
Abstract
Despite significant progress in understanding the homeostatic regulation of energy balance, successful therapeutic options for curbing obesity remain elusive. One potential target for the treatment of obesity is via manipulation of the gut–brain axis, a complex bidirectional communication system that is crucial in maintaining energy homeostasis. Indeed, ingested nutrients induce secretion of gut peptides that act either via paracrine signaling through vagal and non-vagal neuronal relays, or in an endocrine fashion via entry into circulation, to ultimately signal to the central nervous system where appropriate responses are generated. We review here the current hypotheses of nutrient sensing mechanisms of enteroendocrine cells, including the release of gut peptides, mainly cholecystokinin, glucagon-like peptide-1, and peptide YY, and subsequent gut-to-brain signaling pathways promoting a reduction of food intake and an increase in energy expenditure. Furthermore, this review highlights recent research suggesting this energy regulating gut–brain axis can be influenced by gut microbiota, potentially contributing to the development of obesity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Energy homeostasis involves an intricate and complex balance between energy intake and expenditure that is predominantly coordinated by the brain. The central nervous system (CNS) receives constant neural and chemical input regarding the body’s energy state from various peripheral organs, and is responsible for integrating this information and generating appropriate responses to maintain homeostasis. These signals are generally characterized as either more long-term adiposity or ‘tonic’ signals, such as leptin and insulin, which are released continuously to reflect the amount of body fat, or short-term, ‘episodic’ signals that fluctuate depending on the ingestive status of the individual. The gastrointestinal (GI) tract, being the first site of interaction with ingested nutrients, is responsible for the majority of these episodic signals, communicating important information regarding the size and composition of an incoming meal to the brain. This gut–brain axis is vital for the maintenance of energy balance as these gut-derived signals, sometimes referred to as satiation/satiety signals, are not only able to reduce energy intake, but have more recently been demonstrated to control energy expenditure. Ingested nutrients induce secretion of gut peptides, namely cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), and peptide YY (PYY), which through either activation of local neuronal relays or endocrine signaling directly within the CNS, initiate a gut–brain axis. While this gut–brain axis has implications in glucose regulation, as reviewed recently elsewhere [1], the present review will focus on the control of food intake and energy expenditure to maintain energy balance and how this control is disrupted in obesity. Furthermore, we describe how this dysregulation of energy homeostasis in obesity may be due to microbe-induced alterations in gut–brain signaling pathways, given the more recently implicated role of the gut microbiota in influencing host energy metabolism. A better understanding of this gut–brain axis may lead to the development of more targeted treatments that can favorably influence energy balance to reduce adiposity and ameliorate obesity.
The gut–brain axis
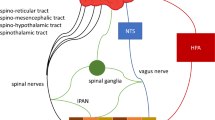
The gut–brain axis represents a bi-directional signaling axis that is vital for metabolic homeostasis. In the GI tract, sensory information is transformed into signals of neural, hormonal, and immunological origin, which are relayed to the CNS. Although emerging evidence links changes in intestinal immune signaling with alterations in gut-mediated energy homeostasis (see below) [2], the majority of established effects of the gut–brain axis on energy balance are a result of neural and hormonal gut-derived signals. Preabsorptive nutrients can generate signals at multiple sites throughout the GI tract, to communicate with the brain regarding not only the caloric value of a meal, but possibly the precise macronutrient composition of ingested calories through individualized nutrient-specific sensory mechanisms [3]. Gut-derived signals are then relayed to a number of brain areas to generate responses that ultimately result in both acute and more chronic changes in energy intake and energy expenditure, to maintain energy homeostasis during both feeding and fasting (Fig. 1).

Gut–brain communication through gut peptides. Gut peptides are released from enteroendocrine cells in response to preabsorptive nutrients and act to relay information regarding incoming energy to the brain. Once released into the subcellular space, gut peptides can act locally on gut peptide receptors expressed on vagal or spinal afferent nerve terminals innervating the gut to activate gut–brain neuronal signaling. Gut peptides might also act indirectly via receptors on intrinsic neurons of the enteric nervous system to relay neuronal signaling to afferent nerves. In contrast, gut peptides can diffuse into the systemic circulation or lymphatics to eventually reach the brain and act on central receptors in an endocrine fashion
Level of the gut
The majority of signaling by intestinal nutrients occurs through the release of gut peptides. Specifically, enteroendocrine cells (EECs) lie within the intestinal epithelium, open to the luminal contents, and express chemosensory machinery on their apical surfaces allowing them to respond to preabsorptive nutrients. Nutrient sensing occurs through G-protein coupled receptors (GPRs), electrogenic solute transporters and/or intracellular metabolism, all of which subsequently lead to calcium influx and gut peptide release into the subcellular space [4]. The various subtypes of EECs are classically characterized by both their localization within the GI tract as well as the peptide(s) they secrete. The stomach contains X/A-like cells that produce ghrelin as well as chief cells that produce gastric leptin, the proximal small intestine contains I cells and K cells that produce CCK and glucose-dependent insulinotropic hormone, respectively, and the distal small intestine contains L cells, which produce GLP-1/2, oxyntomodulin (OXM) and PYY. However, recent findings indicate co-expression of various gut peptides originally thought to be synthesized in distinct EECs, throughout the intestine, arguing that EECs are in fact represented by a single cell type which produces varying spectra of gut peptides depending on the environment [5]. Nonetheless, synthesis and secretion of all of the above gut peptides is induced by an influx of intestinal nutrients, and is mediated by nutrient-specific sensory machinery expressed on the EEC apical membrane. Following release from EECs, gut peptides can enter the circulation to act on peripheral targets, including the brain, in an endocrine fashion. However, there is ample evidence to suggest that gut peptides largely signal to the brain via local, paracrine action on receptors expressed in afferent neurons innervating the gut wall.
The stomach and proximal small intestine, where the majority of digestion and absorption occur, are highly innervated by vagal and splanchnic nerves, with afferents far outnumbering efferents, supporting the pivotal role of neuronal gut-to-brain signaling [6, 7]. Indeed, vagal afferent fibers extend into the lamina propria of the intestinal villi, terminating in close proximity to the basolateral surface of EECs, and express receptors for gut peptides including ghrelin, leptin, CCK, GLP-1, and PYY, whose receptor activation leads to neuronal firing [8]. In addition, gut peptides might also activate vagal and spinal afferents indirectly, via activation of neurons of the enteric nervous system (ENS), which have also been shown to express gut peptide receptors [9–11]. While classically implicated in local neuronal reflexes controlling intestinal function [12], it is plausible that the ENS plays a role in the gut–brain axis by relaying nutrient-derived signals to vagal afferents, given that: intrinsic ENS neurons are positioned in close proximity to both EECs and afferent nerve terminals, intestinal nutrient infusion leads to c-Fos activation in the myenteric plexus [13], and the ENS has been implicated in the activation of vagal afferents in the gut [14]. Nonetheless, while the precise mechanisms remain unclear, nutrient-induced gut peptide secretion activates local afferent signaling to initiate a gut–brain neuronal signaling axis.
Connection to the brain
Vagal afferent neurons terminate in the nucleus tractus solitarius (NTS) of the dorsal vagal complex (DVC) of the brainstem [15], while spinal afferents synapse on neurons of lamina 1 of the spinal dorsal horn which project to the NTS. The NTS integrates both vagal and spinal gut-derived signals, which are subsequently relayed to the hypothalamus [16, 17]. Intestinal nutrient infusion leads to c-Fos activation in the NTS, and this response is attenuated by blockade of gut–brain vagal communication via treatment of intestinal vagal afferents with the neurotoxin capsaicin [18]. This effect is attributed to nutrient-induced gut peptide release, where for example, peripheral CCK administration activates NTS neurons via capsaicin-sensitive vagal afferents [19]. Vagal afferent activation of NTS neurons is mediated by the activation of n-methyl-d-aspartate receptors in afferent neuron terminals, which leads to phosphorylation of ERK 1/2 and synapsin I to stimulate neurotransmitter release from NTS neurons [19]. Various neuronal populations within the NTS are activated by gut peptide-vagal afferent signaling, including POMC and catecholaminergic neurons [20], and NTS melanocortin receptor signaling is required for gut peptide-induced ERK 1/2 activation and suppression of food intake [21]. On the other hand, much less is known about the importance of the ENS or of spinal afferents in mediating the gut–brainstem axis. Nevertheless, studies have implicated spinal afferents in the control of food intake (see below), suggesting that they may represent an additive or redundant pathway of gut–brain signaling.
Interestingly, the brainstem contains motor output neuronal circuitry controlling feeding behaviours, supporting a role for gut–brainstem reflexes to acutely regulate energy balance through alteration of motor controls involved in feeding and energy expenditure. Indeed, studies show that chronic decerebrate rats will suppress food intake in response to intestinal nutrients when only the brainstem remains intact [22]. However NTS neurons additionally project to several higher order brain regions, including the hypothalamus, where they terminate in multiple nuclei that are involved in the control of energy balance, namely the paraventricular (PVN) and arcuate (ARC) nuclei. Further, both the NTS of the brainstem and the ARC of the hypothalamus are juxtaposed to areas that lack a defined blood–brain barrier: the area postrema and median eminence, respectively. Given that plasma levels of gut peptides increase following a meal, there is evidence supporting a model where gut peptides reach the systemic circulation, diffuse through these leaky brain structures, and act directly on brainstem and hypothalamic neurons, as described in the following sections. For example, the Y2 receptor (Y2R), which binds the gut peptide PYY, has been localized in the ARC, and peripheral injection of PYY increases c-Fos immunoreactivity in the ARC [23]. Indeed, endocrine and neural activation of ARC neurons by the gut–brain axis is of particular importance to energy homeostasis, largely through the regulation of the melanocortin system controlled by ARC POMC and agouti-related protein (AgRP) neurons. POMC neurons release α-melanocyte stimulating hormone, which directly activates downstream neuronal melanocortin receptors to inhibit food intake and increase energy expenditure. AgRP neurons release AgRP, which antagonizes melanocortin receptors and neuropeptide Y (NPY), an inhibitory neurotransmitter that stimulates food intake and suppresses energy expenditure. As described below, many gut peptides control energy balance through the regulation of ARC POMC and AgRP neurons and their associated neuropeptides.
In summary, preabsorptive nutrients trigger a gut–brain axis by stimulating the release of gut peptides, which via multiple neural or humoral pathways, activate important metabolic sites in the hindbrain and hypothalamus to regulate energy balance by altering both energy intake and energy expenditure. This axis is nicely illustrated by Vincent et al. who demonstrate that intestinal glucose infusion activates gut peptide secreting cells in the mucosal membrane, neurons of the myenteric plexus (ENS) and nodose ganglion (vagal afferents), as well as neurons in both the NTS and ARC [24].
Gut–brain axis in the control of food intake
When fasted rats are re-fed, food intake decreases within minutes of re-feeding and continues to steadily decline throughout a meal [25], indicating that negative feedback signals are sent to the brain rapidly after food enters the GI tract to prevent an excess of incoming nutrients. One of the first signals generated is that of the mechanoreception of a food bolus entering the stomach. When a gastric cuff is put in place to close off the pyloric sphincter and allow the stomach to fill without emptying, saline loading of the stomach volume-dependently reduces food intake in as quickly as 3 min [26]. Indeed, the vagal and spinal afferents innervating the stomach express stretch-receptive calcium channels [27], and gastric distention causes vagal afferent firing and activation of neurons in the hindbrain [28], supporting a neuronal stomach–brain axis that lowers food intake. Nutrients are emptied into the small intestine via the pyloric sphincter, and the rate of gastric emptying begins to slow upon arrival of nutrients into the small intestine, further increasing the distention of the stomach upon subsequent ingestion of food. Slowing of gastric emptying is accomplished through the release of CCK and GLP-1 [29, 30] and vagal activation, as subdiaphragmatic vagotomy, a surgery that involves severing the afferent fibers of the vagus at the brainstem, attenuates the effects of CCK or GLP-1 on gastric emptying [31, 32].
Although the negative feedback on gastric emptying following nutrient influx contributes to reductions in food intake, direct intestinal infusion of nutrients inhibits sham feeding (a procedure where ingested nutrients are drained from the stomach directly and do not reach the small intestine) [33, 34], indicating that nutrients in the intestine can per se suppress food intake, independent of effects on gastric emptying. Indeed, nutrient-induced release of a number of gut peptides, summarized below, is associated with signaling to the brain to control food intake. Subdiaphragmatic vagotomy or capsaicin treatment completely reverse the acute feeding suppressive effects of intestinal nutrient infusions [35, 36], indicating that a gut–brain neuronal axis involving vagal afferents innervating the small intestine mediates these effects. In addition, celiac-superior mesenteric ganglionectomy, a surgery involving the transection of non-vagal, splanchnic nerves originating from the intestine, also blocks the feeding suppressive effects of intraduodenal nutrients, additionally implicating spinal afferents in the gut–brain axis control of food intake [37]. Interestingly, gut peptide receptor antagonists, such as devazepide, a CCK-1 receptor (CCK-1R) antagonist, block both the suppression of food intake and the neural activation that is associated with nutrient ingestion [14].
CCK
Cholecystokinin is released from I-cells of the small intestine mainly in response to intestinal fatty acids and proteins. CCK is the most well-established satiation signal [38], but it has also been shown to play important roles in activating the gut–brain axis to control gut motility, food intake, energy expenditure, and glucose homeostasis [39]. Co-infusion of an antagonist for the CCK-1R, which is expressed on vagal afferents innervating the intestine [40], attenuates both vagal firing and suppression of food intake following intraduodenal fatty acids and protein [41, 42], implicating CCK as a mediator of fat and protein induced satiation. Exogenous CCK-8 injection, which acutely lowers food intake, has been shown to activate a specific population of neurons in the NTS of the brainstem which project to the PVN of the hypothalamus, and lesion of these neurons blocks the feeding suppressive effects of exogenous CCK-8 [43]. Indeed, the PVN contains anorexigenic thyrotropin releasing hormone neurons which are involved in feeding behaviour [44]. In addition to acting on local vagally expressed receptors, CCK is released into circulation, as the postprandial state is associated with a rise in plasma CCK [45, 46], which potentially reaches the brain to suppress food intake through direct, central action [47, 48]. Indeed, CCK can act directly on CCK-1 receptors in the NTS, as well as in six different regions of the hypothalamus to suppress feeding [49–51]. However, these effects are likely mainly reflective of the actions of centrally produced CCK [52], as most studies demonstrate that chemical and surgical ablation of vagal signaling abolishes the feeding suppressive effects of peripheral CCK, implicating the neuronal gut–brain axis [53]. Further, CCK injections into peripheral arteries supplying the proximal small intestine more potently lower food intake than do systemic CCK injections [54, 55]. Thus, there is sufficient evidence that post-ingestive CCK acts in a paracrine fashion via peripheral CCK-1 receptors in the intestine. This is in line with the more recently established glucoregulatory role of lipid-induced CCK, that acts on the CCK-1R on vagal afferents to lower hepatic glucose production via a neuronal gut–brain–liver axis, which is described in more detail elsewhere [56].
GLP-1
Carbohydrates, lipids and proteins are all potent secretagogues of GLP-1, which is produced mainly within L cells of the distal small intestine and colon [57]. While the effects of endogenous GLP-1 on food intake are more controversial and less understood than those of CCK, an increasing amount of evidence implicates GLP-1 as an important satiation signal [58]. Given that GLP-1 is released within 15 min of nutrient ingestion [59], before nutrients would reach the distal small intestine to directly stimulate L cells, it is hypothesized that nutrients in the proximal small intestine trigger GLP-1 release from the ileum via a neuro-hormonal reflex, involving the vagus [57]. Indeed, when nutrients are infused into the duodenum and not allowed to flow into the distal small intestine, significant GLP-1 secretion is stimulated, and this is reversed when the distal intestine is removed [60, 61]. However, recent evidence has confirmed the presence of GLP-1 expressing EECs in the proximal intestine [62] indicating that direct nutrient sensing by EECs might in fact stimulate early GLP-1 release from the duodenum instead [63], or in addition to, the aforementioned reflex. Nonetheless, GLP-1 is secreted in response to intestinal nutrients, and several studies indicate that endogenous GLP-1 plays a physiological role in suppressing food intake via a paracrine effect [64, 65]. Intestinally secreted GLP-1 is rapidly degraded in circulation, resulting in only 25 % of secreted GLP-1 reaching the hepatoportal circulation and less than 10 % reaching the systemic circulation [66]. Thus, a likely role is supported for local, paracrine action in the intestine. Indeed, vagal afferent neurons express the GLP-1 receptor [67] and GLP-1 directly induces firing of cultured vagal afferent neurons [68]. Further, subdiaphragmatic vagotomy or capsaicin treatment in rodents completely blocks the suppressive effects of intraperitoneal (IP) GLP-1 [69, 70]. In contrast, intravascular infusion of exogenous GLP-1 suppresses food intake and these effects are not reversed by vagotomy or capsaicin [71, 72], implicating a potential role for GLP-1 receptor (GLP-1R) signaling directly within the brain. However, it is unlikely that the doses used in these studies are indicative of endogenous GLP-1 circulating levels, thus it is unlikely that nutrient-induced GLP-1 enters the circulation to act centrally to lower food intake. One other possibility is that rather than entering the portal circulation, GLP-1 is released into the lymph. Indeed, following intestinal glucose or fat infusion, GLP-1 levels dose-dependently increase in the lymph, with levels higher than that of plasma GLP-1, supported by lower levels of dipeptidyl peptidase-4 in the lymph [73, 74]. Further, gastric nutrient infusion causes a greater increase of GLP-1 in the lymph than in the hepatoportal vein [75]. This presents a plausible model where GLP-1 in fact reaches the brain in significant quantities given its transport via the lymph. The GLP-1R is expressed in the DVC of the NTS and in hypothalamic nuclei including the ARC [76, 77], and intracerebroventricular (i.c.v.) GLP-1 acutely and potently suppresses food intake [78, 79], and this is prevented by co-infusion of the antagonist exendin-9 [80]. However, i.c.v. infusion of exendin-9 does not prevent the suppressive effects of IP GLP-1 [80], while IP co-injection of exendin-9 does, indicating that GLP-1R activation on peripheral neurons may be more important in physiological conditions where GLP-1 is released from the gut, while the aforementioned studies may be identifying mechanisms for centrally derived GLP-1 [81]. Although human evidence for a local vagal GLP-1 signaling axis is lacking and difficult to distinguish, patients with pyloroplasty and truncal vagotomy fail to suppress intake following GLP-1 administration, suggesting that vagal signaling is necessary for the short-term effects of peripheral GLP-1 [82]. One important caveat of many studies examining the role of GLP-1 is the use of long-lasting agonists to mimic the effects of endogenous GLP-1. Indeed, GLP-1R agonists, exendin-4 and liraglutide, exhibit a much longer half-life and can cross the blood–brain barrier, thus they are not an ideal representation of GLP-1 that is released in response to nutrients. However, it may be possible that the early satiating effects of these drugs mimic endogenous GLP-1, as subdiaphragmatic vagotomy attenuates short-term effects (but not long-term) on food intake, while CNS GLP-1R antagonism attenuates more long-term effects on food intake [83]. Thus while studies utilizing GLP-1R agonists are useful in determining the mode of action of pharmacological treatments, which can lead to more targeted and improved drug options, further work is required to identify the physiological role of local GLP-1 signaling.
PYY
Peptide YY is also released from L cells, along with GLP-1, in response to intestinal nutrients [84], and given that PYY−/− mice are hyperphagic, and do not respond to the satiating effect of dietary protein [85], it is hypothesized to play an important role in energy homeostasis. Direct nutrient sensing by the distal intestine may promote PYY secretion, as PYY is co-expressed with chemosensors such as those for bitter and sweet taste nutrients in L cells [86]. However, PYY is released within 15 min of food intake, indicating that PYY release involves a reflex arc via proximal intestinal neural or chemical relay, releasing PYY from the distal intestine [87]. Interestingly, plasma PYY levels rise after feeding and stay elevated for several hours, peaking 1–2 h following the onset of food intake [88], suggesting that PYY plays a role in controlling long-term satiety via endocrine signaling, where CCK and possibly GLP-1 are more important in the short-term regulation of satiation. Circulating PYY3–36 is an agonist of Y2R, and while exogenous PYY3–36 lowers food intake in rodents and humans, it fails to do so in Y2R−/− mice [23] or with co-injection of a Y2R antagonist [30]. Receptors for PYY are expressed in the nodose ganglion and possibly on vagal afferent terminals (as demonstrated via axonal transport) [89], and peripheral injection of PYY causes vagal firing as well as neuron activation in the NTS and ARC, which is abolished by vagotomy [89, 90], supporting a role for peripheral PYY action on vagal afferents. However, not all studies support this notion, as peripheral PYY injection can suppress food intake in peripherally capsaicin-treated rats [30], indicating that gut–brain vagal signaling is not imperative for PYY’s effects on food intake. Interestingly, administration of the Y2R antagonist directly into the hypothalamus prevents the satiating effects of peripheral PYY3–36. Indeed, peripheral PYY3–36 increases c-Fos in the ARC of the hypothalamus [23], while direct administration of PYY3–36 into the ARC lowers food intake [23] and mice that lack the hypothalamic Y2R are hyperphagic [91]. Y2R is expressed predominantly by orexigenic NPY neurons in the ARC [92], and peripheral PYY3–36 causes a decrease in NPY mRNA through its action on Y2R [23], while Y2R antagonism increases NPY [93]. Given that NPY neurons of the ARC inhibit anorexigenic POMC neurons to increase food intake, it is not surprising that peripheral PYY3–36 increases c-Fos in POMC neurons [23]. Thus, the current model indicates that PYY3–36, which increases in the plasma postprandially, suppresses food intake through the inhibition of NPY and subsequent activation of POMC neurons, exerting long-term suppressive effects through melanocortin signaling.
Other intestinal factors
In addition to the more understood roles of CCK, GLP-1, and PYY, a number of other intestinally derived hormones and factors have been demonstrated to mediate the gut–brain axis. Gut-derived serotonin (5-HT) is produced within specialized EECs called enterochromaffin cells, is released in response to nutrients [94], and acts locally on receptors expressed by vagal afferents. Interestingly, while 5-HT receptor agonists can suppress food intake [95, 96], and antagonism of 5-HT receptors attenuates the suppressive effects of intestinal nutrients [97], a recent study demonstrates that peripheral 5-HT can paradoxically contribute to the development of obesity [98]. Thus, like many other gut-derived peptides the role of 5-HT in energy balance is likely more complex than originally hypothesized, and requires further investigation.
In addition, a number of non-hormonal mediators within the gut wall can activate the gut–brain axis. One such intestinal factor is chylomicron-derived lipoprotein ApoA-IV, which has been implicated in the control of food intake in response to intestinal lipids. ApoA-IV is a lipoprotein released from enterocytes during lipid absorption. Interestingly, exogenous Apo-IV was found to acutely reduce food intake [99], indicating that this lipoprotein might play a role in signaling to activate the gut–brain axis upon absorption of incoming lipids. In fact, the suppression of food intake in response to intestinal lipid infusion can be attenuated by blocking the formation of chylomicrons with Pluronic L-81 [100]. Pluronic L-81 also blocks the rise in plasma CCK following lipid infusion [101], suggesting that upon release from enterocytes, ApoA-IV signals to stimulate the release of CCK from adjacent EECs. Indeed, the feeding suppressive effects of exogenous ApoA-IV are blocked by CCK-1R antagonism, CCK receptor knockout (KO), or by subdiaphragmatic vagotomy, supporting that Apo-IV from chylomicrons triggers a CCK–CCK-1R-vagal afferent gut–brain axis to suppress food intake [102]. Interestingly, fourth-ventricular infusion of Apo-IV suppresses food intake through activation of neurons in both the NTS and ARC, and this requires CCK-1R receptors, indicating that Apo-IV might reach the circulation to act centrally in a similar CCK-dependent mechanism to suppress food intake [49].
Another group of signaling molecules that has been implicated in the gut–brain axis is membrane lipid-derived endocannabinoids. Given the potent orexigenic effects of exogenous cannabinoids [103], cannabinoid receptor antagonists, such as Rimonabant, were developed for the treatment of obesity and successfully led to modest weight loss despite side effects [104], suggesting that the endocannabinoid system might be important in the physiological regulation of food intake. Indeed, cannabinoid receptor 1 (CB1) is expressed in the nodose ganglia [105] and peripheral CB1 receptor antagonism suppresses food intake through capsaicin-sensitive, likely vagal, neurons [106]. This suggests that endocannabinoids produced in the intestine might play a role in the physiological regulation of food intake, whereby increased production leads to increased food intake during fasting. Indeed, intestinal levels of the endocannabinoid anandamide (AEA) increase following a 24-h fast, and peripheral AEA stimulates short-term feeding, dependent on vagal afferents [106]. CB1 agonists cause an inhibition in vagal afferent firing [107], and the CB1 receptor has been shown to be constitutively active [108], implicating a model where increased intestinal endocannabinoid production and thus vagal afferent CB1 signaling increases food intake during fasting through the suppression of gut-derived satiety signals.
Although the current review focuses mainly on the role of EEC-derived gut–brain signaling, only about 1 % of the intestinal cell population is comprised of EECS, while over 70 % of the body’s immune cells reside in gut-associated tissues, and there is evidence that immune mediators released within the gut wall play a role in gut–brain communication [2]. Vagal afferent terminals express receptors for, and respond to, immune products such as mast cell mediators [109] and macrophage-derived cytokines [110]. In addition, the immune system might indirectly affect the gut–brain axis where intestinal inflammation has been linked to changes in EEC numbers and gut peptide responses [111, 112], and immune mediators have been shown to potentiate vagal responses to gut peptide hormones [113, 114]. It is not surprising, therefore, that changes in intestinal immune factors and inflammation are linked to obesity and metabolic disorder (see [115] for an in-depth review).
Gut–brain axis in the control of energy expenditure
Despite energy intake being a major contributor to the development of obesity, energy homeostasis involves a balance between both intake and expenditure. Energy expenditure can have a profound effect on body weight and several studies have shown that decreased energy expenditure can predict weight gain [116–118]. Energy expenditure consists of three components: basal metabolic rate, thermogenesis, and the energy cost of physical activity [119]. Adaptive thermogenesis, the regulated production of heat, is influenced by environmental temperature and diet [120]. Given that gut peptides can act as dietary intermediates between the GI tract and the CNS to reduce food intake, it is no surprise that gut peptides can alter energy expenditure by activating energy regulation centers of the CNS to initiate signaling pathways that ultimately lead to a decrease in energy expenditure. Indeed, i.c.v. administration of both GLP-1 [121] and OXM, a gut peptide produced from the proglucagon gene [122], increases energy expenditure in rodents. Further, intravenous PYY3–36 has been suggested to increase energy expenditure through an increase in postprandial thermogenesis and resting metabolic rate [123]. However, paracrine regulation of thermogenesis is also possible, as duodenal lipid sensing has been shown to increase brown adipose tissue (BAT) thermogenesis, through a CCK-dependent gut–brain–BAT neuronal axis likely involving vagal afferents [124]. Given that CCK can act through a gut–brain–BAT axis to regulate energy expenditure, it is possible that other gut peptides, such as GLP-1 and OXM, exert their effects on energy expenditure through the activation of this axis. In support of this idea, administration of these peptides has been shown to increase BAT thermogenesis [125], however the activation of a gut–brain–BAT axis in this context remains to be elucidated. Further, a recent study indicates that administration of a gut-restricted FXR agonist enhances the thermogenesis and browning of white adipose tissue, potentially through a similar gut–brain–adipose tissue axis [126]. Studies are warranted to better characterize the mechanisms by which nutrient sensing pathways contribute to the regulation of energy expenditure, as the unveiling of a so-called gut–brain–BAT axis for the regulation of energy expenditure could provide potential therapeutic targets for the treatment and prevention of obesity and its related diseases.
Gut–brain axis in the development of obesity and the role of the gut microbiota
Increased consumption of highly palatable (high-fat, high-sugar, hyper-caloric) foods, and possibly a reduction in energy expenditure in westernized countries, are salient contributors to the rising obesity rates worldwide [127, 128], thus implicating altered gut–brain signaling mechanisms in obesity. Indeed, nutrient sensing is impaired in obese and high fat fed humans and animal models [39] as evidenced by reductions in both postprandial levels of gut peptides [129–131], as well as reduced sensitivity to such peptides [132–134]. For example, obese rats exhibit reduced vagal sensitivity to nutrients [135], as well as CCK [136] and GLP-1 [133], which may promote overeating and weight gain. Although the role of nutrient sensing and vagal signaling in the development of obesity has been extensively reviewed elsewhere [107, 137], recent evidence suggests that the gut microbiota may play a role in energy balance and could be a mediating factor between obesogenic feeding and the impaired nutrient sensing seen in obesity.
Gut microbiota
The gut microbiota is the term for the collective microbial community of the entire GI tract, consisting of over 100 trillion microbes, outnumbering host cells by a factor of 10 [138, 139]. A complex co-evolution allows these microorganisms to colonize and survive within the host gut, forming a symbiotic relationship that provides a nutrient-rich environment for the microbiota, and metabolic, protective, and structural functions for the host. When examining the metabolic impact of the gut microbiota, evidence suggests that it can regulate not only energy extraction from the diet, through the production of short-chain fatty acids (SCFA) from indigestible carbohydrates [140], but it can also influence overall energy intake and storage mechanisms [141, 142]. The effects of the gut microbiota on host metabolism was first shown through the use of germ-free (GF) mice, those lacking a gut microbiota, which display reduced adiposity when compared to normal mice, and exhibit resistance to diet-induced obesity, characteristics likely due, in part, to reduced energy extraction from the diet [140, 143]. While GF animals resemble conditional KO animal models, allowing researchers to examine mechanisms altered by the absence of a gut microbiota or from insertion of a specific microbial population (similar to a selective knock-in performed in KO animals), caution must be raised when interpreting results, as they have clear developmental differences from conventionally raised animals [144]. For example, the small intestine of GF animals is underdeveloped, with a considerably reduced surface area, irregular villi, reduced regeneration of epithelial cells, and slower peristalsis [145]. In addition to intestinal physiology, GF animals exhibit altered development of many body systems including the immune system, the cardiovascular system, and the CNS [144]. As such, studies involving microbiota manipulation of conventionally raised animals, as opposed to those that are completely sterile, may be a more physiologically relevant method for investigating the impact of the gut microbiota on host physiology. For example, high fat feeding can induce drastic and rapid changes in the gut microbiome [146, 147] and obese rodents and humans exhibit significantly altered gut microbiota, with both changes in composition and/or reductions in diversity [148, 149]. Preliminary studies suggested that obesity was associated with an increase in the ratio of bacteria belonging to the Firmicute phylum in comparison to the Bacteroidetes phylum, which decreased following both diet and surgically induced weight loss [150–152]. However, some more recent studies have failed to replicate these findings, and hypothesize that these effects were due more to the diet than the obese phenotype [153, 154]. Nonetheless, it can be argued that despite variations in observed phyla differences, specific changes at the genus and species levels that are responsible for specific metabolic functions are more important. For example, when the microbiota of obese-prone rats and obese-resistant rats was transplanted into GF recipients, researchers identified 25 operational taxonomic units (OTUs) in the obese donors and recipients that were absent in obese-resistant donors and recipients, many of these OTUs belonging to microbial families associated with energy extraction from the diet [155]. Accordingly, studies in mice and humans have shown that obesity is associated with a microbiome enriched in genes encoding enzymes involved in the extraction of calories from indigestible carbohydrates [149, 152]. However, the link between obesity or high fat feeding and microbial energy harvest is not as clear as originally proposed [156]. Furthermore, while abundance of butyrate producing bacteria was positively correlated with BMI, a more abundant network of bacteria labeled as primary degraders was inversely correlated with BMI [157], further suggesting a beneficial role for some, but not all, SCFAs [158–160].
Short-chain fatty acid signaling
The gut microbiota is responsible for the breakdown of indigestible carbohydrates and the production of SCFA, which account for 5–10 % of human energy requirements [161–164]. Manipulation of SCFA production through administration of prebiotics, supplemental indigestible carbohydrates that promote the growth and activity of many microbial species in the gut, promotes weight loss and improves metabolic parameters [165–167]. In addition, SCFA, given both orally or directly into the intestine reduce food intake and body weight in diabetic and healthy rodents and humans [168–170]. SCFAs are produced primarily in the distal GI tract with butyrate, propionate, and acetate making up 90–95 % of the SCFA present in the colon [171]. Butyrate is a major source of energy for the colonic epithelium, while propionate primarily enters the portal circulation to be used in gluconeogenesis and the majority of acetate enters systemic circulation, reaching peripheral tissues [172, 173].
In addition to these functions, one mechanism through which SCFA are thought to influence host energy balance is by activating signaling pathways in the intestinal epithelium, resulting in gut peptide release in both rodents and humans [170, 174]. SCFAs activate the GPRs, FFAR2 and FFAR3, formerly known as GPR43 and GPR41, respectively [175, 176]. Although expressed in many other tissues [177], both receptors have been localized to EECs, with high expression in isolated L cells [178–180], and are responsible for SCFA-induced release of gut peptides [170]. For instance, although SCFAs stimulate GLP-1 release from primary intestinal murine cultures, this effect is lost in FFAR2−/− and FFAR3−/− primary intestinal cultures, and both FFAR2−/− and FFAR3−/− mice have impaired GLP-1 release [181]. Furthermore, release of GLP-1 and PYY following distal intestinal infusion of propionate is absent in FFAR2−/− mice. Although the relevance of intestinal FFAR2/3 signaling in whole body energy homeostasis is debated [182] FFAR3 has recently been localized to the peripheral nervous system [183, 184], further suggesting that SCFAs signal via a gut–brain axis.
Gut microbiota and nutrient sensing
In addition to intestinal SCFA signaling, the gut microbiota can influence gut–brain signaling via alterations in the absorptive and secretory capacity of the intestinal epithelial cells. GF mice exhibit altered levels of glucose transporters and sweet taste receptors, and reduced expression of FFAR2 and FFAR3, as well as long-chain fatty acid receptors GPR40 and GPR120 [185, 186], which are implicated in gut peptide secretion [56]. Accordingly, these mice have reduced intestinal expression of CCK, GLP-1, and PYY, which is associated with increased acceptance of intralipid and sucrose, indicating that decreased nutrient sensing in GF mice promotes increased energy intake [185, 186]. In addition, when GF mice are transplanted with the microbiota of obese-prone or obese-resistant mice, the obese donors and recipients exhibit alterations in intestinal nutrient sensors and gut peptide levels in comparison to obese-resistant donors and recipients [155]. Further, studies indicate that specific bacterial strains can up-regulate GPR120 or down-regulate GLP-1 expression in vitro [187]. Thus, it is plausible that changes in specific bacterial species can alter luminal host nutrient-sensing and gut peptide signaling.
Along these lines, studies with prebiotics have demonstrated the capability of altering the gut microbiota to subsequently improve gut nutrient-sensing mechanisms to reduce food intake and weight gain. Prebiotic treatment has been shown to increase the abundance of Faecalibacterium prausnitzii and Bifidobacterium [188], which improve the gut barrier through a GLP-2-dependent mechanism [189], as well as through increased endocannabinoid signaling [158]. This trophic effect of prebiotic treatment is associated with increased EEC differentiation and a subsequent increase in gut peptide production [112]. In support of this, GLP-1, GIP, and PYY are increased in both rodents and humans in response to prebiotic treatment [190–192], which is associated with increased satiety in humans [193] and decreased food intake and adiposity in rodents [190–192]. Thus, the gut microbiota can influence host luminal nutrient sensing and gut peptide signaling, and it is possible arises that gut microbiota manipulation can additionally alter neuronal signaling and activation of the gut–brain axis (Fig. 2).

Potential influences of the gut microbiota on host gut–brain axis. The gut microbiota has been associated with changes in anorexigenic and orexigenic peptide levels in the brainstem and hypothalamus, as well as with changes in motor control, memory, and anxiety behavior, while the development and activity of the ENS has been shown to be affected by an altered or absent gut microbiota. In addition, the gut microbiota has been associated with changes in EEC differentiation, expression of nutrient receptors, the expression and release of gut peptides, and activation of EECs via SCFAs. CNS central nervous system, ENS enteric nervous system, EEC enteroendocrine cell, SCFA short-chain fatty acid
Gut microbiota and neuronal signaling via gut–brain axis
High fat feeding and obesity are associated with low-grade inflammation, coined metabolic endotoxemia, which is characterized by an increase in plasma lipopolysaccharide (LPS) levels, a pro-inflammatory molecule derived from the cell wall of Gram (−) bacteria [194]. It is hypothesized that changes in the gut microbiota promote gut barrier dysfunction, thus increasing circulating LPS levels (coined metabolic endotoxemia) via a leaky gut, which can then activate pro-inflammatory processes at peripheral sites such as adipose tissue, through activation of its receptor, toll-like receptor-4 (TLR-4) [112, 195]. When challenged with a high fat diet, mice have increased circulating LPS and systemic TLR-4 activation, white adipose tissue inflammation, and reduced insulin sensitivity, all associated with changes in the microbiota composition [196]. In addition to its potential peripheral actions, LPS may act directly on the gut. For example, TLR4 activation is increased in the gut of high fat fed obese rats, and this is associated increased intestinal permeability and circulating LPS [195], while intestinal deletion of MyD88, which is a central adaptor molecule to several TLRs, including TLR4, protects against diet-induced obesity and metabolic endotoxemia [197]. In addition, LPS has been shown to inhibit the pacemaker activity of the interstitial cells of Cajal [198], highlighting the potential for LPS to influence cellular depolarization, possibly in neurons. Indeed, vagal afferents have been shown to express TLR-4 [199] and LPS attenuates the ability of leptin to activate vagal afferent neurons, both in vitro and in vivo [200, 201]. Thus, leptin resistance at the level of vagal afferent neurons may be due to increased LPS from high fat feeding. Given that vagal leptin signaling is hypothesized to promote CCK signaling and subsequent satiation, leptin resistance in vagal afferents could inhibit CCK signaling in diet-induced obese rats, providing a link between altered gut microbiota with CCK resistance in models of high fat feeding and obesity [202].
A recently emerging concept is the ability of the gut microbiota to directly alter CNS signaling. Whether the gut microbiota impacts CNS signaling related to the regulation of energy homeostasis is still relatively unknown, however, ample evidence demonstrates that the gut microbiota can influence CNS-mediated stress and anxiety behaviors. For example, GF mice exhibit differences in motor control, memory, and anxiety behavior, which are associated with changes in brain chemistry [203–205]. Probiotic treatment has been shown to normalize anxiety-like behavior in mice with colitis, possibly through a vagally mediated mechanism that regulates BDNF [206] and probiotic supplementation in humans has been shown to improve cognitive reactivity to sad mood through the reduction of rumination and aggressive thoughts [207]. One mechanism by which probiotics may be altering the gut–brain axis is through improvements in local inflammation and gut barrier integrity, as probiotics have been shown to attenuate the HPA response to acute psychological stress through a mechanism dependent on the prevention of gut barrier impairment and a decrease in circulating LPS levels [208]. While CNS functions related to stress and anxiety are clearly impacted by the gut microbiota, preliminary evidence suggests this may also be true of energy balance. For example, GF mice exhibit differences in anorexigenic and orexigenic peptide levels in the brainstem and hypothalamus, and have an altered response to leptin [209]. Future work should examine how manipulations in the gut microbiota can impact CNS signaling mechanisms related to energy homeostasis, either acutely or possibly at an early age inducing developmental changes. Taken together, these data demonstrate that the gut microbiota can impact both local and central neural signaling, thus possibly influencing host energy balance through a microbiota–gut–brain axis.
Conclusion and perspective
Obesity has become a worldwide social and economic crisis, and to date, modern medicine has struggled to develop effective therapeutic options. Instead, obesity therapy is in a precarious situation, where doctors are relying on therapeutic options without knowing the exact mechanisms for their success. Gastric bypass remains the most effective weight loss treatment available, yet researchers are still uncertain as to how and why gastric bypass achieves both rapid and sustained weight loss. It is interesting to note that favourable alterations in the gut–brain axis may contribute to the weight loss effect of gastric bypass, given that bariatric surgery can increase the number of gut peptide expressing EECs [210] and, consequently, postprandial gut peptide secretion [211]. As such, targeting of the gut–brain axis represents a very promising area of therapy. For example, gut peptide mimetic drugs have proved successful in both rodents [212, 213], as well as clinically [214, 215] in reducing food intake and obesity. Furthermore, increasing energy expenditure may prove an effective strategy, and targeting intestinal mechanisms to increase thermogenesis could be the best option. However, given the redundancy in, and compensatory capacity of, the regulation of energy homeostasis, single target approaches to normalize energy balance may not achieve sustained success. However, the development of monomeric peptide co- and tri-agonists could bypass these challenges. For example, one recently developed tri-agonist can increase glucagon action to promote energy expenditure, while activating the GLP-1R and GIP receptor to reduce food intake and improve glucose control [216]. Therefore, novel drugs targeting the gut to reduce intake while increasing expenditure may prove to be the most efficacious strategies for the treatment of obesity.
Pharmaceutical agents targeting the gut microbiota provide another strategy for battling obesity and associated metabolic disorders, as improvements in metabolic parameters following gastric bypass have also been associated with rapid and sustained shifts in the intestinal microbiota [150, 154, 166, 217]. Interestingly, GF mice colonized with the microbiota of those who have undergone bariatric surgery, show reduced adipose tissue deposition and increased energy expenditure as compared to their control counterpart, indicating that the gut microbiota may play a direct role in the metabolic improvements seen following bariatric surgery possibly through alterations in SCFA production or decreased metabolic endotoxemia via improvements in gut barrier [166, 218, 219]. Consequently, manipulations of the gut microbiota may prove efficacious for the treatment of obesity, although the time period in which intervention is most effective remains to be elucidated, and appears vital for success. For example, studies in mice demonstrate that antibiotic treatment can have beneficial effects on lowering body weight and food intake in high fat fed adult mice [220, 221]. However, when given early in life, agents such as antibiotics that disrupt the microbiota composition and consequently the metabolic activity of the microbiota, can affect host energy balance and can have long-lasting effects on body weight in adulthood [222–225], which is consistent with the role of the microbiota in host development. Indeed, the assembly of the gut microbiota is associated with the development of intestinal immunity and reductions in intestinal defense can lead to metabolic perturbations [224, 226]. Interestingly, breastfeeding, which plays an important role in the development of the gut microbiota, is associated with altered secretion of gut peptides [227], and early life stress, such as maternal separation, leads to gut dysbiosis and subsequent development of anxiety-like behaviour and altered brain chemistry in mice [228]. Therefore, it is possible that the negative metabolic effects associated with early life perturbation of the gut microbiota are due to altered development of the gut–brain axis. As a result, it will be crucial to identify not only which microbes must be manipulated and how, but additionally what time period in an individual’s life would yield the most effective treatment outcome. The delivery of treatments aimed at manipulating the adult gut microbiota is also not straightforward, as fecal microbiota transplantation for the treatment of Clostridium difficile infection has been shown to promote weight gain [229]. However, targeted manipulation in individuals with metabolic dysregulations has been effective, as fecal microbiota transplantation from lean donors to individuals with metabolic syndrome improves insulin sensitivity of recipients [230]. Taken together, this evidence suggests that early gut microbiota changes due to a western diet (high in fat and sugar) can impair the gut–brain signaling axis, both at the level of gut-sensing mechanisms as well as neural relays, ultimately resulting in weight gain and obesity (Fig. 3). However, much remains to be understood as to how and when these interactions between microbe and host occur, in addition to the specific microbial players. Consequently, a better understanding of these principles can lead to the development of therapeutic options targeting the gut microbiota, providing a useful strategy for the treatment of obesity and related diseases.

Effects of an altered gut microbiome on the gut–brain axis potentially contributing to obesity. High fat feeding can alter host gut microbiota to impair gut–brain axis signaling pathways described within the current review, which can lead to increased food intake and weight gain. Detailed are the currently known mechanisms through which the gut microbiota can negatively impact the gut–brain axis control of energy homeostasis, such as changes in both nutrient sensing and gut peptide response, production of bacterial metabolites, namely SCFAs, and via increased intestinal permeability and metabolic endotoxemia. Numerous other mechanisms likely exist but remain to be further explored. Furthermore, perturbations in early life development or use of antibiotics may lead to an aberrant gut microbiota that can promote similar harmful physiological changes. EEC enteroendocrine cell, LPS lipopolysaccharide, SCFA short-chain fatty acid
Abbreviations
- AgRP:
-
Agouti-related protein
- AEA:
-
Anadamide
- ARC:
-
Arcuate nucleus
- BAT:
-
Brown adipose tissue
- BDNF:
-
Brain-derived neurotrophic factor
- CB1 :
-
Cannabinoid receptor 1
- CCK:
-
Cholecystokinin
- CCK-1R:
-
CCK-1 receptor
- CNS:
-
Central nervous system
- DVC:
-
Dorsal vagal complex
- ENS:
-
Enteric nervous system
- EEC:
-
Enteroendocrine cell
- GI:
-
Gastrointestinal
- GF:
-
Germ free
- GLP-1:
-
Glucagon-like peptide 1
- GLP-1R:
-
GLP-1 receptor
- GPR:
-
G-coupled protein receptor
- IP:
-
Intraperitoneal
- KO:
-
Knockout
- LPS:
-
Lipopolysaccharide
- NPY:
-
Neuropeptide Y
- NTS:
-
Nucleus tractus solitarius
- OTU:
-
Operational taxonomic unit
- OXM:
-
Oxyntomodulin
- PVN:
-
Paraventricular nucleus
- PYY:
-
Peptide YY
- TLR:
-
Toll-like receptor
- Y2R:
-
Y2 receptor
References
Duca FA, Bauer PV, Hamr SC, Lam TK (2015) Glucoregulatory relevance of small intestinal nutrient sensing in physiology, bariatric surgery, and pharmacology. Cell Metab 22:367–380
Mayer EA (2011) Gut feelings: the emerging biology of gut–brain communication. Nat Rev Neurosci 12:453–466
Hamr SC, Wang B, Swartz TD, Duca FA (2015) Does nutrient sensing determine how we “see” food? Curr Diab Rep 15:604
Psichas A, Reimann F, Gribble FM (2015) Gut chemosensing mechanisms. J Clin Invest 125:908–917
Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CA, Parker HE, Morley TC, Yeo GS, Reimann F, Gribble FM (2012) Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153:3054–3065
Berthoud HR, Kressel M, Raybould HE, Neuhuber WL (1995) Vagal sensors in the rat duodenal mucosa: distribution and structure as revealed by in vivo DiI-tracing. Anat Embryol (Berl) 191:203–212
Prechtl JC, Powley TL (1990) The fiber composition of the abdominal vagus of the rat. Anat Embryol (Berl) 181:101–115
Dockray GJ (2013) Enteroendocrine cell signalling via the vagus nerve. Curr Opin Pharmacol 13:954–958
Amato A, Cinci L, Rotondo A, Serio R, Faussone-Pellegrini MS, Vannucchi MG, Mule F (2010) Peripheral motor action of glucagon-like peptide-1 through enteric neuronal receptors. Neurogastroenterol Motil 22:664-e203
Patterson LM, Zheng H, Berthoud HR (2002) Vagal afferents innervating the gastrointestinal tract and CCKA-receptor immunoreactivity. Anat Rec 266:10–20
Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, Gribble FM, Reimann F (2014) Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63:1224–1233
Costa M, Brookes SJ, Hennig GW (2000) Anatomy and physiology of the enteric nervous system. Gut 47(Suppl 4):iv15–iv19 (discussion iv26)
Sayegh AI, Covasa M, Ritter RC (2004) Intestinal infusions of oleate and glucose activate distinct enteric neurons in the rat. Auton Neurosci 115:54–63
Ritter RC (2011) A tale of two endings: modulation of satiation by NMDA receptors on or near central and peripheral vagal afferent terminals. Physiol Behav 105:94–99
Norgren R (1978) Projections from the nucleus of the solitary tract in the rat. Neuroscience 3:207–218
Craig AD (1996) An ascending general homeostatic afferent pathway originating in lamina I. Prog Brain Res 107:225–242
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Zittel TT, De Giorgio R, Sternini C, Raybould HE (1994) Fos protein expression in the nucleus of the solitary tract in response to intestinal nutrients in awake rats. Brain Res 663:266–270
Campos CA, Shiina H, Silvas M, Page S, Ritter RC (2013) Vagal afferent NMDA receptors modulate CCK-induced reduction of food intake through synapsin I phosphorylation in adult male rats. Endocrinology 154:2613–2625
Babic T, Townsend RL, Patterson LM, Sutton GM, Zheng H, Berthoud HR (2009) Phenotype of neurons in the nucleus of the solitary tract that express CCK-induced activation of the ERK signaling pathway. Am J Physiol Regul Integr Comp Physiol 296:R845–R854
Sutton GM, Duos B, Patterson LM, Berthoud HR (2005) Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology 146:3739–3747
Seeley RJ, Grill HJ, Kaplan JM (1994) Neurological dissociation of gastrointestinal and metabolic contributions to meal size control. Behav Neurosci 108:347–352
Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR (2002) Gut hormone PYY(3–36) physiologically inhibits food intake. Nature 418:650–654
Vincent KM, Sharp JW, Raybould HE (2011) Intestinal glucose-induced calcium-calmodulin kinase signaling in the gut–brain axis in awake rats. Neurogastroenterol Motil 23:e282–e293
Davis JD, Smith GP (1990) Learning to sham feed: behavioral adjustments to loss of physiological postingestional stimuli. Am J Physiol 259:R1228–R1235
Phillips RJ, Powley TL (1996) Gastric volume rather than nutrient content inhibits food intake. Am J Physiol 271:R766–R769
Raybould HE, Gschossman JM, Ennes H, Lembo T, Mayer EA (1999) Involvement of stretch-sensitive calcium flux in mechanical transduction in visceral afferents. J Auton Nerv Syst 75:1–6
Raybould HE, Gayton RJ, Dockray GJ (1985) CNS effects of circulating CCK8: involvement of brainstem neurones responding to gastric distension. Brain Res 342:187–190
Cooke AR, Clark ED (1976) Effect of first part of duodenum on gastric emptying in dogs: response to acid, fat, glucose, and neural blockade. Gastroenterology 70:550–555
Talsania T, Anini Y, Siu S, Drucker DJ, Brubaker PL (2005) Peripheral exendin-4 and peptide YY(3–36) synergistically reduce food intake through different mechanisms in mice. Endocrinology 146:3748–3756
Wickbom J, Herrington MK, Permert J, Jansson A, Arnelo U (2008) Gastric emptying in response to IAPP and CCK in rats with subdiaphragmatic afferent vagotomy. Regul Pept 148:21–25
Imeryuz N, Yegen BC, Bozkurt A, Coskun T, Villanueva-Penacarrillo ML, Ulusoy NB (1997) Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. Am J Physiol 273:G920–G927
Reidelberger RD, Kalogeris TJ, Leung PM, Mendel VE (1983) Postgastric satiety in the sham-feeding rat. Am J Physiol 244:R872–R881
Gibbs J, Maddison SP, Rolls ET (1981) Satiety role of the small intestine examined in sham-feeding rhesus monkeys. J Comp Physiol Psychol 95:1003–1015
Yox DP, Ritter RC (1988) Capsaicin attenuates suppression of sham feeding induced by intestinal nutrients. Am J Physiol 255:R569–R574
Yox DP, Stokesberry H, Ritter RC (1991) Vagotomy attenuates suppression of sham feeding induced by intestinal nutrients. Am J Physiol 260:R503–R508
Sclafani A, Ackroff K, Schwartz GJ (2003) Selective effects of vagal deafferentation and celiac-superior mesenteric ganglionectomy on the reinforcing and satiating action of intestinal nutrients. Physiol Behav 78:285–294
Gibbs J, Young RC, Smith GP (1973) Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol 84:488–495
Cote CD, Zadeh-Tahmasebi M, Rasmussen BA, Duca FA, Lam TK (2014) Hormonal signaling in the gut. J Biol Chem 289:11642–11649
Moriarty P, Dimaline R, Thompson DG, Dockray GJ (1997) Characterization of cholecystokinin A and cholecystokinin B receptors expressed by vagal afferent neurons. Neuroscience 79:905–913
Brenner L, Ritter RC (1995) Peptide cholesystokinin receptor antagonist increases food intake in rats. Appetite 24:1–9
Moran TH, Ameglio PJ, Schwartz GJ, McHugh PR (1992) Blockade of type A, not type B, CCK receptors attenuates satiety actions of exogenous and endogenous CCK. Am J Physiol 262:R46–R50
Rinaman L (2003) Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci 23:10084–10092
Lechan RM, Fekete C (2006) The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res 153:209–235
Brenner L, Yox DP, Ritter RC (1993) Suppression of sham feeding by intraintestinal nutrients is not correlated with plasma cholecystokinin elevation. Am J Physiol 264:R972–R976
Liddle RA, Green GM, Conrad CK, Williams JA (1986) Proteins but not amino acids, carbohydrates, or fats stimulate cholecystokinin secretion in the rat. Am J Physiol 251:G243–G248
Della-Fera MA, Baile CA (1980) CCK-octapeptide injected in CSF decreases meal size and daily food intake in sheep. Peptides 1:51–54
Schick RR, Stevens CW, Yaksh TL, Go VL (1988) Chronic intraventricular administration of cholecystokinin octapeptide (CCK-8) suppresses feeding in rats. Brain Res 448:294–298
Lo CC, Davidson WS, Hibbard SK, Georgievsky M, Lee A, Tso P, Woods SC (2014) Intraperitoneal CCK and fourth-intraventricular Apo AIV require both peripheral and NTS CCK1R to reduce food intake in male rats. Endocrinology 155:1700–1707
Blevins JE, Stanley BG, Reidelberger RD (2000) Brain regions where cholecystokinin suppresses feeding in rats. Brain Res 860:1–10
Blevins JE, Hamel FG, Fairbairn E, Stanley BG, Reidelberger RD (2000) Effects of paraventricular nucleus injection of CCK-8 on plasma CCK-8 levels in rats. Brain Res 860:11–20
Beinfeld MC (2001) An introduction to neuronal cholecystokinin. Peptides 22:1197–1200
Sayegh AI (2013) The role of cholecystokinin receptors in the short-term control of food intake. Prog Mol Biol Transl Sci 114:277–316
Calingasan N, Ritter S, Ritter R, Brenner L (1992) Low-dose near-celiac arterial cholecystokinin suppresses food intake in rats. Am J Physiol 263:R572–R577
Cox JE, McCown SM, Bridges JM, Tyler WJ (1996) Inhibition of sucrose intake by continuous celiac, superior mesenteric, and intravenous CCK-8 infusions. Am J Physiol 270:R319–R325
Duca FA, Yue JT (2014) Fatty acid sensing in the gut and the hypothalamus: in vivo and in vitro perspectives. Mol Cell Endocrinol 397:23–33
Dube PE, Brubaker PL (2004) Nutrient, neural and endocrine control of glucagon-like peptide secretion. Horm Metab Res 36:755–760
Steinert RE, Beglinger C, Langhans W (2015) Intestinal GLP-1 and satiation-from man to rodents and back. Int J Obes (Lond). [Epub ahead of print]
Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V (1993) Glucagon-like peptide-1 (7–36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. J Endocrinol 138:159–166
Roberge JN, Brubaker PL (1993) Regulation of intestinal proglucagon-derived peptide secretion by glucose-dependent insulinotropic peptide in a novel enteroendocrine loop. Endocrinology 133:233–240
Roberge JN, Gronau KA, Brubaker PL (1996) Gastrin-releasing peptide is a novel mediator of proximal nutrient-induced proglucagon-derived peptide secretion from the distal gut. Endocrinology 137:2383–2388
Theodorakis MJ, Carlson O, Michopoulos S, Doyle ME, Juhaszova M, Petraki K, Egan JM (2006) Human duodenal enteroendocrine cells: source of both incretin peptides, GLP-1 and GIP. Am J Physiol Endocrinol Metab 290:E550–E559
Svendsen B, Pedersen J, Albrechtsen NJ, Hartmann B, Torang S, Rehfeld JF, Poulsen SS, Holst JJ (2015) An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 156:847–857
Hayes MR, De Jonghe BC, Kanoski SE (2010) Role of the glucagon-like-peptide-1 receptor in the control of energy balance. Physiol Behav 100:503–510
Williams DL (2009) Minireview: finding the sweet spot: peripheral versus central glucagon-like peptide 1 action in feeding and glucose homeostasis. Endocrinology 150:2997–3001
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439
Nakagawa A, Satake H, Nakabayashi H, Nishizawa M, Furuya K, Nakano S, Kigoshi T, Nakayama K, Uchida K (2004) Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton Neurosci 110:36–43
Kakei M, Yada T, Nakagawa A, Nakabayashi H (2002) Glucagon-like peptide-1 evokes action potentials and increases cytosolic Ca2+ in rat nodose ganglion neurons. Auton Neurosci 102:39–44
Abbott CR, Monteiro M, Small CJ, Sajedi A, Smith KL, Parkinson JR, Ghatei MA, Bloom SR (2005) The inhibitory effects of peripheral administration of peptide YY(3–36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res 1044:127–131
Hayes MR, Kanoski SE, De Jonghe BC, Leichner TM, Alhadeff AL, Fortin SM, Arnold M, Langhans W, Grill HJ (2011) The common hepatic branch of the vagus is not required to mediate the glycemic and food intake suppressive effects of glucagon-like-peptide-1. Am J Physiol Regul Integr Comp Physiol 301:R1479–R1485
Ruttimann EB, Arnold M, Hillebrand JJ, Geary N, Langhans W (2009) Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology 150:1174–1181
Zhang J, Ritter RC (2012) Circulating GLP-1 and CCK-8 reduce food intake by capsaicin-insensitive, nonvagal mechanisms. Am J Physiol Regul Integr Comp Physiol 302:R264–R273
D’Alessio D, Lu W, Sun W, Zheng S, Yang Q, Seeley R, Woods SC, Tso P (2007) Fasting and postprandial concentrations of GLP-1 in intestinal lymph and portal plasma: evidence for selective release of GLP-1 in the lymph system. Am J Physiol Regul Integr Comp Physiol 293:R2163–R2169
Ohlsson L, Kohan AB, Tso P, Ahren B (2014) GLP-1 released to the mesenteric lymph duct in mice: effects of glucose and fat. Regul Pept 189:40–45
Kohan A, Yoder S, Tso P (2010) Lymphatics in intestinal transport of nutrients and gastrointestinal hormones. Ann N Y Acad Sci 1207(Suppl 1):E44–E51
Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C (1997) Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 77:257–270
Campos RV, Lee YC, Drucker DJ (1994) Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 134:2156–2164
Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, Wilding JP, Smith DM, Ghatei MA, Herbert J, Bloom SR (1996) A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379:69–72
Tang-Christensen M, Larsen PJ, Goke R, Fink-Jensen A, Jessop DS, Moller M, Sheikh SP (1996) Central administration of GLP-1-(7–36) amide inhibits food and water intake in rats. Am J Physiol 271:R848–R856
Williams DL, Baskin DG, Schwartz MW (2009) Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 150:1680–1687
Grill HJ, Hayes MR (2009) The nucleus tractus solitarius: a portal for visceral afferent signal processing, energy status assessment and integration of their combined effects on food intake. Int J Obes (Lond) 33(Suppl 1):S11–S15
Plamboeck A, Veedfald S, Deacon CF, Hartmann B, Wettergren A, Svendsen LB, Meisner S, Hovendal C, Vilsboll T, Knop FK, Holst JJ (2013) The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastrointest Liver Physiol 304:G1117–G1127
Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR (2011) Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 152:3103–3112
Pedersen-Bjergaard U, Host U, Kelbaek H, Schifter S, Rehfeld JF, Faber J, Christensen NJ (1996) Influence of meal composition on postprandial peripheral plasma concentrations of vasoactive peptides in man. Scand J Clin Lab Invest 56:497–503
Batterham RL, Heffron H, Kapoor S, Chivers JE, Chandarana K, Herzog H, Le Roux CW, Thomas EL, Bell JD, Withers DJ (2006) Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab 4:223–233
Rozengurt N, Wu SV, Chen MC, Huang C, Sternini C, Rozengurt E (2006) Colocalization of the alpha-subunit of gustducin with PYY and GLP-1 in L cells of human colon. Am J Physiol Gastrointest Liver Physiol 291:G792–G802
Fu-Cheng X, Anini Y, Chariot J, Castex N, Galmiche JP, Roze C (1997) Mechanisms of peptide YY release induced by an intraduodenal meal in rats: neural regulation by proximal gut. Pflugers Arch 433:571–579
Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS, Ghatei MA, Bloom SR (2003) Inhibition of food intake in obese subjects by peptide YY3–36. N Engl J Med 349:941–948
Koda S, Date Y, Murakami N, Shimbara T, Hanada T, Toshinai K, Niijima A, Furuya M, Inomata N, Osuye K, Nakazato M (2005) The role of the vagal nerve in peripheral PYY3–36-induced feeding reduction in rats. Endocrinology 146:2369–2375
Halatchev IG, Cone RD (2005) Peripheral administration of PYY(3–36) produces conditioned taste aversion in mice. Cell Metab 1:159–168
Sainsbury A, Schwarzer C, Couzens M, Fetissov S, Furtinger S, Jenkins A, Cox HM, Sperk G, Hokfelt T, Herzog H (2002) Important role of hypothalamic Y2 receptors in body weight regulation revealed in conditional knockout mice. Proc Natl Acad Sci USA 99:8938–8943
Broberger C, Landry M, Wong H, Walsh JN, Hokfelt T (1997) Subtypes Y1 and Y2 of the neuropeptide Y receptor are respectively expressed in pro-opiomelanocortin- and neuropeptide-Y-containing neurons of the rat hypothalamic arcuate nucleus. Neuroendocrinology 66:393–408
King PJ, Williams G, Doods H, Widdowson PS (2000) Effect of a selective neuropeptide Y Y(2) receptor antagonist, BIIE0246 on neuropeptide Y release. Eur J Pharmacol 396:R1–R3
Li Y, Wu XY, Zhu JX, Owyang C (2001) Intestinal serotonin acts as paracrine substance to mediate pancreatic secretion stimulated by luminal factors. Am J Physiol Gastrointest Liver Physiol 281:G916–G923
Li B, Shao D, Luo Y, Wang P, Liu C, Zhang X, Cui R (2015) Role of 5-HT3 receptor on food intake in fed and fasted mice. PLoS One 10:e0121473
Halford JC, Lawton CL, Blundell JE (1997) The 5-HT2 receptor agonist MK-212 reduces food intake and increases resting but prevents the behavioural satiety sequence. Pharmacol Biochem Behav 56:41–46
Savastano DM, Hayes MR, Covasa M (2007) Serotonin-type 3 receptors mediate intestinal lipid-induced satiation and Fos-like immunoreactivity in the dorsal hindbrain. Am J Physiol Regul Integr Comp Physiol 292:R1063–R1070
Crane JD, Palanivel R, Mottillo EP, Bujak AL, Wang H, Ford RJ, Collins A, Blumer RM, Fullerton MD, Yabut JM, Kim JJ, Ghia JE, Hamza SM, Morrison KM, Schertzer JD, Dyck JR, Khan WI, Steinberg GR (2015) Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat Med 21:166–172
Fujimoto K, Cardelli JA, Tso P (1992) Increased apolipoprotein A-IV in rat mesenteric lymph after lipid meal acts as a physiological signal for satiation. Am J Physiol 262:G1002–G1006
Sakata Y, Fujimoto K, Ogata S, Koyama T, Fukagawa K, Sakai T, Tso P (1996) Postabsorptive factors are important for satiation in rats after a lipid meal. Am J Physiol 271:G438–G442
Raybould HE, Meyer JH, Tabrizi Y, Liddle RA, Tso P (1998) Inhibition of gastric emptying in response to intestinal lipid is dependent on chylomicron formation. Am J Physiol 274:R1834–R1838
Lo CC, Langhans W, Georgievsky M, Arnold M, Caldwell JL, Cheng S, Liu M, Woods SC, Tso P (2012) Apolipoprotein AIV requires cholecystokinin and vagal nerves to suppress food intake. Endocrinology 153:5857–5865
Williams CM, Rogers PJ, Kirkham TC (1998) Hyperphagia in pre-fed rats following oral delta9-THC. Physiol Behav 65:343–346
Christopoulou FD, Kiortsis DN (2011) An overview of the metabolic effects of rimonabant in randomized controlled trials: potential for other cannabinoid 1 receptor blockers in obesity. J Clin Pharm Ther 36:10–18
Paulino G, Barbier de la Serre C, Knotts TA, Oort PJ, Newman JW, Adams SH, Raybould HE (2009) Increased expression of receptors for orexigenic factors in nodose ganglion of diet-induced obese rats. Am J Physiol Endocrinol Metab 296:E898–E903
Gomez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del Arco I, Cippitelli A, Nava F, Piomelli D, Rodriguez de Fonseca F (2002) A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci 22:9612–9617
Kentish SJ, Page AJ (2015) The role of gastrointestinal vagal afferent fibres in obesity. J Physiol 593:775–786
Fioravanti B, De Felice M, Stucky CL, Medler KA, Luo MC, Gardell LR, Ibrahim M, Malan TP Jr, Yamamura HI, Ossipov MH, King T, Lai J, Porreca F, Vanderah TW (2008) Constitutive activity at the cannabinoid CB1 receptor is required for behavioral response to noxious chemical stimulation of TRPV1: antinociceptive actions of CB1 inverse agonists. J Neurosci 28:11593–11602
Barbara G, Wang B, Stanghellini V, de Giorgio R, Cremon C, Di Nardo G, Trevisani M, Campi B, Geppetti P, Tonini M, Bunnett NW, Grundy D, Corinaldesi R (2007) Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 132:26–37
Watkins LR, Maier SF, Goehler LE (1995) Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci 57:1011–1026
McDermott JR, Leslie FC, D’Amato M, Thompson DG, Grencis RK, McLaughlin JT (2006) Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut 55:492–497
Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, Possemiers S, Van Holle A, Francois P, de Vos WM, Delzenne NM, Schrenzel J, Cani PD (2011) Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60:2775–2786
Bucinskaite V, Kurosawa M, Miyasaka K, Funakoshi A, Lundeberg T (1997) Interleukin-1beta sensitizes the response of the gastric vagal afferent to cholecystokinin in rat. Neurosci Lett 229:33–36
Gaige S, Abou E, Abysique A, Bouvier M (2004) Effects of interactions between interleukin-1 beta and leptin on cat intestinal vagal mechanoreceptors. J Physiol 555:297–310
Pavlov VA, Tracey KJ (2012) The vagus nerve and the inflammatory reflex—linking immunity and metabolism. Nat Rev Endocrinol 8:743–754
Ravussin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG, Boyce V, Howard BV, Bogardus C (1988) Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med 318:467–472
Griffiths M, Payne PR, Stunkard AJ, Rivers JP, Cox M (1990) Metabolic rate and physical development in children at risk of obesity. Lancet 336:76–78
Christiansen E, Garby L (2002) Prediction of body weight changes caused by changes in energy balance. Eur J Clin Invest 32:826–830
Westerterp KR (2004) Diet induced thermogenesis. Nutr Metab (Lond) 1:5
Lowell BB, Spiegelman BM (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404:652–660
Hwa JJ, Ghibaudi L, Williams P, Witten MB, Tedesco R, Strader CD (1998) Differential effects of intracerebroventricular glucagon-like peptide-1 on feeding and energy expenditure regulation. Peptides 19:869–875
Dakin CL, Small CJ, Park AJ, Seth A, Ghatei MA, Bloom SR (2002) Repeated ICV administration of oxyntomodulin causes a greater reduction in body weight gain than in pair-fed rats. Am J Physiol Endocrinol Metab 283:E1173–E1177
Sloth B, Holst JJ, Flint A, Gregersen NT, Astrup A (2007) Effects of PYY1–36 and PYY3–36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. Am J Physiol Endocrinol Metab 292:E1062–E1068
Blouet C, Schwartz GJ (2012) Duodenal lipid sensing activates vagal afferents to regulate non-shivering brown fat thermogenesis in rats. PLoS One 7:e51898
Lockie SH, Heppner KM, Chaudhary N, Chabenne JR, Morgan DA, Veyrat-Durebex C, Ananthakrishnan G, Rohner-Jeanrenaud F, Drucker DJ, DiMarchi R, Rahmouni K, Oldfield BJ, Tschop MH, Perez-Tilve D (2012) Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. Diabetes 61:2753–2762
Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, Atkins AR, Khvat A, Schnabl B, Yu RT, Brenner DA, Coulter S, Liddle C, Schoonjans K, Olefsky JM, Saltiel AR, Downes M, Evans RM (2015) Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 21:159–165
Berteus Forslund H, Lindroos AK, Sjostrom L, Lissner L (2002) Meal patterns and obesity in Swedish women-a simple instrument describing usual meal types, frequency and temporal distribution. Eur J Clin Nutr 56:740–747
Berg C, Lappas G, Wolk A, Strandhagen E, Toren K, Rosengren A, Thelle D, Lissner L (2009) Eating patterns and portion size associated with obesity in a Swedish population. Appetite 52:21–26
Papadimitriou MA, Krzemien AA, Hahn PM, Van Vugt DA (2007) Peptide YY(3–36)-induced inhibition of food intake in female monkeys. Brain Res 1175:60–65
Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ (1994) Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 43:535–539
Baranowska B, Radzikowska M, Wasilewska-Dziubinska E, Roguski K, Borowiec M (2000) Disturbed release of gastrointestinal peptides in anorexia nervosa and in obesity. Diabetes Obes Metab 2:99–103
Xu J, McNearney TA, Chen JD (2011) Impaired postprandial releases/syntheses of ghrelin and PYY(3–36) and blunted responses to exogenous ghrelin and PYY(3–36) in a rodent model of diet-induced obesity. J Gastroenterol Hepatol 26:700–705
Duca FA, Sakar Y, Covasa M (2013) Combination of obesity and high-fat feeding diminishes sensitivity to GLP-1R agonist exendin-4. Diabetes 62:2410–2415
Covasa M, Ritter RC (1998) Rats maintained on high-fat diets exhibit reduced satiety in response to CCK and bombesin. Peptides 19:1407–1415
Donovan MJ, Paulino G, Raybould HE (2009) Activation of hindbrain neurons in response to gastrointestinal lipid is attenuated by high fat, high energy diets in mice prone to diet-induced obesity. Brain Res 1248:136–140
Daly DM, Park SJ, Valinsky WC, Beyak MJ (2011) Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol 589:2857–2870
Duca FA, Covasa M (2012) Current and emerging concepts on the role of peripheral signals in the control of food intake and development of obesity. Br J Nutr 108:778–793
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI (2007) The human microbiome project. Nature 449:804–810
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65
Wostmann BS, Larkin C, Moriarty A, Bruckner-Kardoss E (1983) Dietary intake, energy metabolism, and excretory losses of adult male germfree Wistar rats. Lab Anim Sci 33:46–50
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101:15718–15723
Backhed F, Manchester JK, Semenkovich CF, Gordon JI (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104:979–984
Hoverstad T, Midtvedt T (1986) Short-chain fatty acids in germfree mice and rats. J Nutr 116:1772–1776
Al-Asmakh M, Zadjali F (2015) Use of germ-free animal models in microbiota-related research. J Microbiol Biotechnol 25:1583–1588
Smith K, McCoy KD, Macpherson AJ (2007) Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol 19:59–69
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102:11070–11075
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484
Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, Mariat D, Corthier G, Dore J, Henegar C, Rizkalla S, Clement K (2010) Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes 59:3049–3057
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, Flint HJ (2008) Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 32:1720–1724
Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, Krajmalnik-Brown R (2009) Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA 106:2365–2370
Duca FA, Sakar Y, Lepage P, Devime F, Langelier B, Dore J, Covasa M (2014) Replication of obesity and associated signaling pathways through transfer of microbiota from obese-prone rats. Diabetes 63:1624–1636
Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F, Clarke SF, O’Toole PW, Quigley EM, Stanton C, Ross PR, O’Doherty RM, Shanahan F (2010) Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59:1635–1642
Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, de Vos WM, Zoetendal EG (2013) Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J 7:707–717
Everard A, Cani PD (2013) Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol 27:73–83
Reimann F, Tolhurst G, Gribble FM (2012) G-protein-coupled receptors in intestinal chemosensation. Cell Metab 15:421–431
Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, Gordon JI (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci USA 105:16767–16772
Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, Hardt PD (2010) Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 18:190–195
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461:1282–1286
Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TM, Comelli EM (2014) Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes 4:e121
Fava F, Gitau R, Griffin BA, Gibson GR, Tuohy KM, Lovegrove JA (2013) The type and quantity of dietary fat and carbohydrate alter faecal microbiome and short-chain fatty acid excretion in a metabolic syndrome ‘at-risk’ population. Int J Obes (Lond) 37:216–223
Pan XD, Chen FQ, Wu TX, Tang HG, Zhao ZY (2009) Prebiotic oligosaccharides change the concentrations of short-chain fatty acids and the microbial population of mouse bowel. J Zhejiang Univ Sci B 10:258–263
Liou AP, Paziuk M, Luevano JM Jr, Machineni S, Turnbaugh PJ, Kaplan LM (2013) Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med 5:178ra141
Cani PD, Daubioul CA, Reusens B, Remacle C, Catillon G, Delzenne NM (2005) Involvement of endogenous glucagon-like peptide-1(7–36) amide on glycaemia-lowering effect of oligofructose in streptozotocin-treated rats. J Endocrinol 185:457–465
Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SE, MacDougall K, Preston T, Tedford C, Finlayson GS, Blundell JE, Bell JD, Thomas EL, Mt-Isa S, Ashby D, Gibson GR, Kolida S, Dhillo WS, Bloom SR, Morley W, Clegg S, Frost G (2015) Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 64:1744–1754
den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54:2325–2340
Lin HV, Frassetto A, Kowalik EJ Jr, Nawrocki AR, Lu MM, Kosinski JR, Hubert JA, Szeto D, Yao X, Forrest G, Marsh DJ (2012) Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS One 7:e35240
Puddu A, Sanguineti R, Montecucco F, Viviani GL (2014) Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediators Inflamm 2014:162021
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58:1509–1517
Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, Anastasovska J, Ghourab S, Hankir M, Zhang S, Carling D, Swann JR, Gibson G, Viardot A, Morrison D, Louise Thomas E, Bell JD (2014) The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun 5:3611
Freeland KR, Wolever TM (2010) Acute effects of intravenous and rectal acetate on glucagon-like peptide-1, peptide YY, ghrelin, adiponectin and tumour necrosis factor-alpha. Br J Nutr 103:460–466
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M (2003) Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 278:25481–25489
Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ (2003) The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278:11312–11319
Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I (2015) Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 7:2839–2849
Tazoe H, Otomo Y, Karaki S, Kato I, Fukami Y, Terasaki M, Kuwahara A (2009) Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed Res 30:149–156
Karaki S, Tazoe H, Hayashi H, Kashiwabara H, Tooyama K, Suzuki Y, Kuwahara A (2008) Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J Mol Histol 39:135–142
Karaki S, Mitsui R, Hayashi H, Kato I, Sugiya H, Iwanaga T, Furness JB, Kuwahara A (2006) Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res 324:353–360
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble FM (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61:364–371
Tang C, Ahmed K, Gille A, Lu S, Grone HJ, Tunaru S, Offermanns S (2015) Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med 21:173–177
Nohr MK, Egerod KL, Christiansen SH, Gille A, Offermanns S, Schwartz TW, Moller M (2015) Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience 290:126–137
Nohr MK, Pedersen MH, Gille A, Egerod KL, Engelstoft MS, Husted AS, Sichlau RM, Grunddal KV, Poulsen SS, Han S, Jones RM, Offermanns S, Schwartz TW (2013) GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 154:3552–3564
Duca FA, Swartz TD, Sakar Y, Covasa M (2012) Increased oral detection, but decreased intestinal signaling for fats in mice lacking gut microbiota. PLoS One 7:e39748
Swartz TD, Duca FA, de Wouters T, Sakar Y, Covasa M (2012) Up-regulation of intestinal type 1 taste receptor 3 and sodium glucose luminal transporter-1 expression and increased sucrose intake in mice lacking gut microbiota. Br J Nutr 107:621–630
Fredborg M, Theil PK, Jensen BB, Purup S (2012) G protein-coupled receptor120 (GPR120) transcription in intestinal epithelial cells is significantly affected by bacteria belonging to the Bacteroides, Proteobacteria, and Firmicutes phyla. J Anim Sci 90(Suppl 4):10–12
Dewulf EM, Cani PD, Claus SP, Fuentes S, Puylaert PG, Neyrinck AM, Bindels LB, de Vos WM, Gibson GR, Thissen JP, Delzenne NM (2013) Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 62:1112–1121
Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM (2009) Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58:1091–1103
Neyrinck AM, Van Hee VF, Piront N, De Backer F, Toussaint O, Cani PD, Delzenne NM (2012) Wheat-derived arabinoxylan oligosaccharides with prebiotic effect increase satietogenic gut peptides and reduce metabolic endotoxemia in diet-induced obese mice. Nutr Diabetes 2:e28
Cani PD, Neyrinck AM, Maton N, Delzenne NM (2005) Oligofructose promotes satiety in rats fed a high-fat diet: involvement of glucagon-like Peptide-1. Obes Res 13:1000–1007
Cani PD, Dewever C, Delzenne NM (2004) Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr 92:521–526
Cani PD, Joly E, Horsmans Y, Delzenne NM (2006) Oligofructose promotes satiety in healthy human: a pilot study. Eur J Clin Nutr 60:567–572
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772
de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE (2010) Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299:G440–G448
Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Backhed F (2015) Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab 22:658–668
Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, Denis RG, Cochez P, Pierard F, Castel J, Bindels LB, Plovier H, Robine S, Muccioli GG, Renauld JC, Dumoutier L, Delzenne NM, Luquet S, Backhed F, Cani PD (2014) Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun 5:5648
Zuo DC, Choi S, Shahi PK, Kim MY, Park CG, Kim YD, Lee J, Chang IY, So I, Jun JY (2013) Inhibition of pacemaker activity in interstitial cells of Cajal by LPS via NF-kappaB and MAP kinase. World J Gastroenterol 19:1210–1218
Hosoi T, Okuma Y, Matsuda T, Nomura Y (2005) Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci 120:104–107
de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE (2011) Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab 301:E187–E195
de La Serre CB, de Lartigue G, Raybould HE (2015) Chronic exposure to low dose bacterial lipopolysaccharide inhibits leptin signaling in vagal afferent neurons. Physiol Behav 139:188–194
de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE (2012) Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PLoS One 7:e32967
Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 108:3047–3052
Gareau MG, Wine E, Rodrigues DM, Cho JH, Whary MT, Philpott DJ, Macqueen G, Sherman PM (2011) Bacterial infection causes stress-induced memory dysfunction in mice. Gut 60:307–317
Neufeld KM, Kang N, Bienenstock J, Foster JA (2011) Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol Motil 23(255–264):e119
Bercik P, Verdu EF, Foster JA, Macri J, Potter M, Huang X, Malinowski P, Jackson W, Blennerhassett P, Neufeld KA, Lu J, Khan WI, Corthesy-Theulaz I, Cherbut C, Bergonzelli GE, Collins SM (2010) Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology 139(2102–2112):e2101
Steenbergen L, Sellaro R, van Hemert S, Bosch JA, Colzato LS (2015) A randomized controlled trial to test the effect of multispecies probiotics on cognitive reactivity to sad mood. Brain Behav Immun 48:258–264
Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, Houdeau E, Fioramonti J, Bueno L, Theodorou V (2012) Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 37:1885–1895
Schele E, Grahnemo L, Anesten F, Hallen A, Backhed F, Jansson JO (2013) The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 154:3643–3651
Mumphrey MB, Patterson LM, Zheng H, Berthoud HR (2013) Roux-en-Y gastric bypass surgery increases number but not density of CCK-, GLP-1-, 5-HT-, and neurotensin-expressing enteroendocrine cells in rats. Neurogastroenterol Motil 25:e70–e79
Madsbad S, Krarup T, Deacon CF, Holst JJ (2008) Glucagon-like peptide receptor agonists and dipeptidyl peptidase-4 inhibitors in the treatment of diabetes: a review of clinical trials. Curr Opin Clin Nutr Metab Care 11:491–499
Irwin N, Montgomery IA, Moffett RC, Flatt PR (2013) Chemical cholecystokinin receptor activation protects against obesity-diabetes in high fat fed mice and has sustainable beneficial effects in genetic ob/ob mice. Biochem Pharmacol 85:81–91
Irwin N, Frizelle P, O’Harte FP, Flatt PR (2013) (pGlu-Gln)-CCK-8[mPEG]: a novel, long-acting, mini-PEGylated cholecystokinin (CCK) agonist that improves metabolic status in dietary-induced diabetes. Biochim Biophys Acta 1830:4009–4016
van Bloemendaal L, IJzerman RG, Ten Kulve JS, Barkhof F, Konrad RJ, Drent ML, Veltman DJ, Diamant M (2014) GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes 63:4186–4196
Yang Y, Moghadam AA, Cordner ZA, Liang NC, Moran TH (2014) Long term exendin-4 treatment reduces food intake and body weight and alters expression of brain homeostatic and reward markers. Endocrinology 155:3473–3483
Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, Chabenne J, Zhang L, Habegger KM, Fischer K, Campbell JE, Sandoval D, Seeley RJ, Bleicher K, Uhles S, Riboulet W, Funk J, Hertel C, Belli S, Sebokova E, Conde-Knape K, Konkar A, Drucker DJ, Gelfanov V, Pfluger PT, Muller TD, Perez-Tilve D, DiMarchi RD, Tschop MH (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36
Osto M, Abegg K, Bueter M, le Roux CW, Cani PD, Lutz TA (2013) Roux-en-Y gastric bypass surgery in rats alters gut microbiota profile along the intestine. Physiol Behav 119:92–96
Casselbrant A, Elias E, Fandriks L, Wallenius V (2015) Expression of tight-junction proteins in human proximal small intestinal mucosa before and after Roux-en-Y gastric bypass surgery. Surg Obes Relat Dis 11:45–53
Tremaroli V, Karlsson F, Werling M, Stahlman M, Kovatcheva-Datchary P, Olbers T, Fandriks L, le Roux CW, Nielsen J, Backhed F (2015) Roux-en-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell Metab 22:228–238
Dubos R, Schaedler RW, Costello RL (1963) The effect of antibacterial drugs on the weight of mice. J Exp Med 117:245–257
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57:1470–1481
Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, Chung J, Sohn J, Barber CM, Goldfarb DS, Raju K, Abubucker S, Zhou Y, Ruiz VE, Li H, Mitreva M, Alekseyenko AV, Weinstock GM, Sodergren E, Blaser MJ (2015) Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat Commun 6:7486
Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, Zarate Rodriguez JG, Rogers AB, Robine N, Loke P, Blaser MJ (2014) Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158:705–721
Cox LM, Blaser MJ (2013) Pathways in microbe-induced obesity. Cell Metab 17:883–894
Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, Li H, Alekseyenko AV, Blaser MJ (2012) Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488:621–626
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328:228–231
Tomasik PJ, Sztefko K (2009) The effect of enteral and parenteral feeding on secretion of orexigenic peptides in infants. BMC Gastroenterol 9:92
De Palma G, Blennerhassett P, Lu J, Deng Y, Park AJ, Green W, Denou E, Silva MA, Santacruz A, Sanz Y, Surette MG, Verdu EF, Collins SM, Bercik P (2015) Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat Commun 6:7735
Alang N, Kelly CR (2015) Weight gain after fecal microbiota transplantation. Open Forum Infect Dis 2:ofv004
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143(913–916):e917
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bauer, P.V., Hamr, S.C. & Duca, F.A. Regulation of energy balance by a gut–brain axis and involvement of the gut microbiota. Cell. Mol. Life Sci. 73, 737–755 (2016). https://doi.org/10.1007/s00018-015-2083-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2083-z