
Abstract
During the decade the importance of regulated necrotic cell death in several pathologies has been increasingly recognized. Intensified research has uncovered many pathways involved in regulated necrosis (RN) that has resulted in several neologisms. The best characterized form of RN is induced by receptor-interacting protein kinase-1 and -3, coined as necroptosis. Unfortunately, until now there are no positive and distinguishing markers available for any of these forms of regulated necrosis, let alone a single marker that identifies regulated necrosis from apoptosis. Here we present an overview of different methods that can be used to discriminate apoptosis from necrosis, more in particular necroptosis, both in vivo and in vitro. Methods for necrotic cell death detection are summarized, and potential pitfalls of these methods are briefly discussed. Basically, a sequential approach is proposed to determine the cell death modality by following a decision tree, allowing discrimination between apoptosis and different forms of RN. Finally, pharmacological and transgenic methods for dissecting subroutines in RN are mentioned.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
18.1 Introduction
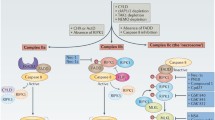
Initially, it was assumed that apoptosis was the only regulated mode of cell death (Kerr et al. 1972), while necrosis according to standard textbooks was considered to be accidental following physicochemical injury and was essentially unregulated (Dave et al. 2012). Hence it was largely disregarded as a phenomenon that could be specifically targeted, until the first evidence of its molecular regulation arose (Laster et al. 1988; Schulze-Osthoff et al. 1993; Vercammen et al. 1998a, b). Later on, the discovery of receptor-interacting protein kinase-1 (RIPK1) and RIPK3 as key mediators of necrotic cell death (Holler et al. 2000; Cho et al. 2009; Zhang et al. 2009), the identification of chemical inhibitors of necrotic cell death and its targets (Degterev et al. 2005, 2008; Wang et al. 2007; You et al. 2008), and the finding of pathologies associated with regulated necrosis (Degterev et al. 2005; You et al. 2008; Takahashi et al. 2012) resulted in a general acceptance of necrosis being also the result of a molecularly regulated process (Vandenabeele et al. 2010). One of the best studied forms of regulated necrosis (RN) is initiated by the kinase activity of RIPK1 and -3 and has been coined necroptosis. However, the discovery of the role of RIPK1 and RIPK3 in necroptosis also allowed the distinction of other types of RN that could not be inhibited by the RIPK1 inhibitor necrostatin-1 (Nec-1), such as H2O2-induced necrosis (Vanden Berghe et al. 2010), MNNG-induced PARP-hyperactivation (parthanatos) (Xu et al. 2010; Sosna et al. 2013), and ferroptosis (Dixon et al. 2012). Due to the complexity of cell death signaling and the existence of many deviations, switches, and overlapping mechanisms, there is currently not a single method that at once discriminates between apoptosis and RN. To determine how cells die, we propose to follow a flow chart (Fig. 18.1), which covers three subsequent steps: (I) determining cell death rate and sensitivity, (II) observing the morphological features, and (III) analyzing the molecular signaling events involved. We discuss different approaches for each of these three main steps in further detail in this chapter. For detailed protocols on these methods, we refer to a more technical methodology paper (Vanden Berghe et al. 2013).

Determination of cell death type following a decision tree. (I) Determine the cell death rate and sensitivity by measuring metabolic activity or by a colorimetric or a fluorometric dye exclusion method (Sect. 2). (II) Analyze the morphology of the dying cells by light or electron microscopy in function of time (Sect. 3.1), and characterize other biochemical events by multiplex methods. aApoptotic morphology is described in Fig. 18.2d–f. bNecrotic morphology is described in Fig. 18.2a–c. (III) Dependent on the activation of CASP3 two different (but partially overlapping) decision trees can be followed (Sects. 3 and 4). The decision tree for apoptosis determines which initiator caspase is involved and whether the apoptosis is dependent on RIPK1 or -3. The decision tree for necrosis analyzes first the involvement of caspase-1 or -11, which could be indicative for pyroptosis. This is followed by determining the involvement of RIPK1, RIPK3, and MLKL, which is indicative for necroptosis. If cell death occurs independent of caspase-1, caspase-11, RIPK1, RIPK3, and MLKL, other mediators of RN (mentioned in Table 18.3) should be analyzed using pharmacological and genetic approaches. #The observation of nuclear condensation and fragmentation during apoptosis should be extended by an approach to analyze DNA fragmentation (Sect. 3.2). *Fluorometric caspase activation assays and FRET/luciferase reporter systems should be interpreted with care, because the catalytic activity of caspases may be more influential in a cellular context than their supposed substrate preference. As such, specificity of the fluorometric activation assay or FRET/luciferase reporter systems should be validated after knockdown of the target caspase in the same cellular model and with the same stimulus
18.2 Three Steps to Determine Cell Death Modality
18.2.1 Detection of Cell Death (Step I)
The first step in analyzing cell death processes is to determine the cell death rate and sensitivity (Fig. 18.1, part I). Traditionally, cell survival, clonogenic, and membrane permeability assays are used to assess the cell death rate. Cell survival assays measure metabolic activity such as mitochondrial succinate dehydrogenase activity using a colorimetric substrate, while clonogenic assays typically stain colonies of cells that expanded from cells that are resistant to or recovered from a cell death challenge and retained the proliferation potential. Therefore, senescent cells or cells arrested in G1 or G2 will not score in a clonogenicity assay, although the cells did not undergo cell death. Regarding metabolic measurements, some drugs or targeted genes may affect metabolism or proliferation without inducing cell death. Also these conditions may lead to misinterpretation using metabolic readouts without really reflecting cell death induction. Therefore, membrane permeability assays are more reliable, as they measure the end stage of the cell death process itself, viz., cell membrane rupture. Generally, the available equipment, cell type, scientific question, and scale of the experiment all contribute to the final choice of cell death assay (Table 18.1).
18.2.1.1 Reduction of Tetrazolium Salts Measured by Spectrophotometry
This assay measures mitochondrial succinate dehydrogenase activity in cells, which reflects cell viability. Succinate dehydrogenase is part of the Krebs cycle and complex II at the inner mitochondrial membrane. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) is a pale yellow substrate that is reduced by succinate dehydrogenase in living cells and reverted to a dark blue formazan product. Alternative substrates, such as (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) (XTT), 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxyme-thoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and water-soluble tetrazolium salts (WSTs), are also often used. They have advantages over the non-soluble MTT in that they can be directly quantified and do not need dissolving in 10 % SDS/HCl. The major disadvantage of these viability assays, especially at the early stages of apoptotic cell death, is that dead cells often still have partially intact succinate dehydrogenase activity, which is erroneously scored as survival. Some conditions and some compounds may also modulate mitochondrial respiration or the Krebs cycle, resulting in increased or decreased succinate dehydrogenase activity, misleadingly appearing to be cell death-modulating conditions. For instance, resveratrol enhances the MTT-reducing ability of cells, although it induces growth arrest (Bernhard et al. 2003). MTT conversion is also affected by cell density and exhaustion of medium, which can result in underestimation of cell viability. Media containing phenol red also absorb light in the same wavelengths as the MTT/MTS/WST products, at ~750 nm (Pick and Keisari 1980). This can elevate background signals and hence reduce the dynamic range of the assay. The latter issue can be circumvented by using media free of phenol red or by subtracting background absorbance measured at 630–690 nm.
18.2.1.2 Dye Exclusion Methods
Trypan blue is a naphthalene sulfonate, impermeable to intact cell membranes and named after its ability to kill trypanosomes in animal models (Wainwright 2010). The negatively charged dye stains dead cells blue, which can be visualized under a light microscope and counted with a hemocytometer. Note that viable cells may stain light blue after more than 5-min exposure to the dye and may even die after 30 min of exposure to trypan blue (Awad et al. 2011). Although this method is most commonly used as a cell viability check during routine cell culture maintenance, devices for (semi)automated measurements of cell death using trypan blue are currently available (Louis and Siegel 2011). A major advantage of this method is its accessibility: a light microscope is all that is required. Unfortunately, the time-dependent toxicity of the dye may result in an overestimation of cell death.
Several types of fluorescent dyes exist that are excluded by an intact cell membrane. Such dyes can intercalate with DNA, can covalently bind to cellular proteins (amine-reactive dyes), or are trapped in viable cells after loading the dye ester and subsequent enzymatic processing (e.g., acetomethoxy-calcein). Although most DNA-intercalating dyes are non-fixable (except ethidium monoazide), they typically display an increase in intensity upon binding to DNA (up to 1,000-fold). As such, they do not require sequential washing steps and can even be added directly to the medium in multiwell-based assays. DNA-intercalating dyes include Sytox® probes, propidium iodide (PI), and 7-amino actinomycin D (7-AAD) and are all ±1 kDa in size. Fixation of PI-stained cells has been reported (Shen et al. 2011) but requires intensive washing before fixation. The amine-reactive dyes, such as LIVE/DEAD® stains, are routinely used with fixation procedures but also require washing steps before fixation and have a lower signal-to-noise ratio (50- to 80-fold increase of signal in dead cells versus living cells) as compared to unfixed DNA-intercalating dyes (Perfetto et al. 2006).
Fluorescent exclusion dyes can be analyzed by image- or flow-based cytometers or fluorescent plate readers. The latter approach may involve risks: certain chemical compounds may quench the fluorescent exclusion dyes or they may be autofluorescent, resulting in, respectively, under- or overestimation of the actual cell death. It is therefore advisable to visually inspect fluorescent cell death assays by light/fluorescent microscopy as well or to complement them by a cell survival assay. Image- or flow-based cytometers have the advantage that they allow to monitor other parameters simultaneously, but they are more complicated to use and to extract data. Cytometric methods will be discussed in more detail in the following sections.
18.2.1.3 Determining Cell Death In Vivo
Few studies address the direct visualization of necrotic cell death in vivo using fluorescent techniques (Cordeiro et al. 2010), because fluorescent light penetrates poorly through tissues of a living animal and because most fluorescent membrane exclusion dyes are not fixable. Therefore, most reports on direct measurement of necrotic cell death in vivo are based on radiolabeling (Table 18.2). These markers have been developed for a clinical setting, so often it is not clear whether they detect cell death in general or specifically apoptosis or necrosis. However, some probes exist that accumulate in regions that correspond with caspase activation in postmortem examination. These either bind to activated caspases (Smith et al. 2008; Cohen et al. 2009; Grimberg et al. 2009; Nguyen et al. 2009) or exposed phosphatidyl serine (Blankenberg et al. 1999). It is important to note that caspase activation also occurs in physiological settings without involvement of cell death, such as inflammation (Lamkanfi et al. 2007; Kuranaga 2012). As such, confirmation of cell death occurring in these regions where caspases are active is required. On the other hand, probes have been described that accumulate in necrotic regions induced by various insults (De Saint-Hubert et al. 2009) but not in cell death regions induced by apoptosis-inducing stimuli (Prinsen et al. 2011). Some of these markers, such as the metalloporphyrins can also be detected with nonradioactive nuclear magnetic resonance (NMR) (Ni et al. 1997). Diffusion-weighted magnetic resonance imaging can also detect cell death without injecting contrast markers (Chiaradia et al. 2013), but no real distinction between apoptotic and necrotic cell regions can be made (Wendland et al. 2008).
Cell death can also be measured in vivo by indirect markers, such as the release of intracellular content in the blood flow. These can be general cell death markers such as the release of cytosolic lactate dehydrogenase, lysosomal hexosaminidase (Kuenzler et al. 2002), and mitochondrial DNA (Zhang et al. 2010; Duprez et al. 2011; Krysko et al. 2011) or biomarkers supposed to be more specific for necrosis such as HMGB1 (Scaffidi et al. 2002), IL-33 (Luthi et al. 2009), IL-1α (Cohen et al. 2010), and cyclophilin A (Christofferson and Yuan 2010). Note that HMGB1 can also be released by macrophages in the absence of cell death (Andersson et al. 2000). Moreover, in the case of massive apoptosis with insufficient phagocytic capacity, apoptotic cells can lose their confinement and evolve to secondary necrosis, during which intracellular content is also released. However, in contrast to necrosis, cells that undergo secondary necrosis did undergo a wave of caspase-mediated proteolysis resulting in the cleavage of certain cellular proteins.
18.2.2 Discrimination of Apoptosis Versus Necrosis (Step II)
Next to the detection of cell death, we want to define the cell death modality. Typically, morphologic examination (Fig. 18.1, part II Morphology), followed by some biochemical approaches (Fig. 18.1, part III Molecular signaling events), will allow to pinpoint whether the cells die by apoptosis, necroptosis, or another form of RN. Generally, it is important to combine several methods from each of the main categories described below in order to reach a valid conclusion. In addition, we want to emphasize that cells should be analyzed at different time points, because these cell death features change over time: absence of apoptotic hallmarks should be confirmed over the entire course of the cell death process for a validation of necrotic cell death. Moreover, some inhibitors may block one cell death modality (necroptosis or apoptosis) and allow a switch to another cell death modality (e.g., the pan-caspase inhibitor zVAD-fmk blocks Fas-induced apoptosis in L929 cells, but allows a switch to necrosis) (Vanden Berghe et al. 2003, 2010). Many more examples of such cell death modality switches have been reported (Vanlangenakker et al. 2011a; Dondelinger et al. 2013; Remijsen et al. 2014). This illustrates that although the extent of cell death may have not been changed at the endpoint, the type of cell death clearly is.
18.2.2.1 Morphologic Analysis by Light Microscopy
Differences in morphology between apoptotic and necrotic cell death were the initial clues that different cellular processes were ongoing (Fig. 18.1, part II). Today, these morphological differences are still the most important telltale signal of necrotic cell death. The typical rounding and swelling of the cells, together with increased cytoplasmic granularity and intact nuclei, are easy to recognize even by light microscopy (Fig. 18.2a, b). These early events are followed by a rapid loss of plasma membrane integrity, which is visualized by the uptake of PI and clear nuclear staining (Fig. 18.2c). In contrast, apoptotic cells shrink, show membrane ruffling and nuclear condensation, form apoptotic bodies (membrane-bound vesicles that contain compacted organelles and nuclear components), and expose phosphatidyl serine (Fig. 18.2d–e) (Wyllie 1981). The morphological necrotic events until today still form the best distinctive marker of necrosis and can be considered as a quick and easy first checkpoint to discriminate apoptotic and necrotic cell death. Apoptotic cells in the absence of phagocytic cells can proceed to secondary necrosis, in which cells have lost plasma membrane integrity, but even at this stage clear morphologic markers of apoptosis can be appreciated such as nuclear fragmentation and presence of apoptotic bodies containing nuclear DNA (compare Fig. 18.2c and f).

Main morphological characteristics of necrotic and apoptotic cell death observed by (fluorescence) light microscopy, illustrated in the L929 model system. Necrosis is characterized by (a) cellular rounding, swelling, and an increased granularity; (b) minor changes to the nucleus and phosphatidyl serine (PS) exposure on the plasma membrane; and, finally, (c) nuclear swelling, continued PS exposure, and rupture of the plasma membrane. Apoptosis is recognized by (d) cellular shrinking, formation of cell membrane ruffles and blebs, (e) nuclear condensation, PS exposure, and (f) nuclear fragmentation, PS exposure, and plasma membrane rupture (secondary necrosis). Notice that secondary necrotic cells still have condensed and fragmented nuclei caused by the apoptotic process, which is in strong contrast to the normal to swollen nuclei observed during primary necrosis
18.2.2.2 Morphologic and Multiparametric Analysis by Multiplex Methods
Considering the dynamic behavior of dying cells, it is important to document the entire process by, e.g., live cell imaging or by high-content cytometry (HCC). This will avoid erroneous categorization of apoptotic cells as necrotic, because they also proceed to a secondary necrotic phase upon plasma membrane rupture in the absence of sufficient phagocytic activity. Notice that these live cell imaging and HCC techniques can be extended by using fluorescent probes to monitor cell death-related biochemical events such as plasma membrane rupture, lysosomal membrane rupture (using lysotracker®), loss of mitochondrial membrane potential (using tetramethylrosamine methyl ester, TMRM), generation of reactive oxygen species (ROS, using carboxy-H2DCFDA or DHR123), phosphatidyl serine exposure (using fluorescently labeled Annexin V), or calcium fluxes (Fluo-3)—all probes are available at Life Technologies (Vanden Berghe et al. 2010). Secondary necrotic cells still retain some apoptotic features, such as condensed and fragmented nuclei (see Fig. 18.2f), loss of chromatin structure (due to caspase-mediated cleavage of lamins), and internucleosomal cleavage (due to the activity of the caspase-activated DNase). Condensation of the nuclei and loss of chromatin structure result in an intense but homogeneous staining with Hoechst or PI. In contrast, primary necrotic cells still retain their chromatin structure and are stained less intensely by Hoechst or PI, initially featuring brightly stained nucleoli after membrane permeabilization (time-lapse movies available at http://www.youtube.com/user/dmbrUPVA?ob = 0).
Cytometers can be either image based or flow based and can measure multiple parameters simultaneously when using fluorescent probes, and this on a cell-by-cell basis. This provides a high resolution at the level of the cell population and thus delivers meaningful insights into intrinsically heterogeneous responses, such as the response of individual cells in a population to a cell death stimulus. Flow cytometry requires the use of cells in suspension, possibly enhancing the risk of artifacts from manipulation when working with adherent cell cultures. However, fluorescence measurements using flow cytometry have a broad dynamic range and allow good separation of positive and negative cell populations. As cellular, nuclear, and organelle morphological aspects are key to get insight into an ongoing cell death modality, high-content imaging systems became extremely useful in the analysis of cell death. These instruments combine multiparameter, automated image acquisition in different cell plate formats and at selective optical resolutions, with automated image analysis algorithms that calculate morphological as well as fluorescence intensity parameters. Depending on the optical resolution and acquisition speed required, data on large cell populations can be processed and analyzed. Assays on high-content imaging systems allow combination of the cell death endpoint measurement with measurements of mitochondrial function, nuclear morphology, lysosomal rupture, PS exposure, autophagy, caspase activity reporters, or any other fluorescence-based parameter.
18.2.2.3 Morphologic Analysis by Electron Microscopy
Although morphologic analysis using light microscopy might be indicative of necrotic cell death, transmission electron microscopy (TEM) is still the most accurate but labor-intensive method for differentiating between apoptosis and necrosis in cell culture, because it allows the visualization of two-dimensional structures at the cellular and subcellular level. Necrotic cells are characterized by swollen organelles and irregular chromatin condensation, while in apoptotic cells, organelles are compacted in the apoptotic bodies and uniformly dense masses of chromatin are distributed against the nuclear envelope (karyopyknosis) (Cummings et al. 1997; Krysko et al. 2003). Recent evolutions in electron microscopy such as focused ion beam/scanning electron microscopy (FIB/SEM) have even rendered 3-dimensional images of tissues at the ultrastructural level (Knott et al. 2011), and the creation of electron-dense protein tags allows direct visualization of proteins of interest in both fluorescent light and EM (Shu et al. 2011). Dying cells that detach during the cell death process may represent a technical challenge for EM: centrifugation of these cells may change their ultrastructural appearance. Fortunately, a method has been developed to trap detached cells by adherent macrophages, thus tethering them to a solid substrate, suitable for fixation (Vanden Berghe et al. 2013). Although TEM presents the most detailed view of morphological changes during cell death, it is a complex and time-consuming method, requiring expert knowledge to recognize subcellular structures, and not suitable for routine examination of cell death processes.
18.2.2.4 DNA Fragmentation Analysis by Electrophoresis, Flow Cytometry, or TUNEL Assay
DNA fragmentation is considered a hallmark of apoptotic cell death and results in fragments of more than 5 kbp and nucleosomal sized fragments of multiples of 200 bp responsible for the typical DNA ladder pattern (Nagata 2000, 2002), which normally does not occur during necrotic cell death. These DNA ladders can be detected by agarose gel electrophoresis (Walker et al. 1999), but this method does not allow a quantitative analysis on the single-cell level, a disadvantage that can be overcome by flow cytometry or terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay.
The flow cytometry method visualizes the DNA fragmentation that occurs during apoptosis but not during necrosis. DNA degradation in apoptotic cells causes generation of small DNA fragments (Nicoletti et al. 1991). The mean intensity of PI seems to drop due to DNA and nuclear fragmentation (referred to as “hypoploid population”). This method will only work properly after permeabilization of all cells, such as achieved by a freeze–thaw cycle, since apoptotic cells maintain membrane integrity after DNA fragmentation. Due to its increase in intensity after binding to DNA, there is no need to remove the PI after staining (Vanden Berghe et al. 2013).
The TUNEL assay is a frequently used histochemical method to demonstrate apoptosis in in vivo-derived samples. In this assay, terminal deoxynucleotidyl transferase recognizes nicks in the DNA and adds dUTPs, modified with bromine, biotin, or fluorophores, which are then directly visualized or indirectly using fluorescent or peroxidase-coupled streptavidin or antibodies (Gavrieli et al. 1992). It is noteworthy that extracellular DNAse activity (Napirei et al. 2004), active gene transcription (Kockx et al. 1998), or even inappropriate processing of samples may also result in a positive TUNEL signal. The same holds true for caspase-independent release of EndoG by ischemic cardiomyocytes (Zhang et al. 2011) and AIF-mediated DNA processing in a caspase-independent manner during hippocampal neuronal cell death (Thal et al. 2011). As such, the TUNEL assay is not sufficient as the only biochemical proof of apoptosis.
18.2.2.5 Analysis of Caspase Activation
Caspases are cysteine-containing aspartate-specific proteases that drive multiple cellular processes such as apoptosis, pyroptosis, proliferation, and differentiation depending on the caspase involved and cellular context (Lamkanfi et al. 2007; Crawford and Wells 2011). Caspases consist of a p20 and a p10 subunit that contain the residues essential for substrate recognition and catalytic activity and a prodomain of variable length. Proteolytic activation results in the physical separation of the p10, p20, and prodomain subunits, forming active heterotrimers in an antiparallel orientation (Kersse et al. 2011). Although all caspases require an aspartate at the P1 site of their substrates, the preference for other upstream amino acid residues (P4–P2) in their substrates varies between different caspases, thus defining different substrate specificities for different caspases. Activation of the two major executioner caspases (caspase-3 and -7) in dying cells is a hallmark of apoptosis and therefore a strong argument in favor. The activity of caspases is transient, and thus analysis in function of time, covering the entire course of the cell death process, is required. Caspase activation can be monitored by fluorometry (fluorescently labeled substrates), antibody-based methods, or reporter assays.
A fluorometric caspase activation assay uses the preferred tetrapeptide substrate for the caspase of interest, where the aspartic residue at P1 is coupled to 7-amino-4-methylcoumarin (AMC) or 7-amino-4-trifluoromethylcoumarin (AFC) by a peptide bond. If the caspase under investigation is active, it hydrolyzes this peptide bond after the P1 aspartic acid, thus releasing free AMC or AFC. Free AMC or AFC can be excited to fluorescence, which can be measured in plate-based fluorometers. Notice that some caspases (such as caspase-3) have higher absolute k cat\K m values in general, presumably due to a higher intrinsic catalytic efficiency (Stennicke et al. 2000), which overrules the supposed specificity of tetrapeptide substrates for the different caspases (Pop et al. 2008; Pop and Salvesen 2009). Therefore researchers should be aware that the limited specificity of these small peptide substrates does not allow conclusion on the identity of caspases involved.
Fortunately, antibody-based techniques for caspase activation became available and allow the direct detection of activated caspases or the products of their proteolytic activity. Western blot is a commonly used technique requiring antibodies raised against epitopes created after proteolytic caspase activation, including both cleaved caspases and their processed substrates. ELISA-based methods require antibodies that recognize epitopes specific for proteolytic cleavage by caspases but are faster and more quantitative than western blots. Activated caspases can also be detected via histochemistry on fixed cells or tissue samples (Nagata 2000). Monitoring activation of the apoptotic executioner caspase-3 and processing of its prototype substrate PARP-1 (116 kDa), generating the characteristic 89 kDa cleavage fragment, is the most reliable approach to confirm or to rule out apoptosis.
A special and very useful case of an antibody-based assay for epithelial cell death involves cytokeratin 18 (CK18), which is cleaved during apoptosis (neo-epitope detectable with M30-Apoptosense® assay) but released in its non-cleaved form during necrosis (detectable by M65® ELISA). Both M30 and M65 are commercialized by Peviva AB (Bromma, Sweden), which also provides standard calibration material to normalize both assays. A high M30:M65 ratio corresponds to induction of apoptosis in cultured cells, while a low M30:M65 ratio is representative for necrosis induction. In vivo detection of caspase-cleaved CK18 is also possible in plasma and by immunohistochemistry (Cummings et al. 2008).
18.2.2.6 Caspase Activity Measurement Using Fluorescence- and Luminescence-Based Reporter Assays
Caspase activity can be monitored by genetically encoded reporter constructs, usually based on Förster resonance energy transfer (FRET) (Rehm et al. 2003; Laussmann et al. 2011). These reporters contain a caspase-cleavage site between both fluorescent proteins that make up the FRET pair; after caspase activation, the fluorescent proteins are physically separated and diffuse apart, thus reducing the signal of the FRET acceptor when exciting with light of the donor-excitation wavelength. Importantly, both fluorescent proteins can be excited by light of their own excitation wavelength, which allows distinguishing proteasomal degradation of the acceptor from bona fide caspase activation. FRET can be measured on live or fixed cells both by fluorescent (confocal) microscopy and flow cytometry. The Casper3-BG vector (FP970; Evrogen) was successfully used to demonstrate cell death switches between apoptosis and necrosis in the L929 model system (Vanlangenakker et al. 2011a). Casper3-BG consists of a blue fluorescent protein connected by a DEVD sequence with a green fluorescent protein. During apoptosis, caspase-3 processes DEVD, eliminates FRET, and increases the blue/green ratio (Fig. 18.3a). However, PI-positive cells should be excluded from the FRET analysis (Fig. 18.3b). Other similar FRET-based probes monitoring caspase-1 (Mahajan et al. 1999), caspase-6/8 (He et al. 2004), or caspase-9 (Figueroa et al. 2011; Wu et al. 2011) activity are also available. Simultaneous monitoring of caspase-8 and caspase-3 activity in single cells is also possible using a dual-FRET system (Kominami et al. 2012). Once a stable cell line has been generated, the FRET technology allows easy, fast, and reliable discrimination between apoptosis and necrosis. A disadvantage is that FRET signals can be quite weak and difficult to detect, requiring stable high expression levels. Fluorescent reporter probes based on a quenching peptide that is removed after activation of caspases have been developed for caspase-6 and -7, but these appear to have a very low signal-to-noise ratio (Wu et al. 2013).

FRET-based reporter system for the detection of caspase activation: a blue fluorescent protein is connected to a green fluorescent protein by a caspase-3 target sequence (DEVD). (a) During apoptosis, activated caspase-3 physically separates both fluorophores of the FRET pair, resulting in an increased blue/green ratio, and, finally, propidium iodide (PI) positivity. After a freeze–thaw cycle, PI intensity also indicates the hypoploidy status of the cell population (not shown). (b) Flow cytometry allows quantification of the blue/green intensity ratio on PI-negative cells
Recently, other reporter constructs for caspase-1 and caspase-3/7 based on luciferase activity have been published, GloSensor and iGLuc (Bartok et al. 2013; Galban et al. 2013). The GloSensor system was created by inserting the caspase-3/7 recognition sequence in the middle of the Photuris pennsylvanica luciferase, which inactivates the luciferase. After processing by caspase-3/7, both parts of the luciferase can reconstitute the active luciferase and a 50- to 100-fold induction of luciferase activity was observed (Bartok et al. 2013). This system was successfully used in a compound screening and to monitor caspase-3/7 activation in an in vivo mouse model of pancreatitis (Galban et al. 2013). The second system, iGLuc, is a fusion between pro-IL-1β and Gaussia luciferase, which forms nonluminescent aggregates. After overexpression of caspase-1 or activation of the inflammasome, pro-IL-1β is processed, releasing the mature IL-1β fused to luciferase from the aggregates and resulting in a 571-fold increase in luminescence (Bartok et al. 2013). iGLuc was successfully used to monitor inflammasome activation in vivo in a mouse infection model with Listeria (Bartok et al. 2013). The system can be modified to report on the activity of other caspases, such as caspase-3, by replacing the caspase-1 substrate recognition site with another one, i.e., DEVDG for caspase-3 (Bartok et al. 2013). Of note, the iGLuc system has a higher signal-to-noise ratio than the GloSensor system, which is probably related to the formation of nonluminescent aggregates in the iGLuc system, effectively reducing the background signal.
The specificity of the genetically encoded reporter probes for the intended target protease versus other off-target protease remains to be evaluated for most reporters. As they are based on the short peptide substrates, the specificity might be an issue especially in conditions of massive caspase-3 activation which has a high catalytic turnover (Pop et al. 2008; Pop and Salvesen 2009). Indeed, a FRET reporter construct with the preferred in vitro substrate recognition sequence of caspase-2 (VDVAD) appears to be cleaved mainly by effector caspases and caspase-8 when transfected in cells (Delgado et al. 2013).
18.2.2.7 Potential New Markers of Necrosis
In contrast to the proteolytic caspase cascade in apoptosis, the necroptotic signaling cascade is a kinase cascade. This kinase cascade is initiated by RIPK1 kinase activity (Degterev et al. 2005, 2008), involves the formation of an RIPK1–RIPK3 containing necrosis complex (“necrosome”) in an RIPK1 activity-dependent manner (Cho et al. 2009), and proceeds with the phosphorylation and activation of MLKL at Thr357 and Ser358 (Sun et al. 2012). Phosphorylation of RIPK1 and RIPK3 is visualized on western blot by the appearance of a second band running slightly above the steady-state band (Cho et al. 2009; He et al. 2009; Zhang et al. 2009). Another as yet to be validated approach of detection of necroptosis-specific phosphorylation events will be the use of phospho-specific antibodies directed against necroptosis-specific phosphorylation events such as the RIPK3 phosphorylation site on MLKL (Thr357 and Ser358) (Sun et al. 2012), the autophosphorylation sites of RIPK1 (Ser14/15, Ser20, Ser161, and Ser166) (Degterev et al. 2008), or RIPK3 (Ser227) (Sun et al. 2012) to probe for RIPK1, RIPK3, or MLKL phosphorylation in multiple applications, such as immunohistochemistry, flow cytometry, or total cell extracts by ELISA, multiplex bead arrays, or western blot. These phospho-specific markers of necroptosis may form the longed-for parameter of necroptosis. However, in view of the role of RIPK3 and MLKL in regulating inflammasome activation in particular conditions of IAP inhibition or absence of caspase-8, rigorous controls may be required again (Vince et al. Immunity 2012; Kang et al. Immunity 2013). Similar to the FRET-based protease reporters, kinase activity reporters (KARs) for RIPK1- or RIPK3-mediated phosphorylation events could be developed (Morris 2013). These KARs are based on a FRET pair flanking a phosphorylation target sequence and a phosphoamino acid-binding domain (PAABD). When kinases phosphorylate the target sequence, the PAABD binding brings the FRET pair in each other’s neighborhood and increased fluorescence is detected (Sipieter et al 2013, Sipieter et al 2014). Similar KARs based on split luciferase have been described as well. These luciferase-based KARs have the advantage that they can be used for in vivo reporting as well (Herbst et al. 2011; Williams et al. 2013).
18.2.2.8 In Vivo Markers of Necrosis
Massive apoptosis saturates the phagocytic system, and secondary necrosis will result in the release of the same marker molecules; confirming the absence of apoptosis by immunohistochemistry of particular tissues or cells is thus necessary to confirm necrotic plasma markers. Postmortem analyses for necrotic cell death is possible by pathologists using hematoxylin- and eosin-stained sections (Kaiser et al. 1995; Gukovskaya et al. 1996). A low M30:M65 ratio of CK18 in plasma (as discussed above), but also in sections of tissue (www.peviva.se and Duan et al. 2003), may also be indicative of necrotic cell death. Although in vivo injection of PI has been applied (You et al. 2008), this is not yet a standardized easily applicable technique. Recently, a fusion of Hoechst with the infrared dye IR-786 (Hoechst-IR) was shown to be cell membrane impermeable and specifically bind to released extracellular DNA (Dasari et al. 2010). Unfortunately, this dye probably also recognizes secondary necrotic tissues in an LPS-GalN-induced hepatitis model (Dasari et al. 2010), underscoring the need for confirming absence of active caspases by immunohistochemistry. The most conclusive postmortem evidence for necrosis can still be obtained by TEM, since organelle swelling, patch-like irregular chromatin condensation, and ruptured plasma membranes can directly be observed. Moreover, in vivo necroptosis is often associated with accumulation of necrotic nuclei in the affected tissue or organs (Gunther et al. 2011).
Real-time and direct in vivo imaging of necrotic cell death is perhaps the most interesting application in a clinical setting. Contrast agents which accumulate in necrotic tissue (necrosis-avid contrast agents) may offer a unique combination of identifying specifically primary necrotic tissue and this in real time (Prinsen et al. 2011). The main disadvantage for this technique is that the equipment and technical staff required for these methods are usually not accessible for small animals in an academic setting.
18.2.3 Identifying Signaling Components of Regulated Necrosis (Step III)
The molecular unraveling of RN has revealed that, similar to apoptosis, it can be activated by many different triggers, which activate partly overlapping pathways, involving RIPK1/RIPK3, PARP-1, ROS, and intracellular Ca2+ release (Table 18.3). Finally, these processes converge on a similar cellular disintegration process, characterized by cellular swelling, lysosomal membrane permeabilization, and cell rupture (Vanden Berghe et al. 2010; Vandenabeele et al. 2010; Vanlangenakker et al. 2012).
18.2.3.1 Pharmacological Inhibition
Currently, the best characterized type of RN is necroptosis. According to its most recent definition (Galluzzi et al. 2012), any cell death that can be inhibited by genetic ablation or knockdown of either RIPK1 or RIPK3 or by chemical inhibition by necrostatin-1 (Nec-1 or 5-(1H-indol-3-ylmethyl)-3-methyl-2-thioxo-4-imidazolidinone) is defined as necroptosis; however, this is not exclusive. Recently, RIPK3-dependent (Dondelinger et al. 2013) and RIPK1 kinase activity-dependent (Wang et al. 2008; Duprez et al. 2012) apoptosis have also been reported in certain cellular settings. This illustrates again the need of using several independent methods, most importantly by checking morphological appearance and confirming absence of caspase activity, cf. the decision tree in Fig. 18.1. More recently, necrosulfonamide (NSA) was identified as a new inhibitor of necroptosis in human cells, which could also be used to confirm the necroptosis typing. This compound covalently binds to Cys86 of MLKL, a crucial substrate of RIPK3 (Sun et al. 2012). In vivo, injection of Nec-1 has been used a lot to prevent cell death in several disease models (Degterev et al. 2005; Smith et al. 2007; You et al. 2008; Duprez et al. 2011). Recently, a more stable and specific variant of Nec-1 was discovered (no off-target effect on immunomodulatory enzyme indoleamine 2,3-dioxygenase), i.e., Nec-1s (Takahashi et al. 2012; Degterev et al. 2013), which is recommended to use in vivo instead of Nec-1 (Vandenabeele et al. 2013).
In addition to Nec-1 and NSA that both block necroptosis (for NSA only in human cells), several pharmacological inhibitors having other targets than RIPK1, RIPK3, or MLKL (Table 18.3) are described to interfere with some form of RN. Often, these pathways occur independent of RIPK1/RIPK3 or the link with RIPK1/RIPK3 is not known yet. In order to study which molecular pathways contribute to the studied form of RN, an approach using pharmacological inhibitors (Table 18.3) can give a first hint. One may also consider to combine inhibitors, as often redundant mechanism turns up upon interference, e.g., depletion or inhibition of both RIPK3 and CypD is required to block ischemia/reperfusion-induced kidney injury (Linkermann et al. 2013).
18.2.3.2 Transgenic Approaches
Ripk1 −/− mice are embryonic lethal (Kelliher et al. 1998), and genetic ablation in vitro causes a shift to apoptosis (Vanlangenakker et al. 2011a), rendering this model unsuitable to demonstrate the involvement of necroptosis. The availability of RIPK1 kinase-dead knock-in mice will probably circumvent these issues. Ripk3 −/− mice develop normally and do not show any obvious spontaneous phenotypes, which allowed phenotyping in a variety of pathological models involving necroptosis (Cho et al. 2009; He et al. 2009; Zhang et al. 2009; Duprez et al. 2011). In addition to RIPK1, RIPK3, and MLKL, several other genes (Table 18.3) contribute to some forms of RN, either independent of necrosome formation or with an as yet unknown link to necroptosis. Typically, a genetic depletion strategy following a pharmacological approach is used to confirm which mechanisms are involved in the studied form of RN. Although this combined pharmacological/genetic approach is an easy and quick method to indicate potential involved mechanisms, one has to be cautious because cell death modality may shift upon interference. For example, blocking apoptosis can shift to necroptosis (Vercammen et al. 1998b), or vice versa (Vanden Berghe et al. 2003; Vanlangenakker et al. 2011a). To detect these possible cell death modality shifts, it is recommended to perform consistently a kinetic analysis to monitor potential transient caspase activation, as mentioned previously. Generally, three major genetic depletion strategies are followed: (1) RNA interference-mediated knockdown, (2) deficient cells derived from transgenic mice, and (3) deficient cells generated by genome editing technologies such as zinc finger nuclease technology (Sigma-Aldrich), TALENs (Transposagen Biopharmaceuticals, Cellectis, and others), or CRISPR (Hsu et al. 2013).
18.3 Conclusions
Research on RN, and in particular of necroptosis, is hampered by a lack of positive markers. Although some markers based on phospho-specific antibodies could have been developed years ago, none have been reported in the literature. Hence, the detection of RN still depends on the combination of morphological observations, biochemical exclusion of caspase activation, and determination of the involved subroutines (Table 18.3). One should examine these elements over the entire course of the cell death process. It is important to combine several independent techniques to identify RN, since several genes (including RIPK1 and RIPK3) are involved in both apoptosis and necrosis. As a consequence, inhibition, knockdown, or genetic ablation of these genes may create a shift in cell death type rather than cell death inhibition. By following the decision tree (Fig. 18.1), using the approaches and pharmacological/genetic targets (Table 18.3) described, researchers should be able to pinpoint more easily the cell death modalities and the major mechanistic pathways involved.
References
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192:565–570
Awad D, Schrader I, Bartok M, Mohr A, Gabel D (2011) Comparative toxicology of trypan blue, brilliant blue G, and their combination together with polyethylene glycol on human pigment epithelial cells. Invest Ophthalmol Vis Sci 52:4085–4090
Baines C, Kaiser R, Purcell N, Blair N, Osinska H, Hambleton M, Brunskill E, Sayen M, Gottlieb R, Dorn G, Robbins J, Molkentin J (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662
Bartok E, Bauernfeind F, Khaminets MG, Jakobs C, Monks B, Fitzgerald KA, Latz E, Hornung V (2013) iGLuc: a luciferase-based inflammasome and protease activity reporter. Nat Methods 10:147–154
Bernhard D, Schwaiger W, Crazzolara R, Tinhofer I, Kofler R, Csordas A (2003) Enhanced MTT-reducing activity under growth inhibition by resveratrol in CEM-C7H2 lymphocytic leukemia cells. Cancer Lett 195:193–199
Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, Kopiwoda S, Abrams MJ, Strauss HW (1999) Imaging of apoptosis (programmed cell death) with 99mTc annexin V. J Nucl Med 40:184–191
Chiaradia M, Baranes L, Van Nhieu JT, Vignaud A, Laurent A, Decaens T, Charles-Nelson A, Brugières P, Katsahian S, Djabbari M, Deux JF, Sobhani I, Karoui M, Rahmouni A, Luciani A (2014) Intravoxel incoherent motion (IVIM) MR imaging of colorectal liver metastases: Are we only looking at tumor necrosis? J Magn Reson Imaging 38:27-40
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137:1112–1123
Christofferson DE, Yuan J (2010) Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ 17:1942–1943
Cohen A, Shirvan A, Levin G, Grimberg H, Reshef A, Ziv I (2009) From the Gla domain to a novel small-molecule detector of apoptosis. Cell Res 19:625–637
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN (2010) Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A 107:2574–2579
Cordeiro MF, Guo L, Coxon KM, Duggan J, Nizari S, Normando EM, Sensi SL, Sillito AM, Fitzke FW, Salt TE, Moss SE (2010) Imaging multiple phases of neurodegeneration: a novel approach to assessing cell death in vivo. Cell Death Dis 1:e3
Crawford ED, Wells JA (2011) Caspase substrates and cellular remodeling. Annu Rev Biochem 80:1055–1087
Cummings J, Ward TH, Greystoke A, Ranson M, Dive C (2008) Biomarker method validation in anticancer drug development. Br J Pharmacol 153:646–656
Cummings MC, Winterford CM, Walker NI (1997) Apoptosis. Am J Surg Pathol 21:88–101
Dasari M, Lee S, Sy J, Kim D, Brown M, Davis M, Murthy N (2010) Hoechst-IR: an imaging agent that detects necrotic tissue in vivo by binding extracellular DNA. Org Lett 12:3300–3303
Dave SR, White CC, Gao X, Kavanagh TJ (2012) Luminescent quantum dots for molecular toxicology. Adv Exp Med Biol 745:117–137
De Saint-Hubert M, Prinsen K, Mortelmans L, Verbruggen A, Mottaghy FM (2009) Molecular imaging of cell death. Methods 48:178–187
De Valck D, Beyaert R, Van Roy F, Fiers W (1993) Tumor necrosis factor cytotoxicity is associated with phospholipase D activation. Eur J Biochem/FEBS 212:491–497
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313–321
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112–119
Degterev A, Maki J, Yuan J (2013) Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ 20:366
Delgado ME, Olsson M, Lincoln FA, Zhivotovsky B, Rehm M (2013) Determining the contributions of caspase-2, caspase-8 and effector caspases to intracellular VDVADase activities during apoptosis initiation and execution. Biochim Biophys Acta 1833:2279–2292
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ (2013) RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 20:1381–1392
Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EA (2003) Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol 199:221–228
Duprez L, Bertrand MJ, Vanden Berghe T, Dondelinger Y, Festjens N, Vandenabeele P (2012) Intermediate domain of receptor-interacting protein kinase 1 (RIPK1) determines switch between necroptosis and RIPK1 kinase-dependent apoptosis. J Biol Chem 287:14863–14872
Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P (2011) RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35:908–918
Festjens N, Kalai M, Smet J, Meeus A, Van Coster R, Saelens X, Vandenabeele P (2006) Butylated hydroxyanisole is more than a reactive oxygen species scavenger. Cell Death Differ 13:166–169
Figueroa RA, Ramberg V, Gatsinzi T, Samuelsson M, Zhang M, Iverfeldt K, Hallberg E (2011) Anchored FRET sensors detect local caspase activation prior to neuronal degeneration. Mol Neurodegener 6:35
Fonge H, Chitneni SK, Lixin J, Vunckx K, Prinsen K, Nuyts J, Mortelmans L, Bormans G, Ni Y, Verbruggen A (2007) Necrosis avidity of (99m)Tc(CO)3-labeled pamoic acid derivatives: synthesis and preliminary biological evaluation in animal models of necrosis. Bioconjug Chem 18:1924–1934
Galban S, Jeon YH, Bowman BM, Stevenson J, Sebolt KA, Sharkey LM, Lafferty M, Hoff BA, Butler BL, Wigdal SS, Binkowski BF, Otto P, Zimmerman K, Vidugiris G, Encell LP, Fan F, Wood KV, Galban CJ, Ross BD, Rehemtulla A (2013) Imaging proteolytic activity in live cells and animal models. PLoS One 8:e66248
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19:107–120
Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493–501
Goossens V, Grooten J, De Vos K, Fiers W (1995) Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A 92:8115–8119
Goossens V, Stange G, Moens K, Pipeleers D, Grooten J (1999) Regulation of tumor necrosis factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron flux through the electron transport chain complex I. Antioxid Redox Signal 1:285–295
Grimberg H, Levin G, Shirvan A, Cohen A, Yogev-Falach M, Reshef A, Ziv I (2009) Monitoring of tumor response to chemotherapy in vivo by a novel small-molecule detector of apoptosis. Apoptosis 14:257–267
Gukovskaya AS, Perkins P, Zaninovic V, Sandoval D, Rutherford R, Fitzsimmons T, Pandol SJ, Poucell-Hatton S (1996) Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology 110:875–884
Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C (2011) Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477:335–339
He L, Wu X, Meylan F, Olson DP, Simone J, Hewgill D, Siegel R, Lipsky PE (2004) Monitoring caspase activity in living cells using fluorescent proteins and flow cytometry. Am J Pathol 164:1901–1913
He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137:1100–1111
Herbst KJ, Allen MD, Zhang J (2011) Luminescent kinase activity biosensors based on a versatile bimolecular switch. J Am Chem Soc 133:5676–5679
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495
Hsu PD, Scott DA, Weinstein J, Ran F, Konermann S, Agarwala V, Li Y, Fine E, Wu X, Shalem O, Cradick T, Marraffini L, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol 31:827–832
Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D, Vandenabeele P, Samson M, Dimanche-Boitrel MT (2012) TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 19:2003–2014
Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML (1995) Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol 269:C1295–C1304
Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D (2013) Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity 38:27–40
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8:297–303
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P (2011) The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci 36:541–552
Kim HJ, Koo SY, Ahn BH, Park O, Park DH, Seo DO, Won JH, Yim HJ, Kwak HS, Park HS, Chung CW, Oh YL, Kim SH (2010) NecroX as a novel class of mitochondrial reactive oxygen species and ONOO(−) scavenger. Arch Pharm Res 33:1813–1823
Kim YS, Morgan MJ, Choksi S, Liu ZG (2007) TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 26:675–687
Knott G, Rosset S, Cantoni M (2011) Focussed ion beam milling and scanning electron microscopy of brain tissue. J Vis Exp (53):e2588
Kockx MM, Muhring J, Knaapen MW, de Meyer GR (1998) RNA synthesis and splicing interferes with DNA in situ end labeling techniques used to detect apoptosis. Am J Pathol 152:885–888
Kominami K, Nagai T, Sawasaki T, Tsujimura Y, Yashima K, Sunaga Y, Tsuchimochi M, Nishimura J, Chiba K, Nakabayashi J, Koyamada K, Endo Y, Yokota H, Miyawaki A, Manabe N, Sakamaki K (2012) In vivo imaging of hierarchical spatiotemporal activation of caspase-8 during apoptosis. PLoS One 7:e50218
Kourtis N, Nikoletopoulou V, Tavernarakis N (2012) Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 490:213–218
Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32:157–164
Krysko DV, Brouckaert G, Kalai M, Vandenabeele P, D’Herde K (2003) Mechanisms of internalization of apoptotic and necrotic L929 cells by a macrophage cell line studied by electron microscopy. J Morphol 258:336–345
Kuenzler KA, Pearson PY, Schwartz MZ (2002) IL-11 pretreatment reduces cell death after intestinal ischemia-reperfusion. J Surg Res 108:268–272
Kuranaga E (2012) Beyond apoptosis: caspase regulatory mechanisms and functions in vivo. Genes Cells 17:83–97
Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P (2007) Caspases in cell survival, proliferation and differentiation. Cell Death Differ 14:44–55
Laster SM, Wood JG, Gooding LR (1988) Tumor necrosis factor can induce both apoptotic and necrotic forms of cell lysis. J Immunol 141:2629–2634
Laussmann MA, Passante E, Dussmann H, Rauen JA, Wurstle ML, Delgado ME, Devocelle M, Prehn JH, Rehm M (2011) Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ 18:1584–1597
Li J, Cona MM, Chen F, Feng Y, Zhou L, Yu J, Nuyts J, de Witte P, Zhang J, Himmelreich U, Verbruggen A, Ni Y (2012) Exploring theranostic potentials of radioiodinated hypericin in rodent necrosis models. Theranostics 2:1010–1019
Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S (2013) Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A 110:12024–12029
Louis KS, Siegel AC (2011) Cell viability analysis using trypan blue: manual and automated methods. Methods Mol Biol 740:7–12
Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, Lavelle EC, Martin SJ (2009) Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31:84–98
Mahajan NP, Harrison-Shostak DC, Michaux J, Herman B (1999) Novel mutant green fluorescent protein protease substrates reveal the activation of specific caspases during apoptosis. Chem Biol 6:401–409
Mariani G, Villa G, Rossettin PF, Spallarossa P, Bezante GP, Brunelli C, Pak KY, Khaw BA, Strauss HW (1999) Detection of acute myocardial infarction by 99mTc-labeled d-glucaric acid imaging in patients with acute chest pain. J Nucl Med 40:1832–1839
Morris MC (2013) Fluorescent biosensors—probing protein kinase function in cancer and drug discovery. Biochim Biophys Acta 1834:1387–1395
Nagata S (2000) Apoptotic DNA fragmentation. Exp Cell Res 256:12–18
Nagata S (2002) Breakdown of chromosomal DNA. Cornea 21:S2–S6
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–658
Napirei M, Wulf S, Mannherz HG (2004) Chromatin breakdown during necrosis by serum Dnase1 and the plasminogen system. Arthritis Rheum 50:1873–1883
Nguyen QD, Smith G, Glaser M, Perumal M, Arstad E, Aboagye EO (2009) Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-3/7 specific [18F]-labeled isatin sulfonamide. Proc Natl Acad Sci U S A 106:16375–16380
Ni Y, Huyghe D, Verbeke K, de Witte PA, Nuyts J, Mortelmans L, Chen F, Marchal G, Verbruggen AM, Bormans GM (2006) First preclinical evaluation of mono-[123I]iodohypericin as a necrosis-avid tracer agent. Eur J Nucl Med Mol Imaging 33:595–601
Ni Y, Petre C, Miao Y, Yu J, Cresens E, Adriaens P, Bosmans H, Semmler W, Baert AL, Marchal G (1997) Magnetic resonance imaging-histomorphologic correlation studies on paramagnetic metalloporphyrins in rat models of necrosis. Invest Radiol 32:770–779
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139:271–279
Perfetto SP, Chattopadhyay PK, Lamoreaux L, Nguyen R, Ambrozak D, Koup RA, Roederer M (2006) Amine reactive dyes: an effective tool to discriminate live and dead cells in polychromatic flow cytometry. J Immunol Methods 313:199–208
Pick E, Keisari Y (1980) A simple colorimetric method for the measurement of hydrogen peroxide produced by cells in culture. J Immunol Methods 38:161–170
Pop C, Salvesen GS (2009) Human caspases: activation, specificity, and regulation. J Biol Chem 284:21777–21781
Pop C, Salvesen GS, Scott FL (2008) Caspase assays: identifying caspase activity and substrates in vitro and in vivo. Methods Enzymol 446:351–367
Prinsen K, Jin L, Vunckx K, De Saint-Hubert M, Zhou L, Cleynhens J, Nuyts J, Bormans G, Ni Y, Verbruggen A (2011) Radiolabeling and preliminary biological evaluation of a (99m)Tc(CO)(3) labeled 3,3′-(benzylidene)-bis-(1H-indole-2-carbohydrazide) derivative as a potential SPECT tracer for in vivo visualization of necrosis. Bioorg Med Chem Lett 21:502–505
Rehm M, Dussmann H, Prehn JH (2003) Real-time single cell analysis of Smac/DIABLO release during apoptosis. J Cell Biol 162:1031–1043
Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, Baekelandt V, Takahashi N, Berghe TV, Vandenabeele P (2014) Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 5:e1004. doi:10.1038/cddis.2013.531. PMID: 24434512
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W (1993) Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J 12:3095–3104
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8:237–248
Shen S, Kepp O, Michaud M, Martins I, Minoux H, Metivier D, Maiuri MC, Kroemer RT, Kroemer G (2011) Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 30:4544–4556
Shinzawa K, Tsujimoto Y (2003) PLA2 activity is required for nuclear shrinkage in caspase-independent cell death. J Cell Biol 163:1219–1230
Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY (2011) A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol 9:e1001041
Sipieter F, Vandame P, Spriet C, Leray A, Vincent P, Trinel D, Bodart JF, Riquet FB, Héliot L (2013) From FRET imaging to practical methodology for kinase activity sensing in living cells. Prog Mol Biol Transl Sci 113:145–216. doi:10.1016/B978-0-12-386932-6.00005-3. PMID:23244791
Sipieter F, Ladik M, Vandenabeele P, Riquet F (2014) Shining light on cell death processes - a novel biosensor for necroptosis, a newly described cell death program. Biotechnol J. PMID:24390900
Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM (2007) Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther 21:227–233
Smith CV (1987) Evidence for participation of lipid peroxidation and iron in diquat-induced hepatic necrosis in vivo. Mol Pharmacol 32:417–422
Smith G, Glaser M, Perumal M, Nguyen QD, Shan B, Arstad E, Aboagye EO (2008) Design, synthesis, and biological characterization of a caspase 3/7 selective isatin labeled with 2-[18F]fluoroethylazide. J Med Chem 51:8057–8067
Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, Herdegen T, Linkermann A, Rittger A, Chan FK, Kabelitz D, Schütze S, Adam D (2014) TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci 71:331–348
Stennicke H, Renatus M, Meldal M, Salvesen G (2000) Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem J 350(Pt 2):563–568
Suffys P, Beyaert R, De Valck D, Vanhaesebroeck B, Van Roy F, Fiers W (1991) Tumour-necrosis-factor-mediated cytotoxicity is correlated with phospholipase-A2 activity, but not with arachidonic acid release per se. Eur J Biochem/FEBS 195:465–475
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227
Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P (2012) Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 3:e437
Thal SE, Zhu C, Thal SC, Blomgren K, Plesnila N (2011) Role of apoptosis inducing factor (AIF) for hippocampal neuronal cell death following global cerebral ischemia in mice. Neurosci Lett 499:1–3
Vanden Berghe T, Grootjans S, Goossens V, Dondelinger Y, Krysko DV, Takahashi N, Vandenabeele P (2013) Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 61:117–129
Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P (2003) Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 278:5622–5629
Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P (2010) Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 17:922–930
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Vandenabeele P, Grootjans S, Callewaert N, Takahashi N (2013) Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ 20:185–187
Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T (2011a) TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2:e230
Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P, Bertrand MJ (2011b) cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 18:656–665
Vanlangenakker N, Vanden Berghe T, Vandenabeele P (2012) Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 19:75–86
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P (1998a) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187:1477–1485
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P (1998b) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188:919–930
Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Häcker G, Silke J, Tschopp J (2012) Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 36:215–227
Wainwright M (2010) Dyes, trypanosomiasis and DNA: a historical and critical review. Biotech Histochem 85:341–354
Walker PR, Leblanc J, Smith B, Pandey S, Sikorska M (1999) Detection of DNA fragmentation and endonucleases in apoptosis. Methods 17:329–338
Wang K, Li J, Degterev A, Hsu E, Yuan J, Yuan C (2007) Structure-activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg Med Chem Lett 17:1455–1465
Wang L, Du F, Wang X (2008) TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133:693–703
Wendland MF, Faustino J, West T, Manabat C, Holtzman DM, Vexler ZS (2008) Early diffusion-weighted MRI as a predictor of caspase-3 activation after hypoxic-ischemic insult in neonatal rodents. Stroke 39:1862–1868
Williams TM, Nyati S, Ross BD, Rehemtulla A (2013) Molecular Imaging of the ATM Kinase Activity. Int J Radiat Oncol Biol Phys 86:969–977
Wu P, Nicholls SB, Hardy JA (2013) A tunable, modular approach to fluorescent protease-activated reporters. Biophys J 104:1605–1614
Wu Y, Wang D, Wang X, Wang Y, Ren F, Chang D, Chang Z, Jia B (2011) Caspase 3 is activated through caspase 8 instead of caspase 9 during H2O2-induced apoptosis in HeLa cells. Cell Physiol Biochem 27:539–546
Wyllie A (1981) Cell death: a new classification separating apoptosis from necrosis. In: Lockshin RA (ed) Cell death in biology and pathology. Chapman & Hall, London, pp 9–34
Xu X, Chua CC, Zhang M, Geng D, Liu CF, Hamdy RC, Chua BH (2010) The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res 1343:206–212
Yamashima T (2000) Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol 62:273–295
Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, Kleinridders A, Wunderlich T, Kashkar H, Utermohlen O, Bruning JC, Schutze S, Kronke M (2009) Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460:1159–1163
Yoshioka A, Yamaya Y, Saiki S, Kanemoto M, Hirose G, Beesley J, Pleasure D (2000) Non-N-methyl-D-aspartate glutamate receptors mediate oxygen–glucose deprivation-induced oligodendroglial injury. Brain Res 854:207–215
You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ (2008) Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 28:1564–1573
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336
Zhang J, Ye J, Altafaj A, Cardona M, Bahi N, Llovera M, Canas X, Cook SA, Comella JX, Sanchis D (2011) EndoG links Bnip3-induced mitochondrial damage and caspase-independent DNA fragmentation in ischemic cardiomyocytes. PLoS One 6:e17998
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–107
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Grootjans, S., Goossens, V., Vandenabeele, P., Vanden Berghe, T. (2014). Methods to Study and Distinguish Necroptosis. In: Shen, HM., Vandenabeele, P. (eds) Necrotic Cell Death. Cell Death in Biology and Diseases. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8220-8_18
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8220-8_18
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8219-2
Online ISBN: 978-1-4614-8220-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)