaptic depression but not in spine shrinkage associated with chemical long-term depression. J Neurophysiol 105:1897–1907. doi:10.1152/jn.00913.2010
Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GCR, Matsuzaki M, Kasai H (2013) GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat Neurosci 16:1409–1416. doi:10.1038/nn.3496
Kopec CD, Li B, Wei W, Boehm J, Malinow R (2006) Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci 26:2000–2009. doi:10.1523/JNEUROSCI.3918-05.2006
Kopec CD, Real E, Kessels HW, Malinow R (2007) GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci 27:13706–13718. doi:10.1523/JNEUROSCI.3503-07.2007
Sdrulla AD, Linden DJ (2007) Double dissociation between long-term depression and dendritic spine morphology in cerebellar Purkinje cells. Nat Neurosci 10:546–548. doi:10.1038/nn1889
Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ (2010) ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci 13:1208–1215. doi:10.1038/nn.2634
Kim CH, Lisman JE (1999) A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci 19:4314–4324
Krucker T, Siggins GR, Halpain S (2000) Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA 97:6856–6861. doi:10.1073/pnas.100139797
Ramachandran B, Frey JU (2009) Interfering with the actin network and its effect on long-term potentiation and synaptic tagging in hippocampal CA1 neurons in slices in vitro. J Neurosci 29:12167–12173. doi:10.1523/JNEUROSCI.2045-09.2009
Soderling SH, Langeberg LK, Soderling JA, Davee SM, Simerly R, Raber J, Scott JD (2003) Loss of WAVE-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc Natl Acad Sci USA 100:1723–1728. doi:10.1073/pnas.0438033100
Meng J, Meng Y, Hanna A, Janus C, Jia Z (2005) Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci 25:6641–6650. doi:10.1523/JNEUROSCI.0028-05.2005
Asrar S, Meng Y, Zhou Z, Todorovski Z, Huang WW, Jia Z (2009) Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56:73–80. doi:10.1016/j.neuropharm.2008.06.055
Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, McConnell SK, Palmer TD (2009) A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci 41:409–419. doi:10.1016/j.mcn.2009.04.005
Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, Sassoè-Pognetto M, Al Banchaabouchi M, Giustetto M, Triller A, Choquet D, Witke W (2010) Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J 29:1889–1902. doi:10.1038/emboj.2010.72
Kang J, Park H, Kim E (2016) IRSp53/BAIAP2 in dendritic spine development, NMDA receptor regulation, and psychiatric disorders. Neuropharmacology 100:27–39. doi:10.1016/j.neuropharm.2015.06.019
Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A (2003) CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38:887–898
Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14:285–293. doi:10.1038/nn.2741
De Rubeis S, Pasciuto E, Li KW, Fernández E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJT, Komiyama NH, Grant SGN, Poujol C, Choquet D, Achsel T, Posthuma D, Smit AB, Bagni C (2013) CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79:1169–1182. doi:10.1016/j.neuron.2013.06.039
Derkach VA, Oh MC, Guire ES, Soderling TR (2007) Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8:101–113. doi:10.1038/nrn2055
Lau CG, Takeuchi K, Rodenas-Ruano A, Takayasu Y, Murphy J, Bennett MVL, Zukin RS (2009) Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem Soc Trans 37:1369–1374. doi:10.1042/BST0371369
Lau CG, Zukin RS (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8:413–426. doi:10.1038/nrn2153
Anggono V, Huganir RL (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol 22:461–469. doi:10.1016/j.conb.2011.12.006
Opazo P, Sainlos M, Choquet D (2012) Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol 22:453–460. doi:10.1016/j.conb.2011.10.010
Ladépêche L, Dupuis JP, Groc L (2014) Surface trafficking of NMDA receptors: gathering from a partner to another. Semin Cell Dev Biol 27:3–13. doi:10.1016/j.semcdb.2013.10.005
Hanley JG (2014) Actin-dependent mechanisms in AMPA receptor trafficking. Front Cell Neurosci 8:1–8. doi:10.3389/fncel.2014.00381
Soltau M, Richter D, Kreienkamp H (2002) The insulin receptor substrate IRSp53 links postsynaptic shank1 to the small G-protein cdc42. Mol Cell Neurosci 21:575–583. doi:10.1006/mcne.2002.1201
Park E, Na M, Choi J, Kim S, Lee J-R, Yoon J, Park D, Sheng M, Kim E (2003) The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem 278:19220–19229. doi:10.1074/jbc.M301052200
Soltau M, Berhörster K, Kindler S, Buck F, Richter D, Kreienkamp H-J (2004) Insulin receptor substrate of 53 kDa links postsynaptic shank to PSD-95. J Neurochem 90:659–665. doi:10.1111/j.1471-4159.2004.02523.x
Kuriu T, Inoue A, Bito H, Sobue K, Okabe S (2006) Differential control of postsynaptic density scaffolds via actin-dependent and -independent mechanisms. J Neurosci 26:7693–7706. doi:10.1523/JNEUROSCI.0522-06.2006
Kerr JM, Blanpied TA (2012) Subsynaptic AMPA receptor distribution is acutely regulated by actin-driven reorganization of the postsynaptic density. J Neurosci 32:658–673. doi:10.1523/JNEUROSCI.2927-11.2012
MacGillavry HD, Song Y, Raghavachari S, Blanpied TA (2013) Nanoscale scaffolding domains within the postsynaptic density concentrate synaptic AMPA receptors. Neuron 78:615–622. doi:10.1016/j.neuron.2013.03.009
Zhang W, Benson DL (2001) Stages of synapse development defined by dependence on F-actin. J Neurosci 21:5169–5181
Allison DW, Gelfand VI, Spector I, Craig AM (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18:2423–2436
Renner M, Choquet D, Triller A (2009) Control of the postsynaptic membrane viscosity. J Neurosci 29:2926–2937. doi:10.1523/JNEUROSCI.4445-08.2009
Czondor K, Mondin M, Garcia M, Heine M, Frischknecht R, Choquet D, Sibarita J-B, Thoumine OR (2012) Unified quantitative model of AMPA receptor trafficking at synapses. Proc Natl Acad Sci USA 109:3522–3527. doi:10.1073/pnas.1109818109
Kusters R, Kapitein LC, Hoogenraad CC, Storm C (2013) Shape-induced asymmetric diffusion in dendritic spines allows efficient synaptic AMPA receptor trapping. Biophys J 105:2743–2750. doi:10.1016/j.bpj.2013.11.016
Jewell JL, Luo W, Oh E, Wang Z, Thurmond DC (2008) Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J Biol Chem 283:10716–10726. doi:10.1074/jbc.M709876200
Jaiswal JK, Rivera VM, Simon SM (2009) Exocytosis of post-golgi vesicles is regulated by components of the endocytic machinery. Cell 137:1308–1319. doi:10.1016/j.cell.2009.04.064
Yang Y, Wang X-B, Frerking M, Zhou Q (2008) Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc Natl Acad Sci USA 105:11388–11393. doi:10.1073/pnas.0802978105
Porat-Shliom N, Milberg O, Masedunskas A, Weigert R (2013) Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell Mol Life Sci 70:2099–2121. doi:10.1007/s00018-012-1156-5
Mooren OL, Galletta BJ, Cooper JA (2012) Roles for actin assembly in endocytosis. Annu Rev Biochem 81:661–686. doi:10.1146/annurev-biochem-060910-094416
Kennedy MJ, Davison IG, Robinson CG, Ehlers MD (2010) Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell 141:524–535. doi:10.1016/j.cell.2010.02.042
Jullie D, Choquet D, Perrais D (2014) Recycling endosomes undergo rapid closure of a fusion pore on exocytosis in neuronal dendrites. J Neurosci 34:11106–11118. doi:10.1523/JNEUROSCI.0799-14.2014
Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JTR (2008) An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 57:872–882. doi:10.1016/j.neuron.2008.01.028
Kelly EE, Horgan CP, McCaffrey MW, Young P (2011) The role of endosomal-recycling in long-term potentiation. Cell Mol Life Sci 68:185–194. doi:10.1007/s00018-010-0516-2
Osterweil E, Wells DG, Mooseker MS (2005) A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J Cell Biol 168:329–338. doi:10.1083/jcb.200410091
Wang Z, Edwards JG, Riley N, Provance DW, Karcher R, Li X-D, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD (2008) Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 135:535–548. doi:10.1016/j.cell.2008.09.057
Huber K, Kayser M, Bear M (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288(5469):1254–1257. doi:10.1126/science.288.5469.1254
Nosyreva E, Huber K (2005) Developmental Switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci 25:2992–3001. doi:10.1523/JNEUROSCI.3652-04.2005
Tanaka J-I, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GCR, Kasai H (2008) Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319:1683–1687. doi:10.1126/science.1152864
Holbro N, Grunditz Å, Oertner TG (2009) Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc Natl Acad Sci USA 106:15055–15060. doi:10.1073/pnas.0905110106
Govindarajan A, Israely I, Huang S-Y, Tonegawa S (2011) The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron 69:132–146. doi:10.1016/j.neuron.2010.12.008
Ramiro-Cortés Y, Israely I (2013) Long lasting protein synthesis- and activity-dependent spine shrinkage and elimination after synaptic depression. PLoS ONE 8:e71155. doi:10.1371/journal.pone.0071155
Lynch G, Kramár EA, Babayan AH, Rumbaugh G, Gall CM (2013) Differences between synaptic plasticity thresholds result in new timing rules for maximizing long-term potentiation. Neuropharmacology 64:27–36. doi:10.1016/j.neuropharm.2012.07.006
Redondo RL, Okuno H, Spooner PA, Frenguelli BG, Bito H, Morris RGM (2010) Synaptic tagging and capture: differential role of distinct calcium/calmodulin kinases in protein synthesis-dependent long-term potentiation. J Neurosci 30:4981–4989. doi:10.1523/JNEUROSCI.3140-09.2010
Redondo RL, Morris RGM (2011) Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci 12:17–30. doi:10.1038/nrn2963
Tam J, Cordier GA, Bálint Š, Sandoval Álvarez Á, Borbely JS, Lakadamyali M (2014) A microfluidic platform for correlative live-cell and super-resolution microscopy. PLoS ONE 9:e115512. doi:10.1371/journal.pone.0115512
Jungmann R, Steinhauer C, Scheible M, Kuzyk A, Tinnefeld P, Simmel FC (2010) Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett 10:4756–4761. doi:10.1021/nl103427w
Jungmann R, Avendaño MS, Woehrstein JB, Dai M, Shih WM, Yin P (2014) Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods 11:313–318. doi:10.1038/nmeth.2835
Tam J, Cordier GA, Borbely JS, Sandoval Álvarez Á, Lakadamyali M (2014) Cross-talk-free multi-color STORM imaging using a single fluorophore. PLoS ONE 9:e101772. doi:10.1371/journal.pone.0101772
Zhang Z, Kenny SJ, Hauser M, Li W, Xu K (2015) Ultrahigh-throughput resolved super-resolution microscopy. Nat Methods. doi:10.1038/nmeth.3528
Meyer D, Bonhoeffer T, Scheuss V (2014) Balance and stability of synaptic structures during synaptic plasticity. Neuron 82:430–443. doi:10.1016/j.neuron.2014.02.031
Xu K, Babcock HP, Zhuang X (2012) Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods 9:185–188. doi:10.1038/nmeth.1841
Galland R, Grenci G, Aravind A, Viasnoff V, Studer V, Sibarita J-B (2015) 3D high- and super-resolution imaging using single-objective SPIM. Nat Methods 12:641–644. doi:10.1038/nmeth.3402
Dani A, Huang B, Bergan J, Dulac C, Zhuang X (2010) Superresolution imaging of chemical synapses in the brain. Neuron 68:843–856. doi:10.1016/j.neuron.2010.11.021
Vangindertael J, Beets I, Rocha S, Dedecker P, Schoofs L, Vanhoorelbeeke K, Hofkens J, Mizuno H (2015) Super-resolution mapping of glutamate receptors in C. elegans by confocal correlated PALM. Sci Rep 5:13532. doi:10.1038/srep13532
Urban NT, Willig KI, Hell SW, Nägerl UV (2011) STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J 101:1277–1284. doi:10.1016/j.bpj.2011.07.027
Hofmann M, Eggeling C, Jakobs S, Hell SW (2005) Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci USA 102:17565–17569. doi:10.1073/pnas.0506010102
Testa I, Urban NT, Jakobs S, Eggeling C, Willig KI, Hell SW (2012) Nanoscopy of living brain slices with low light levels. Neuron 75:992–1000. doi:10.1016/j.neuron.2012.07.028
Kner P, Chhun BB, Griffis ER, Winoto L, Gustafsson MGL (2009) Super-resolution video microscopy of live cells by structured illumination. Nat Methods 6:339–342. doi:10.1038/nmeth.1324
Shao L, Kner P, Rego EH, Gustafsson MGL (2011) Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods 8:1044–1046. doi:10.1038/nmeth.1734
Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ (2013) Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Methods 10:413–420. doi:10.1038/nmeth.2434
Chen B-C, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA 3rd, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Reymann A-C, Bembenek JN, Bohme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E, Reymann A-C, Bohme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E (2014) Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346(6208):1257998. doi:10.1126/science.1257998
Li D, Shao L, Chen B, Zhang X, Zhang M, Moses B, Milkie DE, Beach JR, Hammer J 3rd, Pasham M, Kirchhausen T, Baird MA, Davidson MW, Xu P, Betzig E (2015) ADVANCED IMAGING. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 349(6251):aab3500. doi:10.1126/science.aab3500
Keller PJ, Ahrens MB (2015) Visualizing whole-brain activity and development at the single-cell level using light-sheet microscopy. Neuron 85:462–483. doi:10.1016/j.neuron.2014.12.039
Benson DL, Huntley GW (2012) Synapse adhesion: a dynamic equilibrium conferring stability and flexibility. Curr Opin Neurobiol 22:397–404. doi:10.1016/j.conb.2011.09.011
van Bergeijk P, Adrian M, Hoogenraad CC, Kapitein LC (2015) Optogenetic control of organelle transport and positioning. Nature 518:111–114. doi:10.1038/nature14128
Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ, Tucker CL (2014) An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun 5:4925. doi:10.1038/ncomms5925
Yuste R (2015) From the neuron doctrine to neural networks. Nat Rev Neurosci 16:487–497. doi:10.1038/nrn3962
Markram H, Muller E, Ramaswamy S, Reimann MW, Abdellah M, Sanchez CA, Ailamaki A, Alonso-Nanclares L, Antille N, Arsever S, Kahou GAA, Berger TK, Bilgili A, Buncic N, Chalimourda A, Chindemi G, Courcol J-D, Delalondre F, Delattre V, Druckmann S, Dumusc R, Dynes J, Eilemann S, Gal E, Gevaert ME, Ghobril J-P, Gidon A, Graham JW, Gupta A, Haenel V, Hay E, Heinis T, Hernando JB, Hines M, Kanari L, Keller D, Kenyon J, Khazen G, Kim Y, King JG, Kisvarday Z, Kumbhar P, Lasserre S, Le Bé J-V, Magalhães BRC, Merchán-Pérez A, Meystre J, Morrice BR, Muller J, Muñoz-Céspedes A, Muralidhar S, Muthurasa K, Nachbaur D, Newton TH, Nolte M, Ovcharenko A, Palacios J, Pastor L, Perin R, Ranjan R, Riachi I, Rodríguez J-R, Riquelme JL, Rössert C, Sfyrakis K, Shi Y, Shillcock JC, Silberberg G, Silva R, Tauheed F, Telefont M, Toledo-Rodriguez M, Tränkler T, Van Geit W, Díaz JV, Walker R, Wang Y, Zaninetta SM, DeFelipe J, Hill SL, Segev I, Schürmann F (2015) Reconstruction and simulation of neocortical microcircuitry. Cell 163:456–492. doi:10.1016/j.cell.2015.09.029
Acknowledgments
We would like to thank Dr. Harold D. McGillavry and Dr Laura F. Gumy for helpful comments on the manuscript. We acknowledge financial support from the French Ministry of Research and CNRS, ANR grant Nanomotility, LabEx BRAIN, Conseil Régional Aquitaine, Fondation pour la Recherche Médicale, and from Marie Skłodowska-Curie fellowship to Anaël Chazeau.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chazeau, A., Giannone, G. Organization and dynamics of the actin cytoskeleton during dendritic spine morphological remodeling. Cell. Mol. Life Sci. 73, 3053–3073 (2016). https://doi.org/10.1007/s00018-016-2214-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2214-1