
Abstract
Introduction
G protein-coupled receptors (GPCRs) are transmembrane receptor proteins, which allow the transfer of signals across the membrane. Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovitis and accompanied with inflammatory and abnormal immune response. GPCRs signaling pathways play a significant role in inflammatory and immune response processes including RA.
Findings
In this review, we have focused on the advances in GPCRs signaling pathway implicating the inflammatory and immune response of RA. The signaling pathways of GPCRs–adenylyl cyclase (AC)–cyclic adenosine 3′, 5′-monophosphate (cAMP) include β2 adrenergic receptors (β2-ARs)–AC–cAMP signaling pathways, E-prostanoid2/4 (EP2/4)–AC–cAMP signaling pathways and so on. Regulatory proteins, such as G protein-coupled receptor kinases (GRKs) and β-arrestins, play important modulatory roles in GPCRs signaling pathway. GPCRs signaling pathway and regulatory proteins implicate the pathogenesis process of inflammatory and immune response.
Conclusion
GPCRs–AC–cAMP signal pathways involve in the inflammatory and immune response of RA. Different signaling pathways are mediated by different receptors, such as β2-AR, PGE2 receptor, chemokines receptor, and adenosine receptor. GRKs and β-arrestins are crucial proteins in the regulation of GPCRs signaling pathways. The potential therapeutic targets as well as strategies to modulate GPCRs signaling pathway are new development trends.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
G protein coupled receptors (GPCRs) are the largest families of membrane proteins, which are encoded by more than 800 genes in the human genome. GPCRs contain seven transmembrane helical bundles that provide binding sites for ligands [1]. GPCRs include many of receptors, such as β2-adrenergic receptor (β2-AR), prostaglandin E2 (PGE2) receptor, chemokines receptor, adenosine receptor, and so on. These receptors participate actively in the pathological and physiological process of cells by mediating signal transduction across membranes in response to extracellular stimuli, including light, proteins, peptides, small molecules, hormones, protons and ions [2, 3]. Rheumatoid arthritis (RA) is an autoimmune disease characterized by pain, swelling, and destruction of synovial joints, and resulting in functional disability [4]. Abnormal inflammatory and immune response result in excessive secretion of inflammatory cytokines, growth factors, and matrix metalloproteinases (MMPs), leading to synovitis and joints degradation [5]. This article focuses on major advances on GPCRs signaling pathway implicating in the inflammatory and immune response of RA.
Gα subunits were imbalanced in RA
GPCRs are functionally combined with heterotrimeric G proteins, which composed of Gα, Gβ, and Gγ subunits. Gα binds to guanosine diphosphate (GDP) in inactive state, which promotes Gα to combine with the dimers of Gβ and Gγ. GDP is released when ligand binds to GPCR. GPCR promotes the replacement of GDP with guanosine triphosphate (GTP), which results in the dissociation of Gβ and Gγ from the heterotrimeric complex. GTP-Gα complex initiate signal cascades to downstream effectors [6]. Gα returns to inactive state with the hydrolysis of GTP, allowing the recombination of Gα with Gβγ, and signal is terminated [7]. Gα includes Gαs, Gαi, Gαq, and Gα12/13 protein families [8].
Synoviocytes of RA expressed Gαs-coupled receptors, Gαs-coupled receptor agonists increased TNF-alpha level under hypoxia [9]. Gαi protein-coupled receptors regulate phosphoinositide 3-kinase (PI3K) dependent survival signaling in B lymphoblastoid lines of RA patients [10]. The expressions of Gαq mRNA and Gαq protein were significantly decreased in the peripheral blood lymphocytes of RA patients. Gαq controlled the apoptosis and survival of peripheral blood lymphocytes through mediating the activity of myeloid cell leukemia-1 (Mcl-1) and caspase-3 [11]. The expressions of Gα were imbalanced in the animal models of RA. In collagen-induced arthritis (CIA) rats, the expressions of Gαi and Gαs in fibroblast like synoviocytes (FLS) were abnormal, Gαs mRNA and Gαs proteins were decreased, but Gαi mRNA and Gαi proteins were increased [12]. And compared with normal rats, cyclic adenosine 3′, 5′-monophosphate (cAMP) level and protein kinase A (PKA) activity were reduced because of high level of Gαi [13]. Gαq and Gβγ invole in survival signaling in B lymphocytes of CIA rats by promoting the generation of inositol trisphosphate (IP3) and the activation of PI3 K [7, 14].
GPCRs signal transduction pathways implicate in the inflammatory and immune response of RA
Signal transduction pathways mediated by GPCRs regulate the functions of almost cells and have been widely implicated in human disease [15]. GPCRs are activated by the binding of ligands and mediate classical adenylyl cyclase (AC)–cAMP–PKA signaling pathway [2]. GPCRs–AC–cAMP–PKA signaling pathway transmits signaling from extracellular to intracellular, resulting in a series of physiological and pathological response [16]. β2-ARs, PGE2 receptors, chemokines receptors, adenosine receptors belong to GPCRs, and different GPCRs mediated different GPCRs signaling pathways.
β2 adrenergic receptors (β2-ARs) couple signal transduction pathways are associated with abnormal function of lymphocytes in RA
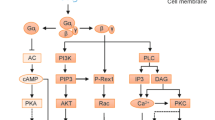
The first GPCR of human to be found was β2-AR, and the crystal structure was first described by Cherezov et al. [2]. As an important link between sympathetic nervous system and immune system, β2-AR is expressed on skeletal and cardiac muscle cells and peripheral blood lymphocytes, including T cells, B cells, and NK cells. The interactions of β2-ARs and Gαs formed the foundation of the ternary complex model of GPCR activation. The expression of β2-AR on target cells is dynamically regulated by ligand availability [17, 18]. β2-ARs bind to ligands and mediate β2-ARs–AC–cAMP transmembrane signal, resulting in trafficking a series of physiological and pathological efforts [19] (Fig. 1).

Schematic diagram of GPCR-AC-cAMP signaling pathway. β2-ARs-AC-cAMP signaling pathway: adrenaline binds to β2-ARs, the Gαs subunit is activated and dissociates from heterotrimeric G proteins. And the activated Gαs initiates downstream signaling cascade cAMP-PKA-CREB by the activation of AC. PGE2-EP2/4-AC-cAMP and PGE2-EP1-PLC signaling pathways: PGE2 binds to EP2/4, Gαs is activated and initiates downstream signaling cascade cAMP-PKA-CREB by the activation of AC. PGE2 binds to EP1, Gαq is activated and initiates the downstream signaling of PLC–IP3–Ca2+–NFAT. Chemokine–chemokine receptors signaling pathway: chemokine binds to its receptor, activated Gα bind to GTP, and free Gβγ initiate signal cascades of PLC–IP3–Ca2+–NFAT and PI3K–AKT–mTOR to downstream effectors. Activated Gα initiates downstream signaling cascade of GEF–RhoA–ROCK-SRF. Adenosine-A3ARs signaling pathways: adenosine binds to A3ARs, Gαi and Gαq subunits are activated, and initiates downstream signaling cascade of PLC–IP3–Ca2+–NFAT. In parallel, Gβγ initiates downstream signaling cascade of PI3K–PKB/Akt-mTOR. GPCR G protein coupled receptor, AC adenylate cyclase, cAMP cyclic adenosine monophosphate, PKA protein kinase A, MAPK mitogen-activated protein kinase, CREB cAMP response element binding protein, PLC phospholipase C, IP3 inositol triphosphate, NFAT nuclear factor of activated T-cells, PIP2 phosphatidylinositol-4,5-bisphosphate, GEF guanine nucleotide exchange factor, RhoA Ras homolog gene family A, ROCK RhoA kinase, SRF serum response factor, PI3K phosphoinositide 3-kinase, PKB/Akt protein kinase B
β2-ARs have been shown to have a role in time-dependent, immuno-modulating effect and are expressed on innate and adaptive immune cells of humans and rodents [20]. Peripheral blood mononuclear cells (PBMCs), specifically monocytes, B cells and CD8+ T cells, from RA patients express lower levels of β2-ARs compared with healthy subjects [21]. In patients with RA, β2-AR on synovial fluid lymphocytes was significantly decreased compared to that on peripheral blood lymphocytes [22]. RA PBMCs are less responsive to norepinephrine (NE) treatment compared with healthy subjects: upon NE administration in vitro [21, 23]. The patients with RA augment cAMP production and suppression of TNF-alpha production by β2-adrenergic agonist (terbutaline) stimulation [24].
β2-AR phosphorylated by PKA (pβ2-ARPKA) is reduced in splenocytes and draining lymph node (DLN) cells of adjuvant arthritis (AA) rats. pβ2-ARPKA impairs Gαs coupling, and enhances Gαi coupling [17]. The studies in our lab showed that the expression of β2-AR in mesenteric lymph node (MLN) lymphocytes was reduced apparently in CIA and AA rats [25, 26]. The cAMP level in synoviocytes and MLNLs of CIA rats was lower than that in normal rats [12, 25].
Prostaglandin E2 (PGE2) receptor signal transduction pathways play roles in synovitis and joint destruction for RA
PGE2 was a potent stimulator of PKA and an inducer of cytokines and proteinase. Biological activities of PGE2 are exerted through four different E-prostanoid (EP) receptors, namely EP1, EP2, EP3, and EP4. These four receptors have seven transmembrane domains and belong to the GPCRs superfamily. Different EPs bind to different G proteins and mediate different downstream signal pathways [27, 28]. PGE2 binds to EP2/4, Gαs is activated and initiates downstream signaling cascade cAMP-PKA-cAMP response element binding protein (CREB) by the activation of AC. PGE2 binds to EP1, Gαq is activated and initiates the downstream signaling of phospholipase C (PLC)-IP3-Ca2+-nuclear factor of activated T-cells (NFAT) (Fig. 1).The four EP receptors are expressed in diverse tissues and play essential roles in variety of pharmacological and biochemical processes [28].
PGE2 contributes to the pathogenesis of RA, and nonsteroidal anti-inflammatory drugs, inhibitors of the synthesis of PGE2, continue to be used in the treatment of RA [29]. PGE2 is important for IL-17 production induced via IL-23 and IL-1β in T cell- antigen presenting cell (APC) co-culture system, and PGE2 enhanced Th17 cell function and differentiation through cAMP and EP2/EP4 receptor signaling [30]. PGE2 is also produced in response to proinflammatory cytokines. PGE2 binding to EP4 stimulates osteoclastogenesis through enhancing RANKL expression. At the same time, PGE2 suppresses osteoclastogenesis by inhibiting macrophage colony-stimulating factor (M-CSF) expression in FLS [31]. PGE2 produced in rheumatoid synovium negatively regulates aberrant synovial overgrowth and the development of osteoclast activity via EP4. EP4-specific agonists significantly inhibited the activities of synovial tissue-derived inflammatory cells in a dose-dependent manner [32]. The small GTPase Rap1 is implicated in a variety of cellar functions. PGE2 activates Rap1 via EP2/EP4 receptors and cAMP-signaling in rheumatoid synovial fibroblasts [33]. PGE2 promotes TNF-alpha-induced the expression of IL-6 mRNA in synovial fibroblasts of RA patients [34]. PGE2 level in FLS of AA rats was elevated significantly [35]. PGE2 could stimulate bone resorption and osteophyte formation in CIA rats and contribute to joint damage by upregulating MMPs, and PGE2 level is associated with the edema and erosion of cartilage and juxta articular bone in RA [13, 27, 36]. EP4 receptor-deficient mice showed decreased incidence and severity of disease. The joint histopathology of EP4 receptor-deficient mice was alleviated in bone destruction, proteoglycan loss, and type II collagen breakdown compared with EP4 receptor -sufficient mice [29]. EP2 and/or EP4 might inhibit the biosynthesis of TNF-alpha and IL-6. The expression of EP2 and EP4 in the synovial tissue of AA rats decreased significantly [27], which suggests that PGE2 contributes to RA progression via binding to the EP2 and EP4 receptor. The modification of EPs signaling may be a new therapeutic strategy for RA.
Chemokines receptors signal transduction pathways are involved in angiogenesis underlying the pathogenesis of RA
Chemokine receptors are GPCR family members and the classification of chemokine receptors is based on the number and arrangement of conserved cysteine residues in the N-terminus of chemokines [8]. In humans, chemokines are classified as CXC (α-chemokines), CC (β-chemokines), XC (δ-chemokines, often referred to as C subfamily), and CX3C (γ-chemokines) [37]. Chemokine binds to its receptor, and initiates downstream signaling cascade of guanine nucleotide exchange factor (GEF)–Ras homolog gene family A (RhoA)–RhoA activates RhoA kinase (ROCK)–serum response factor (SRF), which regulates a variety of cellular responses, such as cytoskeletal rearrangement and cell proliferation [38, 39] (Fig. 1). Chemokine receptors also activate Rho GTPases, resulting in the reorganization of actin cytoskeleton, and cell survival signaling by Gβγ-mediated activation of PI3K-protein kinase B (Akt)-mTOR signaling pathway. Free Gβγ also initiate signal cascades of PLC–IP3–Ca2+–NFAT to downstream effectors [7, 40]. Chemokine receptors signaling pathway could promote Ca2+ stores via PLC and mitogen-activated protein kinase (MAPK) activation [7, 8].
Chemokines and chemokine receptors participate in cellular migration, survival, angiogenesis and leukocyte recruitment of RA and other autoimmune diseases [41]. CCL3, CCL4, CCL5, CCL20, CX3CL1, CXCL13, CXCL9, CXCL10 and CXCL16 are expressed at high levels in inflamed synovium of RA, and CXCL16 acts as a potent chemoattractant for T cells in RA [42, 43]. CCL2 produced by synovial fibroblasts is a pivotal chemokine for the recruitment of monocytes [42]. CCR2 is expressed on regulatory T cells [44, 45]. CCR2 is directly involved in the infiltration of neutrophils in RA. High expression of CCR2 was also demonstrated in neutrophils from early patients with RA and antigen-induced arthritis (AIA) [46]. In CIA model mice, blockade of CCR2 during the initiation phase of disease improved the clinical symptoms. In vitro CCR2 blockade interfered with collagen-specific activation and T cells proliferation [45]. Gene polymorphisms of CCR5 are a genetic risk factor for radiographic severity denoted by modified Sharp score in joint erosion of RA patients [47].
The expression of CCR7 was enhanced in RA synovial tissue lining and endothelial cells compared to normal synovial tissue. CCL21 induces human microvascular endothelial cell (HMVEC) migration in the RA joints and activates PI3 K, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) pathways in HMVECs. Suppression of PI3 K decreases CCL21-induced HMVEC chemotaxis and tube formation. It suggests that CCL21-induced HMVEC chemotaxis and tube formation are mediated through the PI3K/Akt 1 pathway [48]. CCL28 and CCR10 expression is elevated in RA synovial tissue compared with normal synovial tissue. Proinflammatory factors, namely LPS, TNF-α, IL-1β, IL-17 and IL-6, provoke CCL28 production in RA monocytes and endothelial cells [49].
The constitutive chemokine CXCL12 and its receptor CXCR4 are significant in T lymphocyte accumulation. CXCR4 is involved in CXCL12-dependent infiltration of lymphocytes into synovium of RA. Additionally, CXCR1, the receptor for CXCL8, was persistently expressed on neutrophils [43]. CXCR5 is highly expressed in B cells, CD4+ and CD8+ T cells and involved in synovial lymphoid neogenesis underlying arthritis [50]. CXCR6 mediates CXCL16-induced synovitis [37]. T lymphocytes in synovium may express higher CXCR2, CXCR3 and CXCR6 than that in circulating T lymphocytes [51, 52]. CCR4, CCR6 have been implicated in lymphocytes infiltration into joint of RA. XCR1 is expressed on lymphocytes, macrophages and fibroblasts in RA, while CX3CR1 has been detected on macrophages and dendritic cells [37].
CXCL10/CXCR3 mediate FLS invasion in animal model of RA and RA patients [53]. CXCL10 concentrations were increased in synovial fluid of RA patients. CXCL10 augmented nuclear factor kappa-B ligand (RANKL) expression in Jurkat/Hut 78 T cell or CD4+ T cell and was regulated by CXCR3. The results suggested that CXCR3 mediates CXCL10-induced RANKL expression [54].
The anti-inflammatory effect of adenosine/adenosine receptor signal transduction pathways in RA
Adenosine is considered a major regulator of local tissue function, especially when energy supply fails to meet cellular energy demand, and involves the activation of four G protein-coupled adenosine receptors (ARs): A1, A2A, A2B, and A3 [55]. A2AARs couples to Gαs proteins, stimulating cAMP accumulation. cAMP regulates gene expression via PKA and CREB protein [56, 57]. A2AARs signaling increases cAMP levels in immune cells and is crucial in down-regulating pro-inflammatory cytokine and protection from tissue damage [58]. A2AAR agonist suppresses both the basal and LPS-induced release of TNF-alpha and IL-1β in RA patients, while causing an increase in the release of both basal and LPS-induced IL-6 [59]. A2AARs inhibit osteoclast differentiation and regulate bone turnover via PKA-dependent inhibition of nuclear transcription factor-kappa B (NF-κB) nuclear translocation, suggesting a mechanism by which adenosine could target bone destruction in RA [60]. A2AARs agonist CGS 21680 was able to increase IL-10 production in lymphocytes of RA patients. A2AARs up-regulation was gradually decreased with the treatment time [61]. A3ARs couple to classic or G protein-dependent second messenger pathways through activation of both Gαi and Gαq, and initiate downstream signaling cascade of PLC–IP3–Ca2+–NFAT [55]. PLC activity results in the release of Ca2+ from intracellular stores in different cellular models [62–67]. In parallel, Gβγ initiates downstream signaling cascade of PI3K–PKB/Akt-mTOR [56, 68] (Fig. 1).
In early RA patients and MTX-treated RA patients, the up-regulation of A2AAR and A3AR was associated with high levels of TNF-alpha and NF-κB activation. A high density and altered functionality of A2AAR and A3AR was found in early RA patients [69]. A1AR and A2BAR were not implicated in the inflammatory process, whereas stimulation of A2AAR and A3AR was closely associated with a down-regulation of the inflammatory status in human synoviocytes [70].
The A3ARs protein was highly expressed in the synovia, PBMC and DLN tissues of RA patients and AA rats [71–75]. AA rats responded to IB-MECA (highly selective A3AR agonist) treatment with a decrease in clinical and pathological score. IB-MECA de-regulated A3ARs expression and PI3K-NF-κB signaling pathway [73], inhibited the production of TNF-alpha and macrophage inflammatory protein-1alpha (MIP-1α) in vitro, and prevented the development of CIA and AA [76, 77]. A2AAR and A3AR may represent a potential target and biologic markers in RA and other autoimmune diseases [78].
Regulatory proteins for GPCRs signaling pathway play a significant role in inflammatory and immune response for RA
Concomitant with the activation of GPCRs, GPCRs signaling are also attenuate via desensitization, and/or internalization. The desensitization process of GPCRs occurs via receptor phosphorylation by G protein coupled receptor kinases (GRKs) and subsequent binding of β-arrestins. β-arrestins bind to phosphorylated receptors and facilitate receptor internalization [79] (Fig. 2). GRKs and β-arrestins are involved in the regulation of G protein coupled signaling pathway, and play a significant role in inflammatory and immune response processes.

Regulation of GRKs and β-arrestins in GPCR-AC-cAMP signaling pathway. The desensitization process of GPCRs occurs via receptor phosphorylation by GRKs and subsequent binding of β-arrestins. β-arrestins bind to phosphorylated receptors and facilitate receptor internalization and desensitization, resulting in signaling terminal
GRKs expressions are abnormal in RA
GRKs play a crucial role in GPCR internalization, desensitization, dephosphorylation, and recycling. Changes in GRKs expression affect GPCRs signaling [80]. GRKs are grouped into seven subtypes, GRK1-GRK7. Based on sequence similarity and gene structure analysis, vertebrate GRKs are further divided into three subcategories, including visual or rhodopsin–kinases subfamily (comprising of GRK1 and GRK7), β-AR kinases subfamily (comprising of GRK2 and GRK3), and the GRK4 subfamily (comprising of GRK4, GRK5, and GRK6) [81].
In lymphocytes of RA, a significant decrease in GRK activity was detected that was mirrored by a decrease in GRK-2 and GRK-6 expression, whereas GRK-5 protein levels were unchanged. Proinflammatory cytokines induce a declining in GRK-2 in leukocytes of healthy donors [24]. The expression of membrane and plasmatic GRK2 in MLNLs of CIA and AA rats was decreased [25, 26]. GRKs regulate β-ARs functions through β2-ARs phosphorylation at different serines, and pβ2-ARGRK was increased in AA [17]. Lombardi demonstrated that down-regulation of GRK2, GRK3, and GRK6 in splenocytes and MLN cells were founded in AA rats [82]. These findings indicate that the control of GRKs expressions and function might help to affect inflammatory and immune response processes.
The expressions of β-arrestins are increased in animal model of RA
Mammals have four arrestin subtypes, which have over 50% amino acid conservation and similar structures in basal state. Arrestin-1 (also known as visual or rod arrestin) and arrestin-4 (cone arrestin) are predominantly expressed in photoreceptors, whereas arrestin-2 and -3 (also known as β-arrestin-1 and -2) are present in virtually every cell with high expression [80, 83]. β-arrestins are cytosolic proteins and play important roles in the process of homologous desensitization and internalization of GPCRs. Arrestins bind to agonist-activated GPCRs at the plasma membrane, when agonist-activated GPCRs were phosphorylated by GRKs on serine and threonine residues located in the third intracellular loop or carboxyl-terminal tail. The complex of arrestins–GRKs–phosphorylated GPCRs results in the termination of GPCRs signaling, namely desensitization of GPCRs. Formation of stable receptor-beta-arrestin complexes that persist inside cell impedes receptor resensitization. Aberrant formation of these complexes may play a role in GPCR-based diseases [84].β-arrestins also regulate other cellular signaling pathways by serving as multifunctional scaffold/adapter proteins. β2-adaptin and clathrin are recruited to the β-arrestin complex at plasma membrane and initiate inward endocytosis. The endocytotic vesicle with seven transmembrane receptors was targeted for lysosomal degradation, and receptors can be recycled back to the plasma membrane [85, 86]. β-arrestins can also initiate the recruitment and activation of numerous kinases, including c-Src family kinases and MAPKs [26].
β-arrestins are critical regulators of inflammatory response, and the expression of β-arrestins is differentially regulated in immune cells and tissues in response to specific inflammatory stimuli [87]. β-arrestin 1 was increased in splenocytes of AA rats [82]. The expression of the β-arrestin 1, 2 increased in MLN lymphocytes of CIA and AA rats, and mRNA levels of β-arrestin 1 and 2 are increased in FLS of CIA mice [25, 88, 89]. During inflammatory process (d14, d28 after immunization), it was found that a profound up-regulation of β-arrestins in synoviocytes from CIA rats, and returned to baseline levels in remission phase (d42 after immunization) [88]. β-arrestin 1 plays a critical role in the pathogenesis of CIA and Th17 cell differentiation. β-arrestin1 promoted signal transducer and activator of transcription 3 (STAT3) activation through scaffolding the interaction of Janus kinase 1 and STAT3 [90]. β-arrestin 1 promotes TNF-alpha and IL-6 production in FLS of murine models of RA [89].
The expression of β-arrestin 2 increased significantly in human FLS stimulated by IL-1β. β-arrestin 2 expression showed a positive correlation with the FLS proliferation [91]. The interaction of phosphodiesterase-4 (PDE4) with β-arrestin at β-adrenoceptor site can induce receptor switching from Gαs-to-Gαi signaling with subsequent activation of ERK1/2 in RA synovial cells [9].
Conclusions
In sum, GPCRs are involved in inflammatory and immune response of RA and mediate different external stimuli and intracellular signaling cascades. β2-AR, PGE2 receptor, chemokines receptor, and adenosine receptor play important roles in RA. GRKs and β-arrestins are essential proteins in the regulation of GPCRs signaling pathways, and play a significant role in inflammatory and immune response processes in RA. It is very important that to understanding the mechanism of GPCRs signal transduction in inflammatory and immune response for the discoveries of potential diagnostic biomarkers and therapeutic targets. And it also offers a profound foundation and new insight into the development of new RA drugs.
References
Latek D, Modzelewska A, Trzaskowski B, et al. G protein-coupled receptors-recent advances. Acta Biochim Pol. 2012;59:515–29.
Cherezov V, Rosenbaum DM, Hanson MA, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–65.
Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013; 53:531–56.
Toquet S, Nguyen Y, Sabbagh A, et al. Severe apoptotic enteropathy caused by methotrexate treatment for rheumatoid arthritis. Joint Bone Spine. 2015.
Wei Y, Sun X, Hua M, et al. Inhibitory effect of a novel antirheumatic drug T-614 on the IL-6-induced RANKL/OPG, IL-17, and MMP-3 expression in synovial fibroblasts from rheumatoid arthritis patients. Biomed Res Int. 2015; 214683.
Cabrera-Vera TM, Vanhauwe J, Thomas TO, et al. Insights into G Protein structure, function, and regulation. Endocr Rev. 2003;24:765–81.
Billard MJ, Gall BJ, Richards KL, et al. G protein signaling modulator-3: a leukocyte regulator of inflammation in health and disease. Am J Clin Exp Immunol. 2014;3:97–106.
de Munnik Sabrina M, Smit Martine J, Leurs Rob, et al. Modulation of cellular signaling by herpesvirus-encoded G protein-coupled receptors. Front Pharmacol. 2015;6:1–27.
Jenei-Lanzl Z, Zwingenberg J, Lowin T, et al. Proinflammatory receptor switch from Gαs to Gαi signaling by β-arrestin-mediated PDE4 recruitment in mixed RA synovial cells. Brain Behav Immun. 2015;50:266–74.
Tan SY, Xiao L, Pi X, et al. Aberrant Gi protein coupled receptor-mediated cell survival signaling in rheumatoid arthritis B cell lines. Front Biosci. 2007;12:1651–60.
Wang Y, Li Y, He Y, et al. Expression of G protein αq subunit is decreased in lymphocytes from patients with rheumatoid arthritis and is correlated with disease activity. Scand J Immunol. 2012;75:203–9.
Chen Q, Wei W. Effects and mechanisms of glucosides of chaenomeles speciosa on collagen-induced arthritis in rats. Int Immunopharmacol. 2003;3:593–608.
Zhang LL, Wei W, Wang NP, et al. Paeoniflorin suppresses inflammatory mediator production and regulates G protein-coupled signaling in fibroblast -like synoviocytes of collagen induced arthritic rats. Inflamm Res. 2008;57:388–95.
Liu D, Li P, Song S, et al. Pro-apoptotic effect of epigallo-catechin-3-gallate on B lymphocytes through regulating BAFF/PI3K/Akt/mTOR signaling in rats with collagen-induced arthritis. Eur J Pharmacol. 2012;690:214–25.
Zhang P, Mende U. Regulators of G-protein signaling in the heart and their potential as therapeutic targets. Circ Res. 2011;109:320–33.
Serebryany E, Zhu GA, Yan EC, et al. Artificial membrane-like environments for in vitro studies of purified G-protein coupled receptors. Biochim Biophys Acta. 2012;1818:225–33.
Lorton D, Bellinger DL, Schaller JA. Altered sympathetic-to-immune cell signaling via β2-adrenergic receptors in adjuvant arthritis. Clin Dev Immunol. 2013;2013:764395.
Xu B, Arlehag L, Dahlquist SB, et al. β2-Adrenergic receptor gene single-nucleotide polymorphisms are associated with rheumatoid arthritis in northern Swede. Scand J Rheumatol. 2004;33:395–8.
Wahle M, Krause A, Ulrichs T, et al. Disease activity related catecholamine response of lymphocytes from patients with rheumatoid arthritis. Ann NY Acad Sci. 1999;876:287–96.
Elenkov IJ, Wilder RL, Chrousos GP, et al. The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638.
Baerwald C, Graefe C, Muhl C, et al. Beta 2-adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatic diseases. Eur J Clin Invest 1992; 1(Suppl 1):42–6.
Baerwald CG, Laufenberg M, Specht T, et al. Impaired sympathetic influence on the immune response in patients with rheumatoid arthritis due to lymphocyte subset-specific modulation of beta 2-adrenergic receptors. Br J Rheumatol. 1997;36:1262–9.
Wahle M, Neumann RP, Moritz F, et al. Beta2-adrenergic receptors mediate the differential effects of catecholamines on cytokine production of PBMC. J Interferon Cytokine Res. 2005;25:384–94.
Lombardi MS, Kavelaars A, Schedlowski M, et al. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999;13:715–25.
Zhao W, Tong T, Wang L, Li PP, et al. Chicken type II collagen induced immune tolerance of mesenteric lymph node lymphocytes by enhancing beta 2-adrenergic receptor desensitization in rats with collagen-induced arthritis. Int Immunopharmacol. 2011;11:12–8.
Wu H, Wei W, Song L, et al. Paeoniflorin induced immune tolerance of mesenteric lymph node lymphocytes via enhancing beta 2-adrenergic receptor desensitization in rats with adjuvant arthritis. Int Immunopharmacol. 2007;7:662–73.
Xu HM, Wei W, Jia XY, et al. Effects and mechanisms of total glucosides of paeony on adjuvant arthritis in rats. J Ethnopharmacol. 2007;109:442–8.
Chang Y, Wei W, Zhang L, et al. Effects and mechanisms of total glucosides of paeony on synoviocytes activities in rat collagen-induced arthritis. J Ethnopharmacol. 2009;121:43–8.
McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110:651–8.
Boniface K, Bak-Jensen KS, Li Y, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–48.
Akaogi J, Nozaki T, Satoh M, et al. Role of PGE2 and EP receptors in the pathogenesis of rheumatoid arthritis and as a novel therapeutic strategy. Endocr Metab Immune Disord Drug Targets. 2006;6:383–94.
Shibata-Nozaki T, Ito H, Mitomi H, et al. Endogenous prostaglandin E2 in hibits aberrant overgrowth of rheumatoid synovial tissue and thedevelopment of osteoclast activity through EP4 receptor. Arthritis Rheum. 2011;63:2595–605.
Kojima F, Kapoor M, Kawai S, et al. Prostaglandin E2 activates Rap1 via P2/EP4 receptors and cAMP-signaling in rheumatoid synovial fibroblasts: involvement of Epac1 and PKA. Prostaglandins Other Lipid Mediat. 2009;89:26–33.
Kunisch E, Jansen A, Kojima F, et al. Prostaglandin E2 differentially modulates proinflammatory/prodestructive effects of TNF-α on synovial fibroblasts via specific E prostanoid receptors/cAMP. J Immunol. 2009;183:1328–36.
Dai M, Wei W, Shen YX, et al. Glucosides of Chaenomeles speciosa remit rat adjuvant arthritis by inhibiting synoviocyte activities. Acta Pharmacol Sin. 2003;24:1161–6.
Zheng YQ, Wei W, Zhu L, et al. Effects and mechanisms of Paeoniflorin, a bioactive glucoside from paeony root, on adjuvant arthritis in rats. Inflamm Res. 2007;56:182–8.
Szekanecz Z, Vegvari A. Chemokines and chemokine receptors in arthritis. Front Biosci (Schol. Ed.). 2010; 2:153–167.
Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nat Immunol. 2008;9:953–9.
Cotton M, Claing A. G protein-coupled receptors stimulation and the control of cell migration. Cell Signal. 2009;21:1045–53.
Thelen M. Dancing to the tune of chemokines. Nat Immunol. 2001;2:129–34.
Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12:5–13.
Chen X, Oppenheim JJ, Howard OM. Chemokines and chemokine receptors as novel therapeutic targets in rheumatoid arthritis (RA): inhibitory effects of traditional chinese medicinal components. Cell Mol Immunol. 2004;1:336–42.
Filer A, Raza K, Salmon M, et al. The role of chemokines in leucocyte–stromal interactions in rheumatoid arthritis. Front Biosci. 2008;13:2674–85.
Katschke KJ Jr, Rottman JB, Ruth JH, Qin S, et al. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001;44:1022–32.
Bruhl H, Cihak J, Schneider MA, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172:890–8.
Talbot J, Bianchini FJ, Nascimento DC, et al. CCR2 expression in neutrophils plays a critical role in their migration into the joints in rheumatoid arthritis. Arthritis Rheumatol. 2015;67:1751–9.
Han SW, Sa KH, Kim SI, et al. CCR5 gene polymorphism is a genetic risk factor for radiographic severity of rheumatoid arthritis. Tissue Antigens. 2012;80:416–23.
Pickens SR, Chamberlain ND, Volin MV, et al. Role of the CCL21 and CCR7 pathways in rheumatoid arthritis angiogenesis. Arthritis Rheum. 2012;64:2471–81.
Chen Z, Kim SJ, Essani AB, et al. Characterising the expression and function of CCL28 and its corresponding receptor, CCR10, in RA pathogenesis. Ann Rheum Dis. 2015;74:1898–906.
Schmutz C, Hulme A, Burman A, et al. Chemokine receptors in the rheumatoid synovium: upregulation of CXCR5. Arthritis Res Ther. 2005;7:R217–29.
Patel DD, Zachariah JP, Whichard LP, et al. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol. 2001;98:39–45.
Godessart N, Kunkel SL. Chemokines in autoimmune disease. Curr Opin Immunol. 2001;13:670–5.
Laragione T, Brenner M, Sherry B, et al. CXCL10 and its receptor CXCR3 regulate synovial fibroblast invasion in rheumatoid arthritis. Arthritis Rheum. 2011;63:3274–83.
Lee EY, Seo M, Juhnn YS, et al. Potential role and mechanism of IFN-gamma inducible protein-10 on receptor activator of nuclear factorkappa-B ligand (RANKL) expression in rheumatoid arthritis. Arthritis Res Ther.
Borea PA, Varani K, Vincenzi F, et al. The A3 adenosine receptor: history and perspectives. Pharmacol Rev. 2015;67:74–102.
Fredholm BB, IJzerman AP, Jacobson KA, et al. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–52.
Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–64.
Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–20.
Forrest CM, Harman G, McMillan RB, et al. Modulation of cytokine release by purine receptors in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2005;23:89–92.
Mediero A, Perez-Aso M, Cronstein BN. Activation of adenosine A(2A) receptor reduces osteoclast formation via PKA- and ERK1/2-mediated suppression of NF-κB nuclear translocation. Br J Pharmacol. 2013;169:1372–88.
Vincenzi F, Padovan M, Targa M, et al. A(2A) adenosine receptors are differentially modulated by pharmacological treatments in rheumatoid arthritis patients and their stimulation ameliorates adjuvant-induced arthritis in rats. PLoS One. 2013;8:e54195.
Zheng J, Wang R, Zambraski E, et al. Protective roles of adenosine A1, A2A, and A3 receptors in skeletal muscle ischemia and reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293:H3685–91.
Fossetta J, Jackson J, Deno G, et al. Pharmacological analysis of calcium responses mediated by the human A3 adenosine receptor in monocyte-derived dendritic cells and recombinant cells. Mol Pharmacol. 2003;63:342–50.
Kim TH, Kim YK, Woo JS. The adenosine A3 receptor agonist Cl-IBMECA induces cell death through Ca2+/ROS-dependent down regulation of ERK and Akt in A172 human glioma cells. Neurochem Res. 2012;37:2667–77.
Shneyvays V, Leshem D, Zinman T, et al. Role of adenosine A1 and A3 receptors in regulation of cardiomyocyte homeostasis after mitochondrial respiratory chain injury. Am J Physiol Heart Circ Physiol. 2005;288:H2792–801.
Shneyvays V, Zinman T, Shainberg A. Analysis of calcium responses mediated by the A3 adenosine receptor in cultured newborn rat cardiac myocytes. Cell Calcium. 2004;36:387–96.
Panther E, IdzkoM Herouy Y, et al. Expression and function of adenosine receptors in human dendritic cells. FASEB J. 2001;15:1963–70.
Schutle G, Fredholm BB. Signaling pathway from the human adenosine A(3) receptor expressed in Chinese hamster ovary cells to the extracellular signal-regulated kinase 1/2. Mol Pharmacol. 2002;62:1137–46.
Varani K, Massara A, Vincenzi F, et al. Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum. 2009;60:2880–91.
Varani K, Vincenzi F, Tosi A, et al. Expression and functional role of adenosine receptors in regulating inflammatory responses in human synoviocytes. Br J Pharmacol. 2010;160:101–15.
Haskó G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol. 2013;4:85. doi:10.3389/fimmu.2013.00085.
Ochaion A, Bar-Yehuda S, Cohen S, et al. The anti-inflammatory target A(3) adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell Immunol. 2009;258:115–22.
Fishman P, Bar-Yehuda S, Madi L, et al. The PI3K-NF-κB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res Ther. 2006;8:1–9.
Rath-Wolfson L, Bar-Yehuda S, Madi L, et al. IB-MECA, an A3 adenosine receptor agonist prevents bone resorption in rats with adjuvant induced arthritis. Clin Exp Rheumatol. 2006;24:400–6.
Bar-Yehuda S, Silverman MH, Kerns WD, et al. The anti-inflammatory effect of A3 adenosine receptor agonists: a novel targeted therapy for rheumatoid arthritis. Expert Opin Investig Drugs. 2007;16:1601–13.
Szabo C, Scott GS, Virag L, et al. Suppression of macrophage inflammatory protein (MIP)-1alpha production and collagen-induced arthritis by adenosine receptor agonists. Br J Pharmacol. 1998;125:379–87.
Baharav E, Bar-Yehuda S, Madi L, et al. The anti-inflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–76.
Vincenzi F, Targa M, Corciulo C, et al. Pulsed electromagnetic fields increased the anti-inflammatory effect of A2A and A3 adenosine receptors in human T/C-28a2 chondrocytes and hFOB 1.19 osteoblasts. PLoS One. 2013;8:e65561.
Jala VR, Haribabu B. Real time imaging of leukotriene B4 mediated cell migration and BLT1 Interactions with β-arrestin. J Vis Exp. 2010; 23.
Shukla AK, Manglik A, Kruse AC, et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–41.
Sato PY, Chuprun JK, Schwartz M, et al. The evolving impact of G protein coupled receptor kinases in cardiac health and disease. Physiol Rev. 2015;95:377–404.
Lombardi MS. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J Immunol. 2001;166:1635–40.
Palczewski K. Structure and functions of arrestins. Protein Sci. 1994; 1355–1361.
Oakley RH, Laporte SA, Holt JA, et al. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor -beta- arrestin complexs after receptor endocytosis. JBiol Chem. 2001;276:19452–60.
Goodman OB Jr, Krupnick JG, Santini F, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–50.
Lefkowitz RJ, Whalen EJ. β-Arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–8.
Fan H. β-Arrestins 1 and 2 are critical regulators of inflammation. Innate Immun. 2013;20:451–60.
Wang QT, Zhang LL, Wu HX, et al. The expression change of β-arrestins in fibroblast-like synoviocytes from rats with collagen induced arthritis and the effect of total glucosides of paeony. J Ethnopharmacol. 2011;133:511–6.
Li P, Cook JA, Gilkeson GS, et al. Increased expression of beta arrestin 1 and 2 in murine models of rheumatoid arthritis: isoform specific regulation of inflammation. Mol Immunol. 2011;49:64–74.
Li J, Wei B, Guo A, et al. Deficiency of β-arrestin1 ameliorates collagen-induced arthritis with impaired TH17 cell differentiation. Proc Natl Acad Sci USA. 2013;110:7395–400.
Wu HX, Chen JY, Wang QT, et al. Expression and function of β-arrestin 2 stimulated by IL-1β in human fibroblast-like synoviocytes and the effect of paeoniflorin. Int Immunopharmacol. 2012;12:701–6.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81173075, 31100640, 81330081 and 81473223), China Postdoctoral Science Foundation (No. 2013M540509), Anhui Province Postdoctoral Science Foundation (No. 2016B134).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible Editor: John Di Battista.
Rights and permissions
About this article

Cite this article
Shu, J., Zhang, F., Zhang, L. et al. G protein coupled receptors signaling pathways implicate in inflammatory and immune response of rheumatoid arthritis. Inflamm. Res. 66, 379–387 (2017). https://doi.org/10.1007/s00011-016-1011-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-016-1011-5