
Abstract
Objective
Systemic lupus erythematosus (SLE), the most common form of lupus, is a multisystemic rheumatic disease with different clinical features that generally affect women of childbearing age. The common symptoms of SLE are very similar to other autoimmune and non-autoimmune disorders, thereby it is known as a thousand faces disease. In this article, we are going to discuss some of the most updated information about immune system-related factors, cells, and cytokines involved in SLE pathogenesis.
Methods
Different electronic databases, especially PubMed/MEDLINE, Scopus, and Google Scholar, were searched to review and analyze relevant literature on the role of innate and adaptive immune cells and cytokines in the pathogenesis of SLE. A search for relevant literature was accomplished using various keywords including systemic lupus erythematosus, apoptosis, autoantibodies, immunopathogenesis of SLE, adaptive and innate immune cells, inflammatory cytokines, hormones, etc.
Results and conclusion
The most important characteristic of SLE is the production of antibodies against different nuclear autoantigens like double-strand DNA and RNA. The depositions of the immune complexes (ICs) that are generated between autoantibodies and autoantigens, along with aberrant clearance of them, can lead to permanent inflammation and contribute to tissue or organ damage. Related mechanisms underlying the initiation and development of SLE have not been clarified yet. Although, defects in immune tolerance, enhanced antigenic load, hyperactivity of T cells, and inappropriate regulation of B cells contribute to the pathogenic autoantibodies generation. Besides, sex hormones that influence the immune system seem to act as triggers or protectors of SLE development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune rheumatic disorder with different clinical manifestations. The incidence of SLE in females is higher than in males [1], which may be attributable to the existence of different genes on the sex chromosomes and the presence of gender-specific hormones [2]. SLE can involve different systems and organs, such as the skin, joints, central nervous system, heart, and cardiovascular system, and especially, the kidneys known as lupus-induced nephritis (LN).
SLE is characterized by circulating autoantibodies (autoAb) against multiple self-nuclear antigens, especially double-strand DNA (dsDNA), that lead to antigen–antibody complex formation (immune complexes, ICs) [3], and result in the inflammatory response of the immune system [4]. Defects in the apoptosis process [5] and ineffective clearance of apoptotic cells lead to the release of autoantigenic contents of cells that may increase the chance of autoantigens (autoAg) identification by the immune system [6, 7]. It has seemed, dysregulation of innate immunity, e.g., reduced phagocytosis and increased oxidative activity of neutrophils [8, 9], accumulation of plasmacytoid dendritic cells (pDCs) in inflamed tissues [10], as well as the deficiency or mutation in the components of the complement system [11,12,13], has been associated with SLE disorder. Besides, impairment function of the adaptive immunity such as the increased activity of B cells, defect in the removal of auto-reacting B cells [14], and the hyperactivated phenotype of T cells can increase the generation of autoAbs [4, 14]; indicating that, there is a connection between mechanisms of the innate and adaptive immune system in the development of the SLE. Single-cell RNA sequencing (RNS-seq, transcriptomic) analyses on purified immune cells of LN kidneys identified 21 subsets of infiltrating immune cells in different activation state in LN kidney tissues including different clusters of macrophages, natural killer (NK) and DC cells, and B and T lymphocytes which contribute to autoimmune responses and tissue injury in LN patient. While in a normal kidney, no B lymphocytes were identified and only three clusters of immune cells have been detected: a myeloid group and memory T CD4+ lymphocytes [15, 16].
Due to the critical role of inflammation, inflammatory mediators, cytokines, and immune cells’ abnormalities in the etiopathogenesis of SLE, today, most of the treatments are dependent on the consumption of various doses of steroid drugs, nonsteroidal anti-inflammatory drugs (NSAIDs), and B cells targeted biological therapies [17].
Accordingly, this review provides an overview of factors and related mechanisms involved in the pathogenesis of SLE including the correlation of apoptotic process and nucleic acid-sensing, the interaction of different parts of the immune system, and the effect of different hormones on immune system regulation.
Apoptosis and pattern recognition receptors
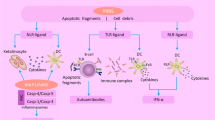
Apoptosis is a normal process of tissue homeostasis and self-tolerance that occurs in physiological and pathological conditions [4]. Normally, phagocytes find early apoptotic cells directly by their receptors or indirectly by serum opsonization through complement components and clear tissue from apoptotic cells before converting to secondary necrotic cells. This process prevents the accumulation of cellular and microbial debris [4, 5]. If early apoptotic debris are not cleared by the immune systems, they can convert to the late apoptotic cells and their containing autoAgs will be released [18,19,20]. Therefore, the clearance of early apoptotic cells is an immunologically silent or anti-inflammatory process, whereas removal of late apoptotic debris is accompanied by pro-inflammatory and autoimmune responses [5, 19]. The clearance of DNA or RNA nucleoprotein complexes (NPCs), derived from late apoptotic cells, by phagocytes, macrophages (MQs), and DCs leads to excess production of pro-inflammatory cytokines (Fig. 1). Antigen-presenting cells (APCs) process and present NPCs by human leukocyte antigen (HLA) classes I and II to the T cells. Subsequently, autoreactive B and T cells are generated, result in the autoAbs production and loss of self-tolerance [4, 19]. With regards to the HLA role in the antigen presentation, HLA genotypes have a substantial role in T-cell activation and are associated with autoimmune disease susceptibility [19, 20].

Schematic review of SLE immunopathogenesis: A In physiologic conditions, early apoptotic cells will be found through APCs (like MQs) and clearance in a silent manner (without inflammation). B The defective function of the complement system or inappropriate clearance causes early apoptotic cells to turn into late apoptotic cells and release their nuclear contents (known as autoAbs). These autoAbs detect through TLR7 and TLR9 in pDCs or other APCs. These autoAbs are presented to the subset of T cells. T cells trigger B cells and migrate to the germinal center. In the germinal center, B cells turn to autoreactive B cell and leave the germinal center as autoreactive plasma cells. Plasma cells produce autoAbs which then form immune complexes (ICs) with nuclear Ag. ICs will be found by immune cells and the pathologic cycle will continue. If ICs are not cleaned properly, they will deposit in different organs and activate the complement system and eventually damaging that organ
Hence, the accelerated apoptosis process and delay in the clearance of apoptosis derbies are contributed to autoAgs exposure and SLE immunopathogenesis [18]. This defect in apoptosis may occur due to some genetic and environmental factors (like smoking, UV light) as well as infections [20]. Cellular debris and danger signals from late apoptotic cells will be recognized by a subset of pattern recognition receptors (PRRs), known as Toll-like receptors (TLRs) as well as cytosolic DNA sensors such as cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase (cGAS) [19, 21, 22].
TLRs have an essential role in the initiation of the innate immune response against cell-derived antigens [19, 21]. Moreover, these receptors have been found to link innate and adaptive immunity and sense pro- or anti-inflammatory signals, through their existence in immune cells like DCs and MQs and non-immune cells like fibroblasts and epithelial cells. Recognition of danger signals by TLRs induces intracellular signaling pathways which contribute to cell proliferation, cytokine production, as well as co-stimulatory molecule expression (Fig. 1) [20, 21].
In the context of SLE, the role of TLR7 and TLR9 is more important than the other TLRs. Specific single-nucleotide polymorphisms and increased expression of TLR7 and 9 have been associated with SLE [23, 24]. TLR7 and 9 are directly interacting with ICs containing single-strand RNA (ssRNA) and unmethylated CpG dsDNA, respectively, and are necessary for the autoAbs production against RNA and DNA [21].
Activation of TLR7 and 9 results in the generation of type I interferon in the immune cells, particularly pDCs. Production of type I interferon contributes to decreased threshold of B-cell receptor (BCR) activation and promotes autoAbs production in a positive-feedback loop [25].
Generally, TLR7 and TLR9 compete with each other for expression on the membranes of endosomes and lysosomes. TLR9 limits TLR7-mediated function, like T-cell activation and expansion as well as the development of the memory T cell. Therefore, the absence of TLR9 results in elevated TLR7 signaling and promotes TLR7-related activity [4, 21, 24]. According to the studies on animal models, in the absence of TLR9, the rate of autoAbs against proteins-related RNAs is raised [19].
In addition to TLRs, mounting evidence suggests that cytosolic DNA can sense by the cGAS–stimulator of interferon genes (STING) pathway [22, 26]. Binding DNA to the cGAS contributes to STING activity and triggers activation of interferon regulatory factor (IRF)-3 [26]. Besides, STING can stimulate the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [27]. Stimulation of IRF3 and NF-κB contributes to the induction of inflammatory genes and the production of inflammatory cytokines, especially type I interferon [22, 27, 28]. The presence of the cGAS–STING pathway is necessary to DCs’ stimulation. Activation of the STING pathway in DCs triggers naive T-cell proliferation and interferon-gamma (IFN-γ) generation, and may accelerate the maturation of naive T cells to become professional APCs [28]. According to the reports, the gain-of-function mutations in STING lead to SLE-like diseases [22, 26]. Besides, peripheral blood mononuclear cells (PBMCs) from SLE individuals represent higher levels of cGAS. However, the role of the cGAS–STING pathway in SLE pathogenesis needs more investigation [22]. Thereby, it seems the cGAS–STING signaling pathway may have a role in SLE pathogenesis.
Innate immune cells and SLE
The innate immune system consists of varied immune cells and mediators that circulate in the blood and reside in the tissues. Activation and recruitment of autoreactive adaptive immune cells require a strong motivation from innate immunity. Besides, severe activation of the innate immune system can independently trigger inflammation and tissue damages [29]. Here, we are going to discuss the function of innate immune cells in SLE pathogenesis. The change in the innate and adaptive immune cells in SLE is summarized in Table 1.
Dendritic cells
DCs are professional APCs that bridge innate and adaptive immunity by producing cytokines and providing co-stimulatory signals as well as presenting antigens by HLA-II molecules [4, 30, 31]. DCs are categorized into three major subpopulations such as myeloid/conventional DC1 and 2 (cDC1/2) and pDCs [32]. Moreover, during inflammation, monocytes can differentiate into monocyte-associated DCs (mo-DCs) [31, 32]. cDCs are able to stimulate T cells, produce IL-6 and IL-8, and promote the inflammatory responses of SLE [33].
pDCs detect ICs presence via their TLRs (7 and 9), and secret a high amount of type I interferon (especially IFN-α) [7]. IFN-α secretion in an autocrine manner enhances pDCs and T-cell activity, indicating adaptive and innate immunity linkage in SLE pathogenesis [6, 7]. Studies about DCs in SLE have reported contradictory results. Normal, reduced [34,35,36,37,38,39,40], and increased [41, 42] number of pDCs have been shown in SLE patients. It has been seemed in the active phase of the SLE; due to the migration and accumulation of pDCs into the inflamed tissues, the number of peripheral pDCs is reduced [36, 38, 43]. Consistent with this idea, pDCs have been detected in kidney and cutaneous lesions of SLE patients [30].
Neutrophils
Neutrophils are considered the most abundant type of granulocytes with a short life span. They provide a substantial part of the innate immune system that promotes inflammation in the site of infection and eliminate pathogens with diverse mechanisms. Neutrophils show diverse abnormalities in SLE including a reduction in phagocytic activities [44], an impairment production of reactive oxygen species (ROS) [45], as well as an enhancement of the neutrophil extracellular traps’ (NETs) generation [46]. Phagocytic activity of neutrophils is required for clearance of apoptotic debris and nuclear material, which has been seemed reduced in SLE patients and contributed to SLE development [8]. It seems that extreme ROS generation and altered redox reaction may be contributing factors in dysregulated apoptosis reaction and SLE flare initiation [47]. Different studies emphasize the substantial role of ROS in dysregulated apoptosis, which leads to an increased rate of apoptosis and delay in apoptotic debris clearance. Interestingly, genome-wide association studies (GWAS) in SLE individuals and animal models indicate the involvement of oxidative stress in SLE pathogenesis [47]. For instance, missense single-nucleotide polymorphisms (SNPs), in the neutrophil cytosol factor 1 (NCF1) and NCF2 genes that code p47phox and p67phox proteins of NADPH oxidase 2 (NOX2) complex, respectively, have been found to be correlated to SLE disorder [48, 49]. According to the study by Olsson Lina M et al., the frequency of a missense SNP located in exon 4 (rs201802880, here denoted NCF1-339T) is higher in patients with SLE disorder [49]. The nucleotide alteration from C to T changes the amino acid from arginine to histidine and leads to diminished extracellular ROS generation in polymorphonuclear (PMN) cells, especially in neutrophils, and elevated expression of type I IFN regulated genes [49, 50]. Another mechanism that is defective in the neutrophils of SLE individuals is NETosis. NETosis is the unique type of cell death and the most important characteristic of neutrophils activity [4, 51]. The NET is a network composed largely of immunogenic DNA, histones, as well as proteins, and it is a rich source of ds-DNA antigens [4, 52]. An increased level of NET deposition has been reported in the skin and the kidneys of SLE patients [51,52,53]. There is an increased rate of NETosis and a reduced capacity to degrade NETs due to the lower rate of serum DNase1 activity, as an endonuclease, in SLE patients [7, 53]. In addition, it seems that the increased rate of the NET formation may be due to disruption of ROS production by neutrophils which has been seen in SLE patients [6]. Therefore, this neutrophils-derived DNA in NETs induces pDCs to produce type I IFN which enhances the formation of NET in a feedback loop and contributes to disease development [51].
Innate-like lymphocytes
Innate lymphoid cells (ILCs) are the innate counterpart of T cells that have lymphoid morphology but lack the BCRs and T-cell receptors (TCR)s, thereby they cannot recognize antigens directly [7]. ILCs are involved in multiple functions including mucosal immunity, tissue remodeling as well as maintenance of homeostasis, and regulate adaptive and innate immunity by producing various cytokines in response to stimulation. The ILCs have been divided into five subpopulations including ILCs1, ILCs2, ILCs3, NK, and lymphoid tissue-inducer (LTi) cells [54]. The ILCs1 and ILCs2, as well as ILCs3 subtype, produce a set of Th1/Th2 and Th17-related cytokines such as IFN-γ, IL-4, and IL-17, respectively. It was found that the percentage of ILCs1 and ILCs3 in the peripheral blood of SLE patients increased, whereas the percentage of the ILCs2 diminished considerably [55]. In contrast, another study indicates that the percentage of ILCs3 in the moderate activity stage of SLE is not considerably different compared to healthy control but in the severe activity stage is diminished [56]. It seemed the ILCs1 becomes a predominant subset of ILCs is SLE disorder. Consistent with this idea, the amount of IL-18 and IL-12, as a dominant cytokine of ILC1, are enhanced in patients and positively correlated to SLE activity. It seems that ILC1 may be a beneficial indicator of disease activity [56].
Natural killer cells
NK cells can produce a wide range of pro-inflammatory cytokines (IFN-γ and tumor necrosis factor-alpha, TNF-α) and chemokines (CCL3, CCL4, and CCL5), in inflammatory responses. In SLE patients, especially in those with renal involvement, the frequency of NK cells in the periphery is lower than healthy controls [57]. The reduced number of NK cells can be due to the high level of IFN-α, a cytokine that induces apoptosis in these cells, or due to the migration of these cells to the disease target organs (especially the kidney) [58, 59]. According to the study by Schepis et al. on SLE patients, in the active stage of the diseases, the cytotoxic activity of NK cells is down-regulated, while the rate of cytokine production of these cells is increased [60]. Dysregulation in cytokine production, like IL-2 as a critical cytokine in NK-cell development, restricts NK cells’ development and leads to the generation of the immature NK (iNK) cells in SLE patients [61]. A high level of IFN-γ production by NK cells is correlated with sustained inflammation in the LN and thereby kidney injury. Moreover, the increased level of the TNF-α is associated with the aberrant activity of NK cells and the formation of iNK cells in patients with SLE disorder [57, 59, 62]. In murine models, contradictory to the reduced level of NK-cell activity in the periphery, the level of IL-15 (another cytokine in NK-cell development) as well as the production of cytotoxic granules is increased and leads to maintaining the NK-cell activity in kidneys and promoting intra-renal inflammation and damage. [59, 61, 62]. It seems that cytokines’ alteration directly affects NK cells activity and changes their maturation process. However, more research is required, to understand how alteration of NK cells can affect SLE progression.
Macrophage
MQs are a type of innate immune cells that may have an immuno-modulatory, inflammatory, and anti-inflammatory role in the immune system. Generally, the polarization of MQs toward to M1 (classically activated) phenotype contributes to the secretion of pro-inflammatory cytokines like TNF-α and IL-12 and promotes the inflammatory reaction. Whereas, polarization toward M2 (alternatively activated) phenotype contributes to the secretion of anti-inflammatory cytokines like IL-10 and IL-4 and promotes the immuno-modulatory reaction (63). According to a previous study, there is no difference between the percentage of MQs in SLE individuals and the healthy control group [63]. While another study represents a diminished percentage of MQs in SLE patients compared with healthy control [64]. Given evidence suggested that M1-MQ is a predominant type of MQs in SLE disorder. Extent production of pro-inflammatory cytokines like IL-1β and IFN-γ, as a pro-inflammatory cytokine, has been seen in SLE patients [65]. In addition, the presence of TNF-α, IFN-γ, and granulocyte–macrophage colony-stimulating factor (GM-CSF), in the sera of SLE patients triggers M1 polarization in a positive-feedback loop [63]. Interestingly, the amount of IL-10, as a prominent cytokine in M2-MQ polarization, is also high and can be detected in the sera of SLE patients [63, 66]. Contradictory to IL-10 anti-inflammatory manner, IL-10 obtains pro-inflammatory function in the presence of a high amount of type I IFNs. Infiltration of monocytes into the renal tissues and differentiation to MQs is particularly associated with LN development and progression [4]. Single-cell transcriptomic analysis revealed a within-tissue differentiation and transition of kidney infiltrated monocyte to phagocytic and eventually M2-like macrophages which may contribute to tissue damage and inflammatory responses [15]. It seems that modulating MQs polarization may have a therapeutic effect in SLE disorder [63].
Basophil
Basophils are the least type of immune cells in the blood that are able to link innate and adaptive immune responses [7, 67]. These cells can trigger CD4+ Th2 cells’ differentiation and prevent Th1 cells’ differentiation. Basophils are responsible for inflammatory reactions and involvement in allergic diseases [7]. They can be stimulated via IgE-dependent and IgE-independent manner to perform relevant activities, and to generate Th2 cells cytokines like IL-4 and IL-6 [67, 68].
The basophils of the SLE individuals represent activated phenotype and overexpress surface markers like CD203c and HLA-DR molecules. Furthermore, the expression of cell adhesion molecules like L-selectin (CD62L) is enhanced on their surface, contributing to the increased migration of these cells to the secondary lymphoid organs (SLOs) [67]. Consistent with this, it has been observed that basophils of the SLE patients aggregate in SLOs, but this abnormal accumulation did not find in healthy individuals. Interestingly, in murine models of SLE, SLO-aggregated basophils can cooperate with B and T cells through the expression of major histocompatibility complex (MHC)-II and membrane-bound form of the B-cell-activating factor (BAFF), contributing to SLE progression. Besides, IgE autoAbs have been detected in the sera of patients with SLE disorder [67]. It seems that IgE autoAbs level is associated with disease activity, especially in patients with LN involvement [68]. The presence of IgE autoAbs leads to basophils activation in SLE disorder, and basophils can trigger autoAbs generation by promoting B cells in a positive-feedback manner. On the other hand, basophils have been obliquely activated with IL-17A and induce Th17 differentiation [68]. It seems that basophils have been involved in the pathogenesis of SLE by supporting autoreactive B and T cells to produce autoAbs, promote inflammatory reactions as well as generation circulating-IC [67].
Adaptive immune cells and SLE
Autoimmunity can be triggered by the activation of specific T lymphocytes that have receptors with reaction affinity to self-antigens and can, in turn, activate autoreactive B cells and innate immune cells. Given that it is impossible to eliminate self-antigens completely, autoimmune adaptive responses sustain and induced inflammatory organ damages [69]. Here, we are going to discuss the function of adaptive immune cells in the pathogenesis of SLE (Table 1).
T cells
Different T-cell subgroups have roles in the development of SLE. Here, we will briefly discuss the subsets of T cells, including follicular helper T (Tfh) cells, Th17, regulatory T cells (Tregs), and γδT cells that participate in SLE pathogenesis.
Tfh cells have a critical role in the germinal center (GC) reactions and contribute to B- and T-cell differentiation, proliferation, and antibody generation by producing IL-21 (Fig. 1) [4]. In SLE disorder, most of the autoAbs have been shown high-affinity IgG form, indicating the involvement of GC reactions. Therefore, dysregulation of the Tfh cell function can contribute to pathogenic autoAbs generation and seems to have a prominent role in SLE development [70]. It seems dysregulated cytokine levels (IL12, IL-23, and transforming growth factor-beta, TGF-β) may lead to abnormal function of circulating follicular regulatory T cells (cTfr) [71]. The enhanced rate of IL-21 in SLE represents an increased number of circulating Tfh (cTfh) cells. Moreover, it has been reported that there is a direct correlation between the level of the anti-dsDNA antibodies and the rate of the cTfh cells in SLE patients [4, 72]. It has been found that the number of cTfr cells was reduced in SLE, which seemed to maybe a proper indication for active disease [72].
Signals through co-stimulatory molecules like inducible costimulator-ligand (ICOS-L) and ICOS have a prominent role in differentiation, activation, migration, as well as the survival of Tfh cells. Therefore, inhibition of ICOS–ICOS-L interaction alleviates SLE symptoms in mice models [73]. Nonetheless, contradictory to the overexpression of ICOS in cTfh cells in patients with active SLE, the expression of ICOS-L by APCs has not been up-regulated [74]. Growing evidence suggests that the interaction between OX40 ligand (OX40L) and OX40 is a more important factor that contributes to the improper response of Tfh cells in SLE disorder [4, 73]. The up-regulation of OX40L on CD14+ myeloid APC in the blood and inflamed tissues of SLE patients is promoted by RNA-containing IC. Therefore, the level of activated Tfh cells associated with the level of circulating OX40L+ myeloid APC [4, 73, 74].
Th17 cells are a subtype of T cells that contribute to the SLE pathogenesis by producing IL-17 pro-inflammatory cytokine which can change the humoral and cellular immune response [75]. Th17 cytokines can trigger antibody generation via B cells and activate DCs and resident cells in inflamed tissues. Besides, Th17 can promote inflammation in the disease target organ and contribute to SLE progression. The frequency of Th17 cells is raised in patients with SLE, especially in those with renal involvement, and is correlated with disease activity [76].
Treg cells have an immuno-modulatory role [77], and their inappropriate activity may lead to autoimmune disorders [78]. The percentage of forkhead box P3 (FoxP3)+ CD4+ CD25+/high Treg cells and their suppressive function are decreased in SLE patients [77]. Down-regulation of FoxP3 in Treg cells results in weakened inhibitory activity in autoreactive T cells of SLE patients [24, 77]. However, some studies have reported a raised number of Treg cells in patients with SLE compared to healthy individuals [77]. Miyara et al. have indicated that CD4+ CD25+/high Treg cell depletion in SLE patients leads to the exacerbation of the disease [79]. Immunosuppressive therapies like glucocorticoids lead to a restoration of functional Treg cells in SLE patients [78].
γδT cells are a subtype of T cells that participate in anti-infection and anti-tumor responses of the immune system and have a role in the immunopathogenesis of autoimmune disorders. It seems that γδT cells in SLE express APC-specific markers like HLA-DR or CD80/86 can activate T cells via antigens’ presentation. Furthermore, the number of these cells is decreased during disease progression [80]. There is a different subtype of γδT cells that produces a set of Th1/Th2 cytokines such as IFN-γ, TNF-α, IL-10, and IL-4 [80, 81] that participates in the SLE pathogenesis. There is another group of γδT cells which is similar to Th17 cells, called Tγδ17 cells, that induces T-cell differentiation and increases B-cell proliferation and antibody generation by IL-17 [80]. Although based on Lu et al.’s study, there is no contrast in the rate of Tγδ17 cells in SLE patients compared to healthy individuals [80, 82]. Some subsets of γδT cells represent FoxP3, CD25, and CD27high, and CD45RAlow, and produce TGF-β, known as regulatory γδT cells, which have an immunoregulatory function [4, 80]. Based on Li et al.’s study, these regulatory cells seemed to be reduced in the peripheral blood of patients during SLE development and were reversely related to disease activity [83].
B cells and autoAbs
B cells are considered a key player in SLE pathogenesis as the precursors of autoAbs producing plasma cells, and antigen-presenting cells [84]. Mature B cells differentiate into B1 or B2 cells and the latter is subsequently differentiated into follicular and marginal zone B cells. The BAFF, known as B lymphocyte stimulator (BLyS), is an essential survival factor that is expressed by B cells’ linage cells [85, 86]. This cytokine is critical for the maintenance and selection of B cells. The expression and the production of BAFF can be promoted by inflammatory cytokines, like TNF-α, IFN-γ, and IL-2 [86]. Mounting evidence suggests that the primitive source of BAFF is DCs that are occasionally settled in mucosal, glandular, and dermal areas. Since the clinical observations indicate that most involvements of SLE patients are in the oral and cutaneous areas, it is sensible to contemplate that the over activity of DCs may be a providing factor for an enhanced level of BAFF and thereby SLE progression [86]. The expression of the BAFF gene is correlated with the serological activity of SLE disorder, and the level of BAFF in the serum of SLE patients is raised and it is associated with the level of anti-dsDNA antibodies [85]. It is suggested that the level of BAFF gene expression is a better marker for tracing SLE activity and progression than the serum level of BAFF protein [87]. The elevated level of BAFF can diminish the stringency of B cells’ clonal selection, leading to the remaining autoreactive B cells peripherally [4]. Therefore, BAFF is a favorable therapeutic target in SLE, and belimumab, an anti-BAFF monoclonal antibody is the first drug that was approved by the FDA for use in treating SLE individuals [4]. According to the reports, In SLE patients, there is an abnormal expansion of the B-cell subpopulation that can produce anti-dsDNA antibodies [25]. In addition, there is a deficiency in the removal of autoreactive B cells in SLE patients even within the inactive phase. Generally, CD19+ B cells’ population includes various subpopulations that are determined by the expression of CD27 and IgD molecules such as naïve B cells (CD27− IgD+), IgM memory (CD27+ IgD+), switched memory (CD27+ IgD−), and late memory (CD27− IgD−) B cells. It has been shown that there is an excessive manner of BCR response and an enhanced rate of class-switched CD27+ IgD− memory B cells in people with SLE. Besides, it has been identified another subtype of memory B cells with CD27− IgD− phenotype in individuals with SLE [88]. In small groups of patients, an enhanced rate of CD27− IgD− memory B cells has been observed which may be linked with disease activity and renal involvement. Furthermore, the existence of CD27− IgD− memory B cells is correlated with the existence of anti-dsDNA, anti-smith, and anti-ribonucleoprotein autoAbs.
Another feature of SLE is an enhanced amount of circulating plasma cells, implying the inappropriate regulation in these cells. Abnormal expansion of plasma cells has been reported in the kidneys of SLE patients. With regards to the role of B cells and related autoAbs in SLE, thereby B-cell depletion may be useful to ameliorate SLE flare. Nevertheless, B-cell depletion therapy has had no satisfactory results, due to the regulatory B cells’ (Breg) elimination. Breg cells regulate B-cell activity via the production of IL-10, hence Breg cells are called B10 cells [89]. Moreover, IL-35 and TGF-β produced by Breg cells have a regulatory role on the rest of the immune cells through inducing Treg cells [89]. Based on Watanabe et al.’s research on murine lupus, it has been shown that splenic transferring of IL-10-producing Breg cells from wild-type NZB/W F1 mice into CD19–/–NZB/W F1 recipients results in reducing symptom in the recipients [89, 90]. On the other side, it has been found that some of Breg cells have a deficiency in IL-10 production as well as their regulatory function both in human and mice models of SLE. Therefore, although the number of Breg cells may be increased in SLE patients, they might produce little amounts of IL-10 [89, 91].
One of the main characteristics of people with SLE disorder is the presence of autoAbs. The existence of anti-dsDNA antibodies contributes to the production of various cytokines like TNFα, IL-1β, IL-6, IL-8, and inducible nitric oxide synthase (iNOS), which leads to chronic inflammation and triggers tissue injury [92]. Therefore, SLE patient, who is positive for anti-dsDNA antibody, is more likely to have vascular kidney involvement [93]. Anti-dsDNA antibodies are composed of different subtypes such as IgG, IgA, IgM, and IgE; however, most of the autoAbs subpopulation is attributed to IgG, and it seems that IgM plays a protective role in SLE due to its role in clearing apoptotic debris [92]. In addition to IgG autoAbs, IgE autoAbs is found in the serum of SLE patients. Growing evidence suggests the activity of IgE is not limited to allergy reactions and might participate in inflammatory and autoimmune reactions (94). A high concentration of IgE-dsDNA in patients with active SLE has been reported. Production of IgE autoAbs (like IgG) stimulates pDCs to produce IFN-α, TNF, and IL-6 [4, 31], and contributes to disease progression [94]. Despite the presence of ICs containing IgE, allergy manifestation is not observed in SLE patients [94, 95]. IgE blocking antibodies lead to reduced production of IFN-α. Besides, IgE deficiency in animal models delays the onset of SLE complications and diminishes inflammatory cell activation and migration [94, 95].
Complement system
The imbalance between the rate of production and clearance of apoptotic debris sometimes is the result of defects in the complement components. Therefore, these deficiencies predispose individuals to the development of infections and some autoimmune diseases like SLE. The complement system is a part of the innate immune system which is responsible for the clearance of apoptotic cells and destroying microbial components [3]. Antibodies-self antigens’ immune complexes will be coated with different components of the complement system like C1q, pentraxin 3, as well as C-reactive protein (CRP) and helping phagocytes to destroy them. Deficiency in complement components is rare but can be in any of the C1q, C1r, C1s, C4, or C2. It has been shown that deficiency in the C1q is associated with SLE development [96] and patients with C1q deficiency represent SLE at a young age [13, 97]. It seems that C1q can modulate DCs function by limiting cytokine production and downregulation of co-stimulatory molecules and thereby decreases T-cell activation [96]. Moreover, C1q can inhibit the generation of type I interferon by pDCs via the inhibition of receptor leukocyte-associated immunoglobulin-like receptor 1 (LAIR-1 or CD305) [3, 98]. Therefore, a defect in C1q is one of the predisposing factors for SLE. This defect may result from genetic polymorphisms, its continued use in the active phase of the diseases [97], and autoAbs generation against it [13, 99]. Studies in SLE patients with renal involvement have been shown a direct correlation between anti-dsDNA and anti-C1q antibodies with disease severity and activity [99].
The underlying cause of the initial disruption that leads to the production of anti-C1q antibodies is not known precisely [3, 99]. It has been suggested that the conformational change in the collagen-like region of C1q may lead to the generation of neoepitopes, known as autoAgs, which is induced anti-C1q antibodies’ production [97, 99, 100]. Moreover, the presence of pathogenic agents with molecular mimicry to C1q structure may trigger an autoimmune response and autoAb production [99]. The production of anti-C1q antibodies leads to a functional impairment of C1q and a failure in binding to ICs and apoptotic debris [99]. Besides, mutations in C1QA and C1QB genes lead to the abnormality in C1q levels in serum [5] and result in impairment clearance of apoptotic bodies [7]. Nevertheless, it has been shown that functional deficiency in C1q can be restored by bone marrow transplantation [4, 5, 101]. Complete homozygous deficiency of C4A or C4B is one of the contributing factors in developing SLE. C4 has a role in the negative selection by inducing anergy in B cells, thereby defects in the whole C4 (C4a and C4b) may increase the number of autoreactive B cells [5, 98]. It seems that an increased copy number of the total C4 (C4A and C4B) gene may have a protective role in SLE [13]. Although in another study, no association was observed between the C4B gene copy number and SLE complication [5]. It has been reported that increased C4A gene expression in the Korean population may have a protective role against SLE, but this finding was not observed in European individuals [5].
Cytokines and SLE
The cytokine balance appears to be impaired in autoimmune diseases. Since the connection between the components of the immune system occurs through cytokines, the role of these proteins in the progression of autoimmune disorders is significant. Here, we considered cytokines’ roles in the pathogenesis of SLE. The cytokines alternations in SLE are summarized in Table 2.
IFN-α
IFN-α belongs to the type I IFN family and it appears to be associated with inadequate clearance of apoptotic debris and inflammation by T cells [102]. IFN-α is the prominent cytokines in SLE disorder that can promote B-cell activation and enhance autoAbs production [4]. IFN-α can affect the expression of some surface molecules (CD80, CD86, and MHC-II) and promote T-cell activity [31]. IFN-α is mainly produced by innate immune cells, especially pDCs. High mobility group box 1 (HMGB1) protein derived from dead cell components is capable to induce IFN-α production [103]. Interferon production mostly is affected by the IRF [17], and various polymorphisms in the IRF locus are linked with an enhanced likelihood of SLE involvement [66]. Studies have shown an enhanced expression of the IRF5 gene in individuals with SLE [4]. SLE patients with a high level of IFN-α in serum have an increased likelihood to develop cutaneous and kidney involvement [100]. Single-cell RNA-seq (transcriptomics) performed on renal and skin biopsies from LN patients revealed an up-regulation of type I IFN-response pathway genes in the tubular cell and keratinocyte of patients compared to healthy controls, indicating a systemic response to the elevated type I IFN in them. It is also found that the score of tubular IFN signatures can predict the response to treatment in LN patients [104, 105] (Table 3).
TNF-α
TNF-α is an inflammatory multipotential cytokine that regulates the immune system. The role of TNF-α in SLE disease is unclear [106, 107]. TNF-α regulates B-cell activity, enhances antibody production, increases the expression of vascular cell adhesion protein-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), and inhibits IFN-α production. [106, 107]. UV radiation stimulates keratinocytes to produce TNF-α. Therefore, it is thought that the use of anti-TNF therapies may ameliorate SLE symptoms. Nevertheless, it has been observed that TNF inhibitors induce the development of a similar lesion in cutaneous lupus [106]. The reason for this phenomenon can be attributed to the inhibitory role of TNF-α on the production of IFN-α in pDCs. Therefore, TNF inhibitors trigger IFN-α generation, which in turn stimulates the production of other inflammatory cytokines, leading to lupus-like cutaneous lesions [106]. However, short-term anti-TNF therapy may be able to prevent lupus-induced nephritis [106].
TGF-β
TGF-β is an immunosuppressive cytokine that induces FoxP3+ Treg cells to prevent the function of Th1 and Th2 cells [108, 109]. TGF-β expression in SLE patients is considerably lower than in healthy individuals [110]; while it has been increased in the urine of patients with lupus nephritis (LN). Studies on murine lupus models have implicated that, besides the TGF-β role in the maintenance of tolerance, it can cause severe organ injury, thereby more studies are required to exploit the TGF-β role in SLE pathogenesis [108].
IL-1
IL-1 superfamily consists of pro-inflammatory and anti-inflammatory cytokines. IL-1α, IL-1β, and IL-18, as a subset of the IL-1 family, have a pro-inflammatory function, and may participate in the pathogenesis of SLE. IL-18 can induce the generation of inflammatory cytokines (IL-1β and TNF-α) in varied immune cells like Th1 cells, monocytes, and NK cells [102, 111]. IL-18 leads to increase expression of co-stimulatory molecules, elevates the level of FasL- and perforin-mediated cytotoxicity by NK cells, and CD8+ T cell. Since all these immune activities are predominant to inflammatory reaction and lead to tissue damage may be a driving factor for developing SLE. There has been an increased level of IL-18 in SLE patients, while no increase in the level of IL-1β has been observed [111, 112] On the other hand, the observations have suggested an association between the IL-18 and lupus-induced nephritis, although this association has not been seen with IL-1β [106, 113]. However, some studies suggest a correlation between IL-1β and SLE disease [106]. Therefore, the role of IL-1β in the pathogenesis of SLE is controversial and more studies are required to better understand the function of this cytokine [112, 114].
IL-2
IL-2 is a growth factor for B and T cells and one of the essential cytokines for Treg cells’ survival and maintenance. Besides, it can suppress the generation of Tfr cells. It seems that IL-2 prevents the function of inflammatory cytokines like IL-17 [4]. Generally, impaired expression of IL-2 and its family (IL-15, IL-21) has been found in SLE patients. IL-2 downregulation results in decreased Treg cell population and reduced activation-induced cell death (AICD) [115]. Besides, reduced IL-2 expression may lead to decreased cytotoxicity activity of the CD8 + T and NK cells [4, 115].
IL-4
IL-4 is commonly known as a cytokine that stimulates antibody production as well as regulating humoral and cellular immunity [102, 106]. Moreover, some studies have suggested a suppressor role for IL-4 on T-cell activity. IL-4 has a confusing function in the pathogenesis of SLE. According to the studies on mouse models, IL-4 inhibits B-cell apoptosis and contributes to autoreactive B cells’ survival [102, 113]. Furthermore, IL-4 knockout mice produce low levels of the IgE and IgG1 antibodies. In SLE patients, who are positive for the anti-dsDNA antibodies, the amount of IL-4 is reduced [102, 116]. However, a reduced amount of IL-4 is involved in the expansion of articular and skin lesions induced by SLE [106, 116]. More evidence is needed to clarify the exact function of IL-4 in the SLE pathogenesis.
IL-6
IL-6 is a pleiotropic cytokine (affect the activity of multiple cell types) that is predominantly produced by MQs, but can be produced by some other immune cells like lymphocytes. IL-6 is an essential cytokine in the B-cell differentiation to the antibody-producing plasma cells in the germinal center[102]. Moreover, in the periphery, it can induce the activation of MQs and promote T-cell function. IL-6 with TGF-β triggers Th17 cell differentiation via activation of Th17-related transcription factors (STAT3, RORγt), contributing to the pathogenic role of Th17 cells in SLE. In addition, IL-6 suppresses Tregs differentiation and activation and thereby SLE progression [102, 111]. It seems that the level of autoAbs, especially anti-dsDNA antibodies, and disease severity are positively associated with IL-6 expression (107). Therefore, it can be used as a marker for monitoring the disease. Besides, SLE patients who have high rates of anti-dsDNA antibody and LN involvement have higher rates of IL-6 in their urine [107].
IL-10
IL-10 is generally produced by activated MQs, although it may be produced by some other immune cells like Treg cells and keratinocytes. IL-10, as an anti-inflammatory cytokine, can inhibit effector T cells function as well as co-stimulatory molecules like CD80, CD86, and CD40. Besides, it is capable to enhance the proliferation and maturation of B cells and promote immunoglobulin (Ig) class switching [102, 111]. The level of IL-10 directly is linked with the level of anti-dsDNA antibodies and conversely associated with the level of C3 and C4 components of complement [102, 106]. It seems that the level of IL-10 in the serum of patients with SLE is higher than IFN-α, so it can be considered as an SLE marker [66, 117]. It appears IL-10 suppression which may limit the renal damage and autoAbs generation in murine models, thereby the blockade of IL-10 may have a therapeutic effect, but more research is needed to design new therapies [102].
IL-17
IL-17 family consists of 6 members (A to F) [118]. IL-17 is a pro-inflammatory cytokine that triggers T-cell activity and induces autoAbs production. IL-17 may cause tissue injury by developing inflammatory cytokines (IL-1 and IL-6) and chemokines (CCL2, CCL7, CCL20) [106]. The amount of IL-17 (produced by Th17) depends on the presence of IL-23 [4]. In addition, IL-23 and its receptor contribute to the pro-inflammatory function of IL-17 by increasing the extrafollicular Tfh cells and enhancing the autoAbs formation [119]. IL-17 is involved in the survival and proliferation of B and plasma cells, antibodies production, and elevated expression of adhesion molecules [107, 119]. Observations have shown that the level of IL-17F is increased in subacute cutaneous lupus erythematosus (SCLE), while the level of IL-17A is similar to the control group [118]. In SLE patients with LN, IL-17 producing T cells, migrate to the kidneys and trigger the inflammatory process [120]. Research on the murine models has suggested that blockade of IL-17 receptors may ameliorate renal complications [121].
Steroid hormones and immune system interactions in SLE
The immune cells are highly responsive to the endocrine signaling pathway following to the presence of hormone receptors on the surface of them. The prevalence of the autoimmune disease is higher in women than men; the underlying mechanism may attribute to the difference in sex hormones. It seems true for SLE disorder, which aggravates during different sexual alterations like puberty, pregnancy, and post-partum periods. Steroid hormones are derived from cholesterol and classified as corticosteroids and sex steroids [122, 123]. Within two classes according to their receptors, they are classified into five different subgroups: glucocorticoids and mineralocorticoids, as corticosteroids and androgens, estrogens, and progestogens as sex steroid groups. They are mainly produced by the adrenal cortex, testes, ovaries, and placenta during pregnancy [4].
Cortisol is the main glucocorticoid, widely known as a stress hormone; it participates in different functions like stress-related responses, regulating metabolism, inflammatory, and immune responses [124]. It seems that cortisol exerts its anti-inflammatory role by inhibiting NF-κB activity [125]. Cortisol is likely to trigger apoptosis in pro-inflammatory T cells and, in physiological dose, may shift immune response from Th1 to Th2 [125]. However, it promotes Treg cells’ survival by activating CD25-related gene coding. Treg cells needed a high level of IL-2 and its associated receptors, known as CD25, to activate and perform regulatory functions [126]. Glucocorticoids prevent antibody production and affect B cells at the differentiation, survival, and proliferation stage [127]. They regulate DCs’ migration and apoptosis and inhibit DCs maturation by converting them to tolerogenic DCs [125, 126]. In addition, glucocorticoids prevent neutrophils rolling, adhesion, and migration by inhibiting adhesion molecules. In contrast, it may suppress apoptosis in these cells by up-regulation of anti-apoptotic mediators like myeloid-cell leukemia (Mcf)-1 and X-linked inhibitor of apoptosis protein (XAIP) [124]. Although the same serum level of cortisol was found in SLE patients and healthy individuals [128], it is reported that the level of awakening cortisol was significantly higher in patients with higher ESR compared to patients with lower ESR [129]. It seems that the hypothalamic–pituitary–adrenal (HPA) axis exerts partial alternations in SLE patients and cannot reduce disease inflammation, while treatment with synthetic glucocorticoids exhibits anti-inflammatory properties in them [130].
The prominent steroid with a mineralocorticoid function is aldosterone. Contrary to glucocorticoid, aldosterone raises NF-κB activity and regulates the expression of inflammatory genes [131, 132]. It induces an inflammatory response that triggers DCs activation, enhances B-cell recruitment, CD4+ T-cell differentiation to Th1 and Th17 cells, and diminish regulatory T cells. According to the Herrada et al.’s study, aldosterone can trigger CD8+ T cells via the DCs-dependent pathway [133]. It seems MQs and T cells infiltrate the kidney, heart, and vascular due to the aldosterone secretion which likely leads to end-organ damage [131]. It was shown that aldosterone receptor blockade leads to reduced kidney damage, diminished levels of serum autoantibodies, and decreased proteinuria in murine lupus nephritis [134].
Many sex hormones that influence the immune system seem to act as triggers or protectors of SLE development. Mounting evidence reveals a correlation between increased risk of SLE development and exposure to estrogen, while progesterone and androgens seem to have a protective role by impeding the function of the estrogen [4].
Estrogen may contribute to the generation of type I interferon through up-regulation of IRF-3, IRF-5, and IRF-7. Estrogen participates in autoreactive B cells’ survival by inhibiting their deactivation or deletion at developmental checkpoints [123, 135]. It seems that up-regulated expression of some molecules like Bcl-2, CD22, vascular cell adhesion molecules (VCAM1), BAFF, and protein tyrosine phosphatase contributes to the survival of autoreactive B cells. Moreover, estrogens augment the antibodies class switching from IgM to IgG by enhancing the expression of activation-induced cytidine deaminase (AICDA) and homeobox protein Hox-C4 (HOXC4) in B cells, contributing to the high-affinity IgG autoAbs development [124, 127]. Estrogen increases the generation of IgG and IgM autoAbs against ds-DNA by PBMCs, contributing to the SLE progression and development. In addition, estrogen participates in the evolution, differentiation, and function of CD4+ T cells. It has been established that the estrogen at a low level can trigger Th1 responses, while at a high level might induce Th2 cell responses [123, 135]. Previous studies in SLE patients document that estrogen triggers T-cell activation by estrogen receptor (ER)-α and ERβ and up-regulates T-cell activation markers like calcineurin and CD40 ligand. Estrogen likely participates in the neutrophils’ apoptosis and NETs formation. Administration of estrogen in SLE-prone women triggers the onset of the disease and promotes anti-dsDNA antibodies’ production [136].
Contrary to estrogens, progesterone and androgens likely have a protective role in SLE disorder. Progesterone seems to have an immune-modulatory role and impede the effects of the estrogens. Progesterone prevents IRF-7 activation and TLR-dependent IFN-α generation in pDCs, leading to relieving the IFN signature and disease severity [122, 135]. Besides, progesterone has a contracting effect in the context of IgM to IgG class switching of B cells by preventing the expression of AICDA, leading to a decrease in pathogenic IgG autoAbs development. In addition, NET formation and NETosis can be impeded by progesterone [123]. Based on studies on animal models, administration of medication that contains progesterone may reduce autoAbs production and ameliorate renal complications in SLE disease [123].
In contrast to estrogens, androgens likely elevated the checkpoints for induction of apoptosis in autoreactive B cells, possibly related to the diminished Bcl-2 expression in B cells [123, 135]. Furthermore, it can diminish IgG autoAbs generation by suppressing antibodies class switching. Androgens impede autoimmune development by increasing the level of TGF-β1. The result from the clinical trial reveals a decreased level of plasma androgen in SLE individuals. According to the Singh et al.’s study, the plasma level of estradiol, as the most common type of estrogens in women of childbearing age, enhanced, and testosterone level diminished in SLE female individuals [137]. In accordance with their report, exposure of the Treg cells to testosterone promotes FoxP3 CD4 + Treg cell generation in SLE female patients. Indeed, the plasma level of testosterone in SLE females is directly associated with FoxP3 expression (137).
Prolactin (PRL), as a peptide hormone, is generated by the anterior pituitary and lymphocytes that participate in immune responses. In the SLE context, it seems to affect the negative selection of autoreactive B cells and dysregulate receptor editing, contributing to the survival and proliferation of the autoreactive B cells, and autoAbs formation [123, 138]. SLE patients with high levels of PRL have raised levels of anti-cardiolipin, anti-dsDNA, anti-La, and anti-Ro antibodies Accordingly, PRL likely up-regulates the expression of INF-γ, CD40L, IL-2, and IL-12, contributing to CD4+ T-cell activation and SLE development [123, 138]. Several reports dedicate the correlation between a low level of PRL with a decreased level of complements, neurological, hematological, and renal complications in SLE patients. In pregnant SLE patients, the presence of anti-PLR antibodies or administration of medicine against PRL may ameliorate SLE complications [136, 138].
Concluding remarks
Alternations in different innate and adaptive immune cells and their cytokines have been observed in SLE patients. According to the immunopathogenesis of SLE’s disorder, the principal reason for the SLE development is attributed to ICs deposition on the walls and surface of arteries and organs. The self-tolerance of the immune system is disrupted for unknown reasons, and the autoreactive B cells’ clones, which are normally removed by negative selection, remain and produce a wide range of autoAbs. Indeed, an increased rate of apoptosis in non-immune cells and a decreased rate of apoptosis in autoreactive clones of immune cells, like a domino, trigger other parts of the immune system. Besides, the innate immune system poses a critical role in the pathogenesis of SLE by the production and promotion of pro-inflammatory cytokines and mediators. These mediators boost local inflammation, trigger self-tolerance breakdown, and further activate the adaptive immune system. In addition, it is well established that the sex hormones and the immune system have an interaction; thereby, an abnormal endocrine alteration may be the cause of the clinical manifestations of autoimmune diseases like SLE. Following the multifactorial nature of SLE and the unknown primary cause of its onset, prevention and treatment of this disease are associated with obstacles. Further studies may lead to the development and advancement of biological therapies with fewer side effects.
Availability of data and materials
Not applicable.
References
Richardson B. Epigenetically altered T cells contribute to lupus flares. Cells. 2019;8(2):127.
Honarpisheh M, Köhler P, von Rauchhaupt E, Lech M. The involvement of microRNAs in modulation of innate and adaptive immunity in systemic lupus erythematosus and lupus nephritis. J Immunol Res. 2018;2018:4126106.
Trouw LA, Pickering MC, Blom AM. The complement system as a potential therapeutic target in rheumatic disease. Nat Rev Rheumatol. 2017;13(9):538–47.
Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–30.
Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis. 2014;73(9):1601–6.
Weidenbusch M, Kulkarni OP, Anders H-J. The innate immune system in human systemic lupus erythematosus. Clin Sci. 2017;131(8):625–34.
Herrada AA, Escobedo N, Iruretagoyena M, Valenzuela RA, Burgos PI, Cuitino L, et al. Innate immune cells’ contribution to systemic lupus erythematosus. Front Immunol. 2019;10:772.
Brandt L, Hedberg H. Impaired phagocytosis by peripheral blood granulocytes in systemic lupus erythematosus. Scand J Haematol. 1969;6(5):348–53.
Alves CM, Marzocchi-Machado CM, Louzada-Junior P, Azzolini AE, Polizello AC, de Carvalho IF, et al. Superoxide anion production by neutrophils is associated with prevalent clinical manifestations in systemic lupus erythematosus. Clin Rheumatol. 2008;27(6):701–8.
Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159(1):237–43.
Lipsker DM, Schreckenberg-Gilliot C, Uring-Lambert B, Meyer A, Hartmann D, Grosshans EM, et al. Lupus erythematosus associated with genetically determined deficiency of the second component of the complement. Arch Dermatol. 2000;136(12):1508–14.
Paul E, Pozdnyakova OO, Mitchell E, Carroll MC. Anti-DNA autoreactivity in C4-deficient mice. Eur J Immunol. 2002;32(9):2672–9.
Macedo AC, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol. 2016;7:55.
Yap DYH, Chan TM. B cell abnormalities in systemic lupus erythematosus and lupus nephritis-role in pathogenesis and effect of immunosuppressive treatments. Int J Mol Sci. 2019;20(24):6231.
Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019;20(7):902–14.
Rao DA, Arazi A, Wofsy D, Diamond B. Design and application of single-cell RNA sequencing to study kidney immune cells in lupus nephritis. Nat Rev Nephrol. 2020;16(4):238–50.
Javinani A, Ashraf-Ganjouei A, Aslani S, Jamshidi A, Mahmoudi M. Exploring the etiopathogenesis of systemic lupus erythematosus: a genetic perspective. Immunogenetics. 2019;71(4):283–97.
Munoz LE, van Bavel C, Franz S, Berden J, Herrmann M, Van Der Vlag J. Apoptosis in the pathogenesis of systemic lupus erythematosus. Lupus. 2008;17(5):371–5.
Liphaus BL, Kiss MHB. The role of apoptosis proteins and complement components in the etiopathogenesis of systemic lupus erythematosus. Clinics. 2010;65(3):327–33.
Poon IK, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. 2010;17(3):381–97.
Lorenz G, Lech M, Anders HJ. Toll-like receptor activation in the pathogenesis of lupus nephritis. Clin Immunol. 2017;185:86–94.
Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS–STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. 2018;77(10):1507–15.
Lyn-Cook BD, Xie C, Oates J, Treadwell E, Word B, Hammons G, et al. Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol. 2014;61(1):38–43.
Devarapu SK, Anders H-J. Toll-like receptors in lupus nephritis. J Biomed Sci. 2018;25(1):35.
Karrar S, Graham DSC. Abnormal B cell development in systemic lupus erythematosus: what the genetics tell us. Arthritis Rheumatol (Hoboken, NJ). 2018;70(4):496.
Murayama G, Chiba A, Kuga T, Makiyama A, Yamaji K, Tamura N, et al. Inhibition of mTOR suppresses IFNα production and the STING pathway in monocytes from systemic lupus erythematosus patients. Rheumatology. 2020;59(10):2992–3002.
Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–70.
Thim-Uam A, Prabakaran T, Tansakul M, Makjaroen J, Wongkongkathep P, Chantaravisoot N, et al. STING mediates lupus via the activation of conventional dendritic cell maturation and plasmacytoid dendritic cell differentiation. Iscience. 2020;23(9):101530.
Lang KS, Burow A, Kurrer M, Lang PA, Recher M. The role of the innate immune response in autoimmune disease. J Autoimmun. 2007;29(4):206–12.
Chan VS-F, Nie Y-J, Shen N, Yan S, Mok M-Y, Lau C-S. Distinct roles of myeloid and plasmacytoid dendritic cells in systemic lupus erythematosus. Autoimmun Rev. 2012;11(12):890–7.
Klarquist J, Zhou Z, Shen N, Janssen EM. Dendritic cells in systemic lupus erythematosus: from pathogenic players to therapeutic tools. Mediators Inflamm. 2016;2016:5045248.
Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154(1):3–20.
Choi SC, Morel L. B cell contribution of the CD4(+) T cell inflammatory phenotypes in systemic lupus erythematosus. Autoimmunity. 2017;50(1):37–41.
Scheinecker C, Zwolfer B, Koller M, Manner G, Smolen JS. Alterations of dendritic cells in systemic lupus erythematosus: phenotypic and functional deficiencies. Arthritis Rheum. 2001;44(4):856–65.
Gill MA, Blanco P, Arce E, Pascual V, Banchereau J, Palucka AK. Blood dendritic cells and DC-poietins in systemic lupus erythematosus. Hum Immunol. 2002;63(12):1172–80.
Robak E, Smolewski P, Wozniacka A, Sysa-Jedrzejowska A, Robak T. Clinical significance of circulating dendritic cells in patients with systemic lupus erythematosus. Mediators Inflamm. 2004;13(3):171–80.
Blomberg S, Eloranta ML, Magnusson M, Alm GV, Ronnblom L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-alpha by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Rheum. 2003;48(9):2524–32.
Fiore N, Castellano G, Blasi A, Capobianco C, Loverre A, Montinaro V, et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol. 2008;45(1):259–65.
Vallin H, Blomberg S, Alm GV, Cederblad B, Ronnblom L. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-alpha) production acting on leucocytes resembling immature dendritic cells. Clin Exp Immunol. 1999;115(1):196–202.
Migita K, Miyashita T, Maeda Y, Kimura H, Nakamura M, Yatsuhashi H, et al. Reduced blood BDCA-2+ (lymphoid) and CD11c+ (myeloid) dendritic cells in systemic lupus erythematosus. Clin Exp Immunol. 2005;142(1):84–91.
Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus. 2008;17(7):654–62.
Hagberg N, Ronnblom L. Systemic lupus erythematosus–a disease with a dysregulated type I interferon system. Scand J Immunol. 2015;82(3):199–207.
Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008;58(1):251–62.
Wu S, Yeh K, Lee W, Yao T, Kuo M, Huang B, et al. Impaired phagocytosis and susceptibility to infection in pediatric-onset systemic lupus erythematosus. Lupus. 2013;22(3):279–88.
Bengtsson AA, Pettersson Å, Wichert S, Gullstrand B, Hansson M, Hellmark T, et al. Low production of reactive oxygen species in granulocytes is associated with organ damage in systemic lupus erythematosus. Arthritis Res Ther. 2014;16(3):1–8.
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187(1):538–52.
Zhong J, Olsson LM, Urbonaviciute V, Yang M, Bäckdahl L, Holmdahl R. Association of NOX2 subunits genetic variants with autoimmune diseases. Free Radical Biol Med. 2018;125:72–80.
Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci. 2012;109(2):E59–67.
Olsson LM, Johansson ÅC, Gullstrand B, Jönsen A, Saevarsdottir S, Rönnblom L, et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis. 2017;76(9):1607–13.
Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang S-Y, et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet. 2017;49(3):433–7.
de Bont CM, Boelens WC, Pruijn GJ. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16(1):19–27.
Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med. 2017;23(7):615–35.
He Y, Yang F-Y, Sun E-W. Neutrophil extracellular traps in autoimmune diseases. Chin Med J. 2018;131(13):1513.
Huang C, Li F, Wang J, Tian Z. Innate-like lymphocytes and innate lymphoid cells in asthma. Clin Rev Allergy Immunol. 2020;59(3):359–70.
Hou M, Liu S. Innate lymphoid cells are increased in systemic lupus erythematosus. Clin Exp Rheumatol. 2019;37(4):676–9.
Guo C, Zhou M, Zhao S, Huang Y, Wang S, Fu R, et al. Innate lymphoid cell disturbance with increase in ILC1 in systemic lupus erythematosus. Clin Immunol. 2019;202:49–58.
Hervier B, Beziat V, Haroche J, Mathian A, Lebon P, Ghillani-Dalbin P, et al. Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-γ production in patients with active disease. Arthritis Rheum. 2011;63(6):1698–706.
Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, et al. Mechanisms of interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002;21(8):1251–62.
Spada R, Rojas JM, Barber DF. Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J Leukoc Biol. 2015;98(4):479–87.
Schepis D, Gunnarsson I, Eloranta ML, Lampa J, Jacobson SH, Kärre K, et al. Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus. Immunology. 2009;126(1):140–6.
Hudspeth KL, Shu W, Wang J, Rahman S, Smith MA, Casey KA, et al. NK cell phenotype and proliferation in systemic lupus erythematosus. Am Assoc Immnol. 2016;196:194.
Henriques A, Teixeira L, Inês L, Carvalheiro T, Gonçalves A, Martinho A, et al. NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol. 2013;32(6):805–13.
Ma W-T, Gao F, Gu K, Chen D-K. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front Immunol. 2019;10:1140.
Li Y, Lee PY, Sobel ES, Narain S, Satoh M, Segal MS, et al. Increased expression of FcγRI/CD64 on circulating monocytes parallels ongoing inflammation and nephritis in lupus. Arthritis Res Ther. 2009;11(1):1–13.
Umare V, Pradhan V, Nadkar M, Rajadhyaksha A, Patwardhan M, Ghosh KK, et al. Effect of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) on clinical manifestations in Indian SLE patients. Mediat Inflamm. 2014;2014:1–8.
Agha-Hosseini F, Moosavi M-S, Tabrizi MH. Comparison of oral lichen planus and systemic lupus erythematosus in interleukins level. Arch Iran Med (AIM). 2015;18(10):703.
Pellefigues C, Charles N. The deleterious role of basophils in systemic lupus erythematosus. Curr Opin Immunol. 2013;25(6):704–11.
Dossybayeva K, Abdukhakimova D, Poddighe D. Basophils and systemic lupus erythematosus in murine models and human patients. Biology. 2020;9(10):308.
Janeway C, Travers P, Capra JD, Walport M. Immunobiology: the immune system in health and disease. Current Biology Publications; 1999.
Kurata I, Matsumoto I, Sumida T. T follicular helper cell subsets: a potential key player in autoimmunity. Immunol Med. 2020;44:1–9.
Lai NS, Koo M, Yu CL, Lu MC. Immunopathogenesis of systemic lupus erythematosus and rheumatoid arthritis: the role of aberrant expression of non-coding RNAs in T cells. Clin Exp Immunol. 2017;187(3):327–36.
Xu B, Wang S, Zhou M, Huang Y, Fu R, Guo C, et al. The ratio of circulating follicular T helper cell to follicular T regulatory cell is correlated with disease activity in systemic lupus erythematosus. Clin Immunol. 2017;183:46–53.
Blanco P, Ueno H, Schmitt N. T follicular helper (Tfh) cells in lupus: activation and involvement in SLE pathogenesis. Eur J Immunol. 2016;46(2):281–90.
Jacquemin C, Schmitt N, Contin-Bordes C, Liu Y, Narayanan P, Seneschal J, et al. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity. 2015;42(6):1159–70.
Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(2):R53.
Suarez-Fueyo A, Bradley SJ, Tsokos GC. T cells in systemic lupus erythematosus. Curr Opin Immunol. 2016;43:32–8.
Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45(2):344–55.
Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. 2014;2014:419029.
Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, et al. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol (Baltimore). 2005;175(12):8392–400.
Wu M, Yang J, Li X, Chen J. The role of gammadelta T cells in systemic lupus erythematosus. J Immunol Res. 2016;2016:2932531.
Lu Z, Su D, Wang D, Li X, Feng X, Sun L. Elevated apoptosis and impaired proliferation contribute to downregulated peripheral γδ T cells in patients with systemic lupus erythematosus. Clin Dev Immunol. 2013;2013:1–3.
Lu Z, Su D, Wang D, Li X, Feng X, Sun L. Elevated apoptosis and impaired proliferation contribute to downregulated peripheral gamma delta T cells in patients with systemic lupus erythematosus. Clin Dev Immunol. 2013;203:405395.
Li X, Kang N, Zhang X, Dong X, Wei W, Cui L, et al. Generation of human regulatory γδ T cells by TCRγδ stimulation in the presence of TGF-β and their involvement in the pathogenesis of systemic lupus erythematosus. J Immunol. 2011;186(12):6693–700.
Dörner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis Res Ther. 2011;13(5):243.
Gottschalk TA, Tsantikos E, Hibbs ML. Pathogenic inflammation and its therapeutic targeting in systemic lupus erythematosus. Front Immunol. 2015;6:550.
Cancro MP, D’Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Investig. 2009;119(5):1066–73.
Zollars E, Bienkowska J, Czerkowicz J, Allaire N, Ranger AM, Magder L, et al. BAFF (B cell activating factor) transcript level in peripheral blood of patients with SLE is associated with same-day disease activity as well as global activity over the next year. Lupus Sci Med. 2015;2(1):e000063.
Wang T, Mei Y, Li Z. Research progress on regulatory B cells in systemic lupus erythematosus. BioMed Res Int. 2019;209:1–7.
Cai X, Zhang L, Wei W. Regulatory B cells in inflammatory diseases and tumor. Int Immunopharmacol. 2019;67:281–6.
Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, et al. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol. 2010;184(9):4801–9.
Matsushita T. Regulatory and effector B cells: friends or foes? J Dermatol Sci. 2019;93(1):2–7.
Wang X, Xia Y. Anti-double stranded DNA antibodies: origin, pathogenicity, and targeted therapies. Front Immunol. 2019;10:1667.
Speyer CB, Costenbader KH. Cigarette smoking and the pathogenesis of systemic lupus erythematosus. Expert Rev Clin Immunol. 2018;14(6):481–7.
Bayry J. Lupus pathogenesis: role of IgE autoantibodies. Cell Res. 2016;26(3):271.
Augusto J-F, Truchetet M-E, Charles N, Blanco P, Richez C. IgE in lupus pathogenesis: friends or foes? Autoimmun Rev. 2018;17(4):361–5.
Moudi B, Salimi S, Farajian Mashhadi F, Sandoughi M, Zakeri Z. Association of FAS and FAS ligand genes polymorphism and risk of systemic lupus erythematosus. Sci World J. 2013;2013:1–3.
Toubi E, Vadasz Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun Rev. 2019;18:306.
Ramsey-Goldman R, Li J, Dervieux T, Alexander RV. Cell-bound complement activation products in SLE. Lupus Sci Med. 2017;4(1):e000236.
Hristova M, Stoyanova V. Autoantibodies against complement components in systemic lupus erythematosus–role in the pathogenesis and clinical manifestations. Lupus. 2017;26(14):1550–5.
Pickering MC, Botto M. Are anti-C1q antibodies different from other SLE autoantibodies? Nat Rev Rheumatol. 2010;6(8):490–3.
Bao L, Cunningham PN, Quigg RJ. Complement in lupus nephritis: new perspectives. Kidney Dis (Basel). 2015;1(2):91–9.
Rojas M, Rodríguez Y, Leon KJ, Pacheco Y, Acosta-Ampudia Y, Monsalve DM, et al. Cytokines and inflammatory mediators in systemic lupus erythematosus. Rheumatology. 2018.
Panda SK, Kolbeck R, Sanjuan MA. Plasmacytoid dendritic cells in autoimmunity. Curr Opin Immunol. 2017;44:20–5.
Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol. 2019;20(7):915–27.
Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, et al. Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI Insight. 2017;2(9):e93009.
Robinson ES, Werth VP. The role of cytokines in the pathogenesis of cutaneous lupus erythematosus. Cytokine. 2015;73(2):326–34.
Yap DY, Lai KN. The role of cytokines in the pathogenesis of systemic lupus erythematosus—from bench to bedside. Nephrology (Carlton). 2013;18(4):243–55.
Okamoto A, Fujio K, Okamura T, Yamamoto K. Regulatory T-cell-associated cytokines in systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011:463412.
Mirkazemi S, Akbarian M, Jamshidi AR, Mansouri R, Ghoroghi S, Salimi Y, et al. Association of STAT4 rs7574865 with susceptibility to systemic lupus erythematosus in Iranian population. Inflammation. 2013;36(6):1548–52.
Sanjabi S, Oh SA, Li MO. Regulation of the immune response by TGF-beta: from conception to autoimmunity and infection. Cold Spring Harb Perspect Biol. 2017. https://doi.org/10.1101/cshperspect.a022236.
Davis LS, Hutcheson J, Mohan C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31(10):781–9.
Mende R, Vincent FB, Kandane-Rathnayake R, Koelmeyer R, Lin E, Chang J, et al. Analysis of serum interleukin (IL)-1beta and IL-18 in systemic lupus erythematosus. Front Immunol. 2018;9:1250.
Pacheco Y, Barahona-Correa J, Monsalve DM, Acosta-Ampudia Y, Rojas M, Rodriguez Y, et al. Cytokine and autoantibody clusters interaction in systemic lupus erythematosus. J Transl Med. 2017;15(1):239.
Tahmasebi Z, Akbarian M, Mirkazemi S, Shahlaee A, Alizadeh Z, Amirzargar AA, et al. Interleukin-1 gene cluster and IL-1 receptor polymorphisms in Iranian patients with systemic lupus erythematosus. Rheumatol Int. 2013;33(10):2591–6.
Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J Biomed Biotechnol. 2010. https://doi.org/10.1155/2010/740619.
Dong C, Fu T, Ji J, Li Z, Gu Z. The role of interleukin-4 in rheumatic diseases. Clin Exp Pharmacol Physiol. 2018;45(8):747–54.
Rezaei N, Aghamohammadi A, Mahmoudi M, Shakiba Y, Kardar GA, Mahmoudi M, et al. Association of IL-4 and IL-10 gene promoter polymorphisms with common variable immunodeficiency. Immunobiology. 2010;215(1):81–7.
Speeckaert R, Lambert J, Grine L, Van Gele M, De Schepper S, van Geel N. The many faces of interleukin-17 in inflammatory skin diseases. Br J Dermatol. 2016;175(5):892–901.
Dai H, He F, Tsokos GC, Kyttaris VC. IL-23 limits the production of IL-2 and promotes autoimmunity in lupus. J Immunol. 2017;199(3):903–10.
Miyake K, Akahoshi M, Nakashima H. Th subset balance in lupus nephritis. J Biomed Biotechnol. 2011;2011:980286.
Paley MA, Strand V, Kim AH. From mechanism to therapies in systemic lupus erythematosus. Curr Opin Rheumatol. 2017;29(2):178–86.
Shepherd R, Cheung AS, Pang K, Saffery R, Novakovic B. Sexual dimorphism in innate immunity: the role of sex hormones and epigenetics. Front Immunol. 2021;11:3559.
Pan Q, Chen X, Liao S, Chen X, Zhao C, Xu Y-Z, et al. Updated advances of linking psychosocial factors and sex hormones with systemic lupus erythematosus susceptibility and development. PeerJ. 2019;7:e7179.
Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34(9):518–30.
Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol. 2010;120(2–3):69–75.
Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, et al. The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev. 2011;10(6):305–10.
Thau L, Gandhi J, Sharma S. Physiology, cortisol. StatPearls [Internet]. 2021.
Gutiérrez MA, Garcia ME, Rodriguez JA, Rivero S, Jacobelli S. Hypothalamic-pituitary-adrenal axis function and prolactin secretion in systemic lupus erythematosus. Lupus. 1998;7(6):404–8.
van der Goes MC, Bossema ER, Hartkamp A, Godaert GLR, Jacobs JWG, Kruize AA, et al. Cortisol during the day in patients with systemic lupus erythematosus or primary sjögren’s syndrome. J Rheumatol. 2011;38(2):285–8.
Porta S, Danza A, Arias Saavedra M, Carlomagno A, Goizueta MC, Vivero F, et al. Glucocorticoids in systemic lupus erythematosus. Ten questions and some issues. J Clin Med. 2020. https://doi.org/10.3390/jcm9092709.
Ferreira NS, Tostes RC, Paradis P, Schiffrin EL. Aldosterone, inflammation, immune system, and hypertension. Am J Hypertens. 2021;34(1):15–27.
Leroy V, De Seigneux S, Agassiz V, Hasler U, Rafestin-Oblin M-E, Vinciguerra M, et al. Aldosterone activates NF-κB in the collecting duct. J Am Soc Nephrol. 2009;20(1):131–44.
Herrada AA, Campino C, Amador CA, Michea LF, Fardella CE, Kalergis AM. Aldosterone as a modulator of immunity: implications in the organ damage. J Hypertens. 2011;29(9):1684–92.
Monrad SU, Killen PD, Anderson MR, Bradke A, Kaplan MJ. The role of aldosterone blockade in murine lupus nephritis. Arthritis Res Ther. 2008;10(1):R5.
Bereshchenko O, Bruscoli S, Riccardi C. Glucocorticoids, sex hormones, and immunity. Front Immunol. 2018;9:1332.
Gonzalez DA, Diaz BB. Rodriguez Perez Mdel C, Hernandez AG, Chico BN, de Leon AC. Sex hormones and autoimmunity Immunol Lett. 2010;133(1):6–13.
Singh RP, Bischoff DS. Sex hormones and gender influence the expression of markers of regulatory T cells in SLE patients. Front Immunol. 2021;12:408.
Legorreta-Haquet MV, Flores-Fernández R, Blanco-Favela F, Fuentes-Pananá EM, Chávez-Sánchez L, Hernández-González R, et al. Prolactin levels correlate with abnormal B cell maturation in MRL and MRL/lpr mouse models of systemic lupus erythematosus-like disease. Clin Dev Immunol. 2013. https://doi.org/10.1155/2013/287469.
Funes SC, Ríos M, Gómez-Santander F, Fernández-Fierro A, Altamirano-Lagos MJ, Rivera-Perez D, et al. Tolerogenic dendritic cell transfer ameliorates systemic lupus erythematosus in mice. Immunology. 2019;158(4):322–39.
Mok MY. Tolerogenic dendritic cells: role and therapeutic implications in systemic lupus erythematosus. Int J Rheum Dis. 2015;18(2):250–9.
Ritprajak P, Kaewraemruaen C, Hirankarn N. Current paradigms of tolerogenic dendritic cells and clinical implications for systemic lupus erythematosus. Cells. 2019;8(10):1291.
Hasni S, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Joyal E, et al. Safety and tolerability of Omalizumab: a randomized clinical trial of humanized anti-IgE monoclonal antibody in systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(7):1135–40.
Yildirim-Toruner C, Diamond B. Current and novel therapeutics in the treatment of systemic lupus erythematosus. Journal of Allergy and Clinical Immunology. 2011;127(2):303–12.
Barnado A, Crofford LJ, Oates JC. At the bedside: neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J Leukoc Biol. 2016;99(2):265–78.
Salemme R, Peralta LN, Meka SH, Pushpanathan N, Alexander JJ. The role of netosis in systemic lupus erythematosus. J Cell Immunol. 2019;1(2):33.
Baxter AG, Smyth MJ. The role of NK cells in autoimmune disease. Autoimmunity. 2002;35(1):1–14.
Vukelic M, Li Y, Kyttaris VC. Novel treatments in lupus. Front Immunol. 2018;9:2658.
La Cava A. Anticytokine therapies in systemic lupus erythematosus. Immunotherapy. 2010;2(4):575–82.
Postal M, Costallat LT, Appenzeller S. Biological therapy in systemic lupus erythematosus. Int J Rheumatol. 2012. https://doi.org/10.1155/2012/578641.
Mok MY, Shoenfeld Y. Recent advances and current state of immunotherapy in systemic lupus erythematosus. Expert Opin Biol Ther. 2016;16(7):927–39.
Zhu L-J, Yang X, Yu X-Q. Anti-TNF-alpha therapies in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:465898.
Metawie SA, ElRefai RM, ElAdle SS, Shahin RMH. Transforming growth factor-β1 in systemic lupus erythematosus patients and its relation to organ damage and disease activity. Egypt Rheumatol. 2015;37(4):S49–54.
He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2020;79(1):141–9.
Zhang H, Bernuzzi F, Lleo A, Ma X, Invernizzi P. Therapeutic potential of IL-17-mediated signaling pathway in autoimmune liver diseases. Mediat Inflamm. 2015;2015:436450.
Parikh SV, Rovin BH. Current and emerging therapies for lupus nephritis. J Am Soc Nephrol. 2016;27(10):2929–39.
Alunno A, Bartoloni E, Bistoni O, Nocentini G, Ronchetti S, Caterbi S, et al. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: the old and the new. Clin Dev Immunol. 2012. https://doi.org/10.1155/2012/823085.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
NB, MA, and STF contributed to acquisition and interpretation of data and drafting the article. EF*, RM, AJ, and MM contributed to the conception and design of the study and revising the article critically for important intellectual content. All authors contributed to manuscript revision, read, and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bolouri, N., Akhtari, M., Farhadi, E. et al. Role of the innate and adaptive immune responses in the pathogenesis of systemic lupus erythematosus. Inflamm. Res. 71, 537–554 (2022). https://doi.org/10.1007/s00011-022-01554-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-022-01554-6