
Abstract
Necroptosis has been extensively studied recently, and the receptor-interacting kinase 3 (RIP3 or RIPK3) and its substrate, the pseudokinase mixed lineage kinase domain-like protein, have been discovered to be core components of this process. Classical necroptosis requires RIP1 (or RIPK1) for the activation of RIP3 through the induction of RIP1/RIP3 necrosomes. Increasing evidence from genetic and pharmacological studies has been expanding the view that necroptosis plays important roles in the etiology and/or progression of many human diseases, such as pancreatitis, ischemic injury, and neurodegenerative diseases, among others. Ongoing progress in translational research about necroptosis has highlighted the increasingly important need for the identification of biomarkers for use in disease diagnosis, monitoring, and drug development. This review presents a discussion of the current status of biomarkers that can be used to detect necroptosis both in vitro and in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
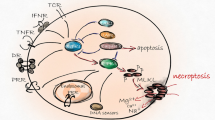
Necroptosis signaling
Necrotic cell death characterized by disrupted plasma membranes has been observed in a variety of physiological and pathological processes, including mammalian development and tissue damage [1–3]. Receptor-interacting protein kinase-3 (RIP3 or RIPK3) [4–6] and its substrate, the mixed lineage kinase domain-like protein (MLKL) [7, 8], are emerging as core molecules that regulate necroptosis. This regulated form of necrosis is induced by multiple triggers, including the tumor necrosis factor (TNF) family of cytokines, such as TNF, TRAIL, and FasL; the activated Toll-like receptors (TLRs), such as TLR3 and TLR4 [9, 10]; type I interferons [11]; and certain pathogens [12–14]. The best characterized necroptosis pathway is triggered by TNF, and this process requires the protein kinase RIP1 (or RIPK1) [15, 16] (Fig. 1). In TNF-triggered necrosis, RIP1 and RIP3 form a protein complex, termed necrosome [17], that is formed through the RIP homotypic interaction motif (RHIM) domains of both proteins [18]. Necrosome formation leads to the activation of RIP3. Deubiquitination of RIP1 by cylindromatosis (CYLD) is known to be critical for necrosome assembly and activation [19, 20]. Activated human RIP3 (hRIP3) is phosphorylated at Ser227, and can subsequently phosphorylate hMLKL at its Thr357 and Ser358 sites. The phosphorylation of MLKL triggers oligomerization and membrane localization of MLKL, eventually resulting in the induction of membrane rupture [21–23]. Similar to the situation in RIP1 activation, RHIM-containing proteins, such as TRIF, DAI, and ICP6, have been shown to associate with RIP3 in response to the initiating signals triggered by, respectively, TLR3/4 [9], M45/vIRA mutant murine cytomegalovirus (MCMV) [24], and human herpes simplex virus type 1 (HSV-1) [13, 14]. This type of RHIM-dependent interaction is essential for RIP3 kinase activation. Thus, RIP3 is a central signal-transducing element in necroptosis that receives upstream signals and, further, transduces the necrosis signal by phosphorylating MLKL.

Signaling pathway of necroptosis. In TNF-triggered necroptosis of human cells, RIP1 is deubiquitinated by CYLD and disassociated from TNF receptor 1 (TNFR1). RIP1 subsequently binds to RIP3 to form a protein complex, termed a necrosome, leading to the activation of both protein kinases. This process results in autophosphorylation of RIP1 at the S14/15 and S166 sites, and autophosphorylation of RIP3 at S227. Activated RIP3 recruits MLKL to the necrosome and phosphorylates MLKL at the T357 and S358 sites. Phosphorylated MLKL oligomerizes and translocates to the plasma membrane. 7N-1 and GSK’963 inhibit RIP1 kinase activity and necroptosis. GSK’872 and GSK’843 block RIP3 kinase activity and RIP3-dependent necrosis. NSA blocks MLKL-dependent necrosis by preventing the membrane translocation of MLKL; NSA has no effect on MLKL phosphorylation or oligomerization
Necroptosis-associated diseases
A number of studies assessing the in vivo significance of necroptosis have been conducted using RIP3-deficient mice and MLKL-deficient mice. Despite the neonatal lethality of RIP1 knockout mice [25], a RIP1 kinase inhibitor necrostatin-1 (Nec-1) has been widely utilized as a valuable tool to dissect the role of RIP1-dependent necroptosis in vivo [26].
Necroptosis has been shown to play prominent roles in various pathophysiological conditions, including acute pancreatitis [4, 6, 27], ischemic injury [16, 28–30], retinal detachment [31], inflammatory bowel disease (IBD) [32–34], atherosclerosis [35, 36], liver injury [37, 38], systemic inflammatory syndrome [39], retinal degeneration [40, 41], and neuron degeneration [42, 43]. Increased RIP3 protein levels have been detected in the damaged tissue of mouse models for acute pancreatitis [4], IBD [32], myocardial ischemia [30], dsRNA-induced retinal degeneration [41], and ethanol-induced liver injury [38]. The levels of both RIP3 and MLKL are elevated in inflamed tissues of IBD patients [34]. A recent study showed that the levels of both RIP1 and the phosphorylated forms of RIP1 are elevated in a mouse model of oligodendrocyte degeneration [43]. Of note, activation of RIP1, RIP3, and MLKL are associated with their phosphorylation. These phosphorylation events are key steps in the activation of necroptosis. Thus, activated forms of RIP1, RIP3, and MLKL represent optimal biomarkers for both the analysis of necroptosis and the diagnosis of diseases associated with necrotic injury.
Biomarkers of necroptosis for in vitro analysis
To date, biomarker research in the necroptosis field has been primarily focused on the critical molecular events involved in this form of cell death (Table 1).
Phosphorylation of RIP1, RIP3, and MLKL are the hallmarks of the induction of necroptosis. Several autophosphorylation sites of RIP1 have been identified in in vitro kinase assays, including Ser14/15, Ser20, Ser161, and Ser166 [26]. Recently, two anti-phospho-RIP1 antibodies have been developed as makers for the activation of RIP1; these antibodies specifically recognize the phospho-Ser166 and phospho-Ser14/15 of hRIP1, respectively [43, 44]. The anti-phospho-Ser166 RIP1 antibody has been verified by phospho-RIP1 ELISA [44] and immunoblotting analysis [43]. Upon the activation of necroptosis, autophosphorylation of RIP1 at Ser166 is induced and detected by the anti-phospho-Ser166 RIP1 antibody. This phosphorylation event is prevented by 7-Cl-O-Nec-1(7N-1), an optimized analog of Nec-1 [43]. Moreover, immunoblotting analysis has shown that both anti-phospho-Ser166 and anti-phospho-Ser14/15 RIP1 antibodies detect increased signals in brain samples from patients with multiple sclerosis. Given that the anti-phospho-Ser14/15 RIP1 antibody also recognizes the phospho-Ser14/15 of mouse RIP1 (mRIP1), it has been used to measure the elevated phosphorylation of Ser14/15 RIP1 in the mouse model of oligodendrocyte degeneration [43]. In this disease model, 7N-1 can block autophosphorylation of Ser14/15 of RIP1 and prevent oligodendrocytes from undergoing necroptosis. In addition to Nec-1, other RIP1 kinase inhibitors that can block autophosphorylation of RIP1 have been found. These include GSK’963 [45], ponatinib [46], and pazopanib [46]. The activation of hRIP3 is marked by autophosphorylation at Ser227 [7], and this event is conserved in mRIP3 (Ser 232). Furthermore, Thr231 of mRIP3 has been found to be phosphorylated during the activation of necroptosis [47]. RIP3 kinase inhibitors, such as GSK’872 [10], GSK’843 [10], dabrafenib [48], and ponatinib [46, 49], can block RIP3 autophosphorylation. The phosphorylation of Ser227 in hRIP3 and the phosphorylation of Thr231/Ser232 in mRIP3 are required for their recruitment of the corresponding MLKL [7, 47]. A specific antibody recognizing the phospho-Ser227 of hRIP3 has been successfully used as a biomarker for necrosis that was triggered by TNF or the forced polymerization of RIP3 [50]. Recently, an anti-phospho-Ser232 mRIP3 antibody has been shown to recognize the activation of mRIP3 in both TNF- and TLR-induced necrosis [36]. Activated hRIP3 directly phosphorylates hMLKL at the Thr357 and Ser358 sites, while the activated form of mRIP3 phosphorylates mMLKL at Ser358 [7]. Anti-phospho-Thr357/Ser358 hMLKL and anti-phospho-Ser358 mMLKL antibodies have been developed; they exhibit specific recognition of phosphorylated hMLKL and mMLKL, respectively, upon the activation of necroptosis [22, 36]. All of these antibodies that specifically detect phosphorylated RIP1, RIP3, or MLKL have been recognized as useful biomarkers for the characterization of necroptosis in vitro.
In addition, the interaction between RIP3 and a RHIM-containing protein, such as RIP1, and the formation of the RIP3/MLKL complex are essential events for the activation of necrosis. During necroptosis, the RIP1/RIP3 necrosome forms an amyloid-like signaling complex through interactions that depend on the RHIM domains of both proteins [29]. Nec-1 is able to prevent the formation and activation of RIP1/RIP3 complexes [4, 26]. Therefore, the status of the RIP1/RIP3 complex can also be understood as a biomarker for necroptosis. Furthermore, it is well known that RIP3/MLKL complex formation is induced upon the activation of RIP3. RIP3-mediated phosphorylation of MLKL drives oligomerization of MLKL, which elicits MLKL translocation to membranes to mediate necrotic cell death [21–23]. During necroptosis, the oligomerized MLKL can also be detected at a large size of around 160–250 kDa on non-reducing SDS-PAGE gels, indicating the activation of necroptosis [21–23]. The MLKL inhibitor necrosulfonamide (NSA), which targets Cys86 of MLKL, has no effect on MLKL phosphorylation and oligomerization, but significantly inhibits its membrane translocation [7, 22]. Recently, a small molecule has been found to interact with MLKL and can block MLKL membrane translocation [51]. In addition to the formation of the RIP3/MLKL complex and phosphorylation of MLKL, both the oligomerization of MLKL and the membrane localization of MLKL are indicative of the activation of necroptosis and can thus be considered as markers for necroptosis in vitro.
Biomarkers of necroptosis for disease diagnosis
Immunohistochemistry (IHC)-based biomarkers have been extensively used in animal models and clinical studies. The execution of necroptosis is dependent on a kinase cascade that results in the specific phosphorylation on RIP1, RIP3, and MLKL. Therefore, IHC-based assays using antibodies that recognize these specific phosphorylation sites would be ideal for detecting necroptosis in vivo. However, most of the available antibodies recognize these specific phosphorylation sites are only suitable for immunoblotting. Luckily, a monoclonal antibody against phospho-Thr357/Ser358 of hMLKL has been shown to be very useful in IHC-based measurement of necroptosis in vivo; this antibody has been especially useful in monitoring human disease tissues exhibiting necroptosis. With this antibody, Wang et al. found that strong IHC staining of phospho-MLKL occurred with hepatocytes of liver biopsy tissue from patients with drug-induced liver injury (DILI) [22]. Recently, this monoclonal antibody has also been used to show the activation of necroptosis in the cortical lesions of samples from human multiple sclerosis patients [43]. In addition, in an animal model of atherosclerosis disease, the antibody recognizing phospho-Ser232 of mRIP3 has been used to show increased necroptosis in the non-cellular cavity areas of atherosclerotic plaques in apolipoprotein E (ApoE)-knockout mice [36]. Taken together, these phospho-specific antibodies for IHC could be useful biomarkers for assessing necroptosis in human and animal tissues.
Summary
In the past few years, the combined use of multiple tools, including RIP1 inhibitors, and RIP3 and MLKL knockout mice, has contributed greatly to the identification of necroptosis-related pathologies. However, due to nonspecific effects of RIP1 inhibitors and the pleiotropic roles of RIP1 and RIP3 in immunology and apoptosis, there is an obvious need for more biomarkers with increased specificity that can be used to definitively evaluate the contribution of necroptosis to the etiology and/or progression of these pathologies. To this end, increasing efforts have been focused on both the development of phospho-specific antibodies for the detection of the activated forms of RIP1, RIP3, and MLKL, and the development of specific inhibitors of these regulators of necroptosis. Further studies to identify novel biomarkers of necroptosis and to develop ideal tools for the precise characterization of necroptosis in various animal models and human disease samples will provide crucial insight into the diagnosis, treatment, and monitoring of necroptosis-associated diseases.
References
Zhou W, Yuan J (2014) Necroptosis in health and diseases. Semin Cell Dev Biol 35:14–23
Sun L, Wang X (2014) A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem Sci 39:587–593
Jouan-Lanhouet S et al (2014) Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 35:2–13
He S et al (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137:1100–1111
Cho YS et al (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137:1112–1123
Zhang DW et al (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336
Sun L et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227
Zhao J et al (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 109:5322–5327
He S et al (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 108:20054–20059
Kaiser WJ et al (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288:31268–31279
Robinson N et al (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13:954–962
Upton JW, Kaiser WJ, Mocarski ES (2010) Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7:302–313
Wang X et al (2014) Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA 111:15438–15443
Huang Z, Wu SQ, Liang Y (2015) RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17:229–242
Holler N et al (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495
Degterev A et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112–119
Declercq W, Vanden Berghe T, Vandenabeele P (2009) RIP kinases at the crossroads of cell death and survival. Cell 138:229–232
Sun X et al (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 277:9505–9511
Hitomi J et al (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323
Moquin DM, McQuade T, Chan FK (2013) CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8:e76841
Cai Z et al (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16:55–65
Wang H et al (2014) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54:133–146
Chen X et al (2014) Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24:105–121
Upton JW, Kaiser WJ, Mocarski ES (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–297
Kelliher MA et al (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8:297–303
Degterev A et al (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313–321
Wu J et al (2013) Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23:994–1006
Linkermann A et al (2012) Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81:751–761
Oerlemans MI et al (2012) Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol 107:270
Luedde M et al (2014) RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res 103:206–216
Trichonas G et al (2010) Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA 107:21695–21700
Welz PS et al (2011) FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477:330–334
Gunther C et al (2011) Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477:335–339
Pierdomenico M et al (2014) Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol 109:279–287
Lin J et al (2013) A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep 3:200–210
Meng L, Jin W, Wang X (2015) RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci USA 112:11007–11012
Ramachandran A et al (2013) Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 58:2099–2108
Roychowdhury S et al (2013) Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57:1773–1783
Duprez L et al (2011) RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35:908–918
Murakami Y et al (2012) Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci USA 109:14598–14603
Murakami Y et al (2014) Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ 21:270–277
Re DB et al (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81:1001–1008
Ofengeim D et al (2015) Activation of necroptosis in multiple sclerosis. Cell Rep 10:1836–1849
Berger SB et al (2014) Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol 192:5476–5480
Berger SB et al (2015) Characterization of GSK′963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov 1:15009
Fauster A et al (2015) A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 6:e1767
Chen W et al (2013) Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem 288:16247–16261
Li JX et al (2014) The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5:e1278
Najjar M et al (2015) Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep 10:1850–1860
Li D et al (2015) A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA 112:5017–5022
Hildebrand JM et al (2014) Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111:15072–15077
Acknowledgments
The authors want to thank Dr. Xiaodong Wang for his continuous support. This work was supported by National Basic Science 973 Grants (2013CB910102 to S.H., 2013CB530805 to Z.S. and 2012CB837502 to S.H.), the National Natural Science Foundation of China (31222036, 31471303, 81571385), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Beijing Nova Program (Z121102002512076) to H.S and Special Research Foundation of State Key Laboratory of Medical Genomics.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
He, S., Huang, S. & Shen, Z. Biomarkers for the detection of necroptosis. Cell. Mol. Life Sci. 73, 2177–2181 (2016). https://doi.org/10.1007/s00018-016-2192-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2192-3