
Abstract
Short-chain fatty acids (SCFAs), 2–4 carbon monocarboxylates including acetate, propionate and butyrate, are known to have a variety of physiological and pathophysiological effects on the intestine. Previously, we reported that the SCFA receptor, G-protein coupled receptor 43 (GPR43), is expressed by enteroendocrine and mucosal mast cells in the rat intestine. In the present study, expression and localization of GPR43 were investigated in the human large intestine. Gene and protein expression of GPR43 in the human ascending colon was analyzed by reverse transcriptase/polymerase chain reaction and Western blotting, respectively. In addition, localization of GPR43 was investigated by immunohistochemistry. In RT-PCR analysis, GPR43 mRNA was detected in whole wall mRNA samples. Western blotting analysis revealed the expression of GPR43 protein in whole wall and scraped mucosa protein samples, but not in muscle or submucosa. GPR43 immunoreactivity was observed in the intracellularly in enterocytes and in the peptide YY-immunoreactive enteroendocrine cells. These results indicate that the short chain fatty acid receptor, GPR43 is expressed by enteroendocrine L cells containing peptide YY in the human large intestine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Short-chain fatty acids (SCFAs), specifically acetate, propionate and butyrate, are the major anions (present at about 100 mM) in the lumen of the large intestine. They are produced by bacterial fermentation of undigested carbohydrates. Luminal SCFAs are not only absorbed as nutrients across the intestinal epithelium, but also influence various physiological and pathophysiological functions of the gastrointestinal (GI) tract (Cummings et al. 1995; Topping and Clifton 2001; Andoh et al. 2003; Kles and Chang 2006). For example, SCFAs stimulate colonic blood flow (Mortensen and Hielsen 1995), fluid/electrolyte uptake (Binder and Mehta 1989; Lutz and Scharrer 1991; Vidyasagar and Ramakrishna 2002), smooth muscle contraction (Yajima 1985; Mitsui et al. 2005a, b), transepithelial chloride secretion (Yajima 1988; Hubel and Russ 1993; Diener et al. 1996), exert proliferative stimuli of colonic epithelial cells (Sakata 1987; Kripke et al. 1989; Scheppach et al. 1992), and prevent colorectal cancer (D’Argenio et al. 1996). It is also known that the dietary fiber and SCFAs are possible to have efficacy for the inflammatory bowel disease (IBD), e.g. by inhibition of proinflammatory cytokine-induced NF-κB activation (Andoh et al. 2003; Galvez et al. 2005; Tedelind et al. 2007). Thus, dietary fiber and SCFAs are considered to have much potential for clinical use. In addition to their local effects, luminal SCFAs in the terminal ileum and colon have been reported to reduce motility in proximal GI regions, an inhibitory response probably mediated by peptide YY (PYY) release from the mucosa, and frequently called “ileal brake” (Cuche and Malbert 1999; Cuche et al. 2000). It is therefore important to investigate the mechanism for SCFA-induced responses in the GI.
The effects of SCFAs in the intestinal lumen are considered to be induced via the activation of specific receptors and/or via absorption in epithelial cells, however, the sensing mechanism of SCFAs in the intestinal lumen is currently unclear. In 2003, two different groups, Brown et al. (2003) and Le Poul et al. (2003), simultaneously reported that the SFCA receptors were identified from orphan G-protein coupled receptors (GPCRs), specifically GPR41 and GPR43. They also reported that both receptors are coupled with Gq and Gi/o, and their activation induces an increase in intracellular Ca2+ concentration and a decrease in intracellular cyclic adenosine monophosphate (cAMP). The potencies of respective SCFAs for the GPR41 and the GPR43 activation-induced decrease in intracellular cAMP differ as follows: propionate > butyrate >> acetate in GPR41 and acetate = propionate = butyrate in GPR43. In addition, GPR41 and GPR43 have been reported to be expressed in adipocytes and immune cells, respectively (Brown et al. 2003).
We recently reported that GPR43 is expressed in the rat intestine by PYY-containing enteroendocrine cells and 5-hydroxytryptamine (5-HT)-containing mucosal mast cells (Karaki et al. 2006). This immunohistochemical evidence suggests that SCFAs in the intestinal lumen activate GPR43 SCFA receptor in enteroendocrine cells releasing PYY and in mucosal mast cells releasing 5-HT, a result consistent with physiological data mentioned above. As there are no published data to date regarding GPR43 expression in the human intestine, in this study we investigate GPR43 SCFA receptor expression in the human colon.
Materials and methods
Human tissue preparation
Segments of human ascending colon were obtained (following informed consent) from 80-year old female, 58-year old male and 76-year old female patients undergoing colectomy for carcinoma. This study was approved by the Institutional Review Board of Shizuoka General Hospital and the University of Shizuoka. A nonpathological region was cut from the surgical specimen, placed in ice-cold Krebs-Ringer solution saturated with 95%O2–5%CO2, and transported to the laboratory.
RT-PCR analysis
Small tissue sections (about 5 mg) of whole wall specimen were immersed immediately in RNAlater RNA Stabilization Reagent (Qiagen, Tokyo, Japan), transferred to new 2.0-ml Eppendorf tubes and then freeze-ground utilizing a grinding mill (SK-100; Tokken Inc., Kashiwa, Japan). Bullet-like crushers were put in the tubes with samples. Three tubes were set in a holder, and frozen in liquid nitrogen. Then, the holder was put in a pipe-like case, and shaken to grind the samples. Total RNA was isolated by RNeasy Micro Kit (Qiagen). Two sets of primers for RT-PCR of human GPR43 (A and B, see Table 1) were based on the partial human GPR43 mRNA sequence (Genbank Accession No.: BC096200). RT-PCR was performed by using the Qiagen OneStep RT-PCR Kit (Qiagen). Reaction mixtures of the isolated RNA were first incubated at 50°C for 30 min for reverse transcription followed by initial PCR activation and reverse transcriptase denaturing at 95°C for 15 min. PCR cycles consisted of denaturing at 94°C for 1 min, annealing at 60°C for 1 min, and extension at 72°C for 1 min. The reactions were repeated for 35 cycles, followed by extension at 72°C for 10 min. Finally, amplification products were stored at 4°C until use. Amplification products and DNA ladder (GeneRulerTM 100 bp DNA Ladder Plus, Fermentas, Burlington, Ontario, Canada) were separated by electrophoresis on 1.7% agarose gel in 0.5 × TRIS-borate-EDTA buffer and stained with ethidium bromide.
Western blot analysis
Segments of the human colonic specimen were cut and divided into whole wall, mucosa, submucosa, and muscle layer by a surgical knife. They were frozen in liquid nitrogen, crushed into powder, and dissolved in a lysate buffer consisting of 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.05% Triton X-100, and 1% protease inhibitor cocktail (P8340, Sigma, St. Louis, MO, USA). Samples were centrifuged at 10,000 rpm for 10 min at 4°C, and the supernatant was taken. Protein concentrations were measured utilizing a Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA). The same amount of a 2× sample buffer consisting of 4% sodium dodecyl sulfate (SDS), 12% β-mercaptoethanol, and 20% glycerol in 50 mM Tris–HCl, pH 6.8, with a small amount of bromophenol blue was added to each sample. Samples were incubated at 65°C for 15 min and were stocked at −20°C.
Equal amounts of protein and pre-stained standards (Bio-Rad Laboratories) were separated by electrophoresis in 10% SDS-polyacrylamide gels. Separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Hybond-P, Amersham Biosciences, Piscataway, NJ, USA). PVDF membranes were then incubated with 0.3% skim-milk in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 (T-PBS) for 1 h to suppress non-specific binding of immunoglobulins. The pre-blocked membranes were incubated with anti-GPR43 (RY1505) antibody diluted 1:30,000 in T-PBS containing 0.3% skim milk at 4°C overnight, washed in T-PBS (3 × 10 min), incubated with horseradish-peroxidase (HRP)-labeled secondary antibody (1:30,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in T-PBS containing 0.3% skim-milk for 2 h at room temperature, and washed in T-PBS (3 × 10 min). Bands of GPR43 proteins were detected by Amersham ECL Plus Western Blotting Detection System (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA), and the images were taken by a luminescent image analyzer (LAS-3000mini, Fuji Photo Film Co., Ltd, Tokyo, Japan). Anti-GPR43 antiserum (RY1505) was prepared by the investigators during a previous study (Karaki et al. 2006).
To check the specificity of the anti-GPR43 antiserum, an absorption test was performed. Antigen solution (1:1,000; see Karaki et al. (2006)) and antiserum (1:30,000) were mixed, and the PVDF membrane was stained using the antiserum-antigen mixture.
Immunohistochemistry
Segments of the human colonic specimen were immersed in Zamboni’s fixative (2% formaldehyde and 15% saturated picric acid in 0.1 M phosphate buffer, pH 7.4) at 4°C overnight. Fixed tissues were washed in PBS (3 × 10 min) and stored in PBS containing 0.1% sodium azide at 4°C, the PBS being changed each day for 3 days. After washing, the tissues were further stored in PBS containing 30% sucrose and 0.1% sodium azide at 4°C for cryoprotection. Cryoprotected tissues were rapidly frozen with optimal cutting temperature (OCT) compound (TissueTek, Sakura Finetechnical, Tokyo, Japan) in liquid nitrogen, and stored at −80°C until use. Frozen blocks of tissue were cut into 10-μm sections in a cryostat (CM1100, Leica Microsystems, Wetzlar, Germany), and sections were placed on glass slides and then dried. Sections were washed in PBS (3 × 10 min) to remove OCT compound, and incubated with 10% normal donkey serum and 0.3% Trion-X 100 in PBS at room temperature for 30 min to suppress non-specific binding of antibodies. Preblocked sections were incubated with primary antibodies (Table 2) with 0.3% Triton X-100 in PBS at 4°C overnight, washed in PBS (3 × 10 min), and incubated with secondary antibodies (Table 2) with 0.3% Triton X-100 and 4′,6-Diamidino-2-phenylindole (DAPI) solution (1:200, Dojindo Laboratories, Kumamoto, Japan) for 1 h at room temperature. Sections were then washed in PBS (3 × 10 min), and cover-slipped with a mounting medium (DakoCytomation, Glostrup, Denmark). Immunoreactivity was visualized and captured by using a fluorescence microscope and a cooled charge-coupled device digital camera system (BZ-8000, Keyence, Osaka, Japan). In order to remove out-of focus fluorescence, captured images were deconvoluted using image analyzing software (BZ Analyzer, Keyence).
To check the specificity of secondary antibody, a section incubated without primary antibody was stained by secondary antibodies as a negative control. Additionally, to check the specificity of antiserum for GPR43, an absorption test was performed. Neat antigen solution (see Karaki et al. (2006)) was mixed with neat anti-GPR43 serum 1:1, the mixture equilibrated overnight at 4°C, and was then diluted with PBS containing 0.3% Triton X-100 to a working concentration of GPR43 (1:10,000). Sections were incubated with this diluted mixture of antibody and antigen in the immunohistochemistry procedure.
Results
RT-PCR analysis
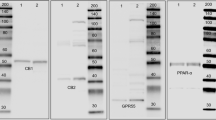
Messenger RNA for GPR43 was detected in extract of human ascending colon with both sets of primers (Fig. 1). Expected sizes of PCR products utilizing primer sets A and B are 172 and 209 base pair (bp), respectively. Detected bands were consistent with said expectation thereby indicating that the GPR43 gene is expressed in the human colon.

Analysis of GPR43 mRNA expression in the human colon by RT-PCR. Two sets of primers (A and B) for human GPR43 were used (see Table 1). Arrows indicate the expected sizes of PCR products. RT+ or – indicate whether reverse transcription was completed or not
Western blot analysis
A band of about 25 kD considered to be GPR43 protein was detected in extracts of whole wall and in the separated mucosa from the human ascending colon, but was not present in extracts of submucosa and muscle layers (Fig. 2A). Although the same amounts of proteins were found in the various samples, the band density of the separated mucosa sample was clearly higher than that of whole tissue samples. In the absorption test, the single band thought to be GPR43 protein was almost completely abolished (Fig. 2B). It is therefore suggested that the single band is specific for GPR43 protein.

Analysis of expression of GPR43 protein in the human colon by Western blots probed with anti-GPR43 antiserum. The same volumes of extracted proteins were separated by SDS-PAGE, and transferred to PVDF membrane. Pre-blocked PVDF membranes were stained by anti-GPR43 (1:30,000) (A), by anti-GPR43 (1:30,000) and antigen (1:1,000) mixture as absorption control (B)
Immunohistochemistry
GPR43-immunoreactivities were observed as dotted staining in the cytoplasm of almost all enterocytes (Figs. 3A and 4A), and also in many lamina proprial cells (Fig. 3A). This staining by anti GPR43 antibody was stronger at surface epithelium than at the bottom of the crypt (Fig. 3A). Apical membranes of enterocytes were only slightly stained by anti-GPR43 antibody. In negative control staining utilizing secondary antibody alone (Fig. 3B) and in absorption control (Fig. 3C), such staining was not observed. However, many lamina proprial cells were stained in absorption control. Thus, staining in the enterocytes was specific for GPR43, but almost all staining of lamina proprial cells seemed to be non-specific.

Immunohistochemistry for GPR43 in the human colon. Sections (10 μm-thick) of human colon were stained with rabbit anti-GPR43 antiserum as primary antibody, Alexa594-labeled anti-rabbit IgG antibody as secondary antibody (red), and DAPI indicating nuclei (blue). (A): Specific GPR43 immunoreactivity was observed in enterocyte cytoplasm. The staining of many cells in lamina propria seemed to be non-specific since they were observed also by negative control (C). (B): Negative control without anti-GPR43 antiserum. (C): Absorption control using the mixture of anti-GPR43 anti-serum and the antigen

Morphology of GPR43-IR cells. (A): GPR43-IR enterocytes. Dotted GPR43 immunoreactivities (red) were observed in the cytoplasm at the upper site of the nucleus (blue) in enterocytes. (B): GPR43-IR enteroendocrine cell. Immunostaining (red) indicates GPR43 immunoreactivity, and DAPI (blue) indicates nuclei
GPR43-immunoreactive (IR) enteroendocrine cells were found in the crypt (Fig. 4B) and were open-type enteroendocrine cells, with thin cell bodies that extended to the lumen surface. In rat intestine, GPR-IR enteroendocrine cells often have processes that extend from their bases and run beneath adjacent epithelial cells (Karaki et al. 2006), but such is not the case in human ascending colon.
Our previous immunohistological study of GPR43 in rat intestine found complete co-localization of immunoreactivity for GPR43 and PYY (Karaki et al. 2006). In the present study for the human colon, all GPR43-IR enteroendocrine cells were PYY-IR, and vice-versa (Fig. 5A–C). Moreover, GPR43 immunoreactivity was not colocalized with 5-HT (Fig. 5D–F) as was demonstrated in our previous study in the rat intestine (Karaki et al. 2006).

Colocalization of GPR43 and PYY, and lack of colocalization of GPR43 and 5-HT in enteroendocrine cells. (A–C): Triple-staining for GPR43 (A, red), PYY (B, green) and DAPI (blue) in the crypt cut in the round. Colocalization appears yellow (C). (D–F): triple-staining for GPR43 (D, red), 5-HT (E, green) and DAPI (blue) in the crypt cut in round. Colocalization would appear yellow, but there is no colocalization between GPR43 and 5-HT in enteroendocrine cells (F)
Discussion
The current study is the first to demonstrate expression and localization of the SCFA receptor, GPR43 in the human ascending colon. RT-PCR analysis detected GPR43 mRNA (Fig. 1) as did Western blot analysis where GPR43 protein was detected in mucosal preparation, but not muscle nor submucosal layers (Fig. 2). In immunohistochemistry, spotted GPR43-immunoreactivity was observed in cytoplasm looking like endoplasmic reticulum or Golgi apparatus (Figs. 3A and 4A), and flat cytoplasmic staining was observed in a few open-type enteroendocrine cells (Fig. 4B). The colocalization study demonstrated that GPR43-immunoreactive enteroendocrine L cells contained PYY-, but were not colocalized with 5-HT (Fig. 5).
The presence of SCFAs in the colonic lumen is reported to induce inhibition of upper gastrointestinal motility mediated by PYY, (sometimes described as “colonic brake” (Ropert et al. 1996)) as well as ileal brake. The present morphological data further supports the previously reported physiological study (Ropert et al. 1996). Therefore, colonic brake induced by SCFAs in the colonic lumen seems to be mediated by the activation of SCFA receptor, GPR43 expressed in enteroendocrine L cells. PYY is also known to be an important appetite control hormone, inhibiting food intake by as a satiety signal (Wren and Bloom 2007). Furthermore, it has been reported that SCFAs stimulate leptin expression in adipocytes (Xiong et al. 2004). Leptin is known as an anorexigenic hormone inhibiting food intake through the receptors in the brain (Cohen et al. 2001). Therefore, it is considered that SCFAs may influence appetite control by several mechanisms.
The function of GPR43 expressed in the cytoplasm in a dotted manner is not clear, but might initiate in the Golgi-apparatus prior to transport to apical sites. GPR43 has been reported to couple with Gq and Gi/o proteins to increase intracellular Ca2+ and to decrease cAMP concentrations (Brown et al. 2003; Le Poul et al. 2003). Both intracellular Ca2+ and cAMP are known to function as second messengers to induce synergistic fluid secretion (Karaki and Kuwahara 2004). Therefore, under certain conditions such as when GPR43 is present on the apical membrane, it is possible that SCFAs modulate colonic secretory function with GPR43 functioning as a luminal chemical stimuli. However to clarify the hypothesis, it is necessary to perform more precise study, e.g., using endoplasmic reticulum and/or Golgi apparatus markers. In addition, it has been reported that SCFAs induce acute phosphorylation of the p38 mitogen-activated protein kinase (MAPK)/heat shock protein 27 pathway via GPR43 in the MCF-7 human breast cancer cell line (Yonezawa et al. 2007). Therefore, SCFAs may play a role of stress management in the epithelial cells.
We previously reported that consumption of a fiber-free diet induces changes in neural and muscular functions and decreases the amount of 5-HT-containing enterochromaffin (EC) cells in the rat colon (Mitsui et al 2006). In this study, it has been demonstrated that the animal group fed with a fermented fiber, guar gum-containing diet demonstrates partially prevent such changes of the neural and muscular functions, and loss of 5-HT-containing EC cells. This suggests that chemical stimulation by SCFAs, as well as mechanical stimuli caused by fecal bulk (the cellulose-fed group in that study), are important for maintenance of intestinal integrity. The present study suggests that the several responses induced by SCFAs are possibly due to the presence of GPR43 SCFA receptors. However, Dass et al. (2007) have recently shown that SCFAs inhibit the electrical field stimulation (EFS)-induced contraction of circular muscle in the mouse colon consistent with activity at the GPR43, but the effect is not altered in GPR43 gene knockout mice. Moreover, they have shown that the SCFA-induced inhibitory effect on the contraction by EFS is independent of the presence and absence of mucosa. Therefore, they concluded that these effects are not involved in the activation of GPR43. On the other hand, although the effect is consistent with activity at GPR41 rather than GPR43, Mitsui et al. (2005b) have reported that propionate increases the frequency of spontaneous contractions in the rat longitudinal muscle strip with mucosa, but not without mucosa. Ono et al. (2004) have also reported that acetate, which can activate GPR43 without GPR41, decreases the frequency of spontaneous contractions. Therefore, luminal SCFAs may modulate the frequency of spontaneous contractions through the activation of mucosal GPR43 and/or GPR41, though further study is necessary to clarify the physiological role of GPR43 and/or GPR41 expressed in enterocytes.
In conclusion, the present study of SCFA receptor expression in human colon may prove to be clinically significant in the area of functional and inflammatory bowel disease prevention and the appetite control.
References
Andoh A, Tsujikawa T, Fujiyama Y (2003) Role of dietary fiber and short-chain fatty acids in the colon. Curr Pharm Des 9:347–358
Binder HJ, Mehta P (1989) Short-chain fatty acids stimulate active sodium and chloride absorption in vitro in the rat distal colon. Gastroenterology 96:989–996
Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ (2003) The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278:11312–11319
Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM (2001) Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 108:1113–1121
Cuche G, Malbert CH (1999) Short-chain fatty acids present in the ileum inhibit fasting gastrointestinal motility in conscious pigs. Neurogastroenterol Motil 11:219–225
Cuche G, Cuber JC, Malbert CH (2000) Ileal short-chain fatty acids inhibit gastric motility by a humoral pathway. Am J Physiol Gastrointest Liver Physiol 279:G925–G930
Cummings JH, Rombeau JL, Sakata T (1995) Physiological and clinical aspects of short-chain fatty acids. Cambridge University Press, Cambridge, UK
D’Argenio G, Cosenza V, Delle Cave M, Iovino P, Delle Valle N, Lombardi G, Mazzacca G (1996) Butyrate enemas in experimental colitis and protection against large bowel cancer in a rat model. Gastroenterology 110:1727–1734
Dass NB, John AK, Bassil AK, Crumbley CW, Shehee WR, Maurio FP, Moore GB, Taylor CM, Sanger GJ (2007) The relationship between the effects of short-chain fatty acids on intestinal motility in vitro and GPR43 receptor activation. Neurogastroenterol Motil 19:66–74
Diener M, Vujicic Z, Scharrer E (1996) Neuronally mediated anion secretion induced by short-chain fatty acids in the rat distal small intestine. Acta Physiol Scand 157:33–40
Galvez J, Rodriguez Cabezas ME, Zarzuelo A (2005) Effects of dietary fiber on inflammatory bowel disease. Mol Nutr Food Res 49:601–608
Hubel KA, Russ L (1993) Mechanisms of the secretory response to luminal propionate in rat descending colon in vitro. J Auton Nerv Syst 43:219–229
Karaki SI, Kuwahara A (2004) Regulation of intestinal secretion involved in the interaction between neurotransmitters and prostaglandin E2. Neurogastroenterol Motil 16 Suppl 1:96–99
Karaki S, Mitsui R, Hayashi H, Kato I, Sugiya H, Iwanaga T, Furness JB, Kuwahara A (2006) Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res 324:353–360
Kles KA, Chang EB (2006) Short-chain fatty acids impact on intestinal adaptation, inflammation, carcinoma, and failure. Gastroenterology 130:S100–S105
Kripke SA, Fox AD, Berman JM, Settle RG, Rombeau JL (1989) Stimulation of intestinal mucosal growth with intracolonic infusion of short-chain fatty acids. J Parenter Enteral Nutr 13:109–116
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M (2003) Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 278:25481–25489
Lutz T, Scharrer E (1991) Effect of short-chain fatty acids on calcium absorption by the rat colon. Exp Physiol 76:615–618
Mitsui R, Ono S, Karaki S, Kuwahara A (2005a) Neural and non-neural mediation of propionate-induced contractile responses in the rat distal colon. Neurogastroenterol Motil 17:585–594
Mitsui R, Ono S, Karaki S, Kuwahara A (2005b) Propionate modulates spontaneous contractions via enteric nerves and prostaglandin release in the rat distal colon. Jpn J Physiol 55:331–338
Mitsui R, Karaki SI, Kubo Y, Sugiura Y, Kuwahara A (2006) Fibre-free diet leads to impairment of neuronally mediated muscle contractile response in rat distal colon. Neurogastroenterol Motil 18:1093–1101
Mortensen FV, Hielsen H (1995) In vivo and in vitro effects of short-chain fatty acids on intestinal blood circulation. In: Cummings JH, Rombeau JL, Sakata T (eds) Physiological and clinical aspects of short-chain fatty acids. Cambridge University Press, Cambridge, UK Cambridge, pp 391–400
Ono S, Karaki S, Kuwahara A (2004) Short-chain fatty acids decrease the frequency of spontaneous contractions of longitudinal muscle via enteric nerves in rat distal colon. Jpn J Physiol 54:483–493
Ropert A, Cherbut C, Roze C, Le Quellec A, Holst JJ, Fu Cheng X, Bruley des Varannes S, Galmiche JP (1996) Colonic fermentation and proximal gastric tone in humans. Gastroenterology 111:289–296
Sakata T (1987) Stimulatory effect of short-chain fatty acids on epithelial cell proliferation in the rat intestine: a possible explanation for trophic effects of fermentable fibre, gut microbes and luminal trophic factors. Br J Nutr 58:95–103
Scheppach W, Bartram P, Richter A, Richter F, Liepold H, Dusel G, Hofstetter G, Ruthlein J, Kasper H (1992) Effect of short-chain fatty acids on the human colonic mucosa in vitro. J Parenter Enteral Nutr 16:43–48
Tedelind S, Westberg F, Kjerrulf M, Vidal A (2007) Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J Gastroenterol 13:2826–2832
Topping DL, Clifton PM (2001) Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 81:1031–1064
Vidyasagar S, Ramakrishna BS (2002) Effects of butyrate on active sodium and chloride transport in rat and rabbit distal colon. J Physiol 539:163–173
Wren AM, Bloom SR (2007) Gut hormones and appetite control. Gastroenterology 132:2116–2130
Xiong Y, Miyamoto N, Shibata K, Valasek MA, Motoike T, Kedzierski RM, Yanagisawa M (2004) Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc Natl Acad Sci USA 101:1045–1050
Yajima T (1985) Contractile effect of short-chain fatty acids on the isolated colon of the rat. J Physiol 368:667–678
Yajima T (1988) Luminal propionate-induced secretory response in the rat distal colon in vitro. J Physiol 403:559–575
Yonezawa T, Kobayashi Y, Obara Y (2007) Short-chain fatty acids induce acute phosphorylation of the p38 mitogen-activated protein kinase/heat shock protein 27 pathway via GPR43 in the MCF-7 human breast cancer cell line. Cell Signal 19:185–193
Acknowledgements
This work was partly supported by a grant-in-aid from the Japan Society for Promotion of Science (No. 18590207), and by the Smoking Research Foundation (to A.K.); and by the Promotion of Health and Nutrition from the Danone Institute (to S. K.). The fluorescence microscope and image analyzing system (BZ-8000) was kindly borrowed from KEYENCE Corporation (Osaka, Japan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karaki, Si., Tazoe, H., Hayashi, H. et al. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J Mol Hist 39, 135–142 (2008). https://doi.org/10.1007/s10735-007-9145-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10735-007-9145-y