
Abstract
Cell signaling is considered a part of a network for communication that regulates basic cellular activities. The ability of cells to communicate correctly to the surrounding environment has an important role in development, tissue repair, and immunity as well as normal tissue homeostasis. Dysregulated activation and crosstalk between many intracellular signaling pathways are implicated in the pathogenesis of rheumatoid arthritis (RA), such as the Janus Kinase/signal transducers and activators of transcription (JAK/STAT), Toll-like receptor/nuclear factor kappa B (TLR/NF-κB), phosphatidylinositide-3Kinase/protein kinase B/mammalian target of rapamycin (PI-3K/AKT/mTOR), the stress activated protein kinase/mitogen-activated protein kinase (SAPK/MAPK), and spleen tyrosine kinase (SYK) pathways. Other interrelated pathways that can be targeted to halt the inflammatory status in the disease are purinergic 2X7 receptor (P2X7R)/nucleotide binding oligomerization domain-like receptor family pyrin domain containing 3 or inflammasome (NLRP-3)/NF-κB and Notch pathways. In this review, we will show the orchestrated modulation in the pathogenesis of RA via the crossregulation between dysregulated signaling pathways which can mediate a sustained loop of activation for these signaling pathways as well as aggrevate the inflammatory condition. Also, this review will highlight many targets that can be useful in the development of more effective therapeutic options.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic, progressive, inflammatory disease associated with articular, extra-articular, and systemic effects affecting about 1% of the population with a female:male ratio of 3:1. It is characterized by polyarthritis with often progressive joint damage, disability, immunologic abnormalities, systemic inflammation, increased co-morbidity, and premature mortality (Branimir and Miroslav 2014). This complex immune disease is characterized by synovial and vascular proliferation with cellular infiltration and pannus formation which damage articular cartilage and adjacent bone. Extra-articular manifestations of the disease including fever, weight loss, anemia, rheumatoid nodules, pleuritis, pericarditis, and vasculitis can affect the hypothalamic pituitary adrenal axis, resulting in fatigue and depression (Choy 2012). It was found that various dysfunctional crosstalking intracellular signaling pathways played an important role in the pathogenesis of RA, such as Janus Kinase/signal transducers and activators of transcription (JAK/STAT), Toll-like receptor/nuclear factor kappa B (TLR/NF-κB), phosphatidylinositide-3Kinase/protein kinase B/mammalian target of rapamycin (PI-3K/AKT/mTOR), the stress activated protein kinase/mitogen-activated protein kinase (SAPK/MAPK), spleen tyrosine kinase (SYK), and Notch pathways as well as purinergic 2X7 receptor (P2X7R) and nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 or inflammasome (NLRP-3).
JAK/STAT pathway
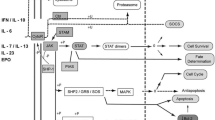
There are four members of the JAK tyrosine kinase family; JAK-1, JAK-2, JAK-3, and tyrosine kinase-2 (Tyk-2). They are associated with different kinds of cytokine receptors in identical or different pairs. Upon receptor activation, JAK kinases phosphorylate tyrosine cytokine receptor subunits. Then STATs, which are a family of transcription factors consists of seven proteins: STAT-1, STAT-2, STAT-3, STAT-4, STAT-5a, STAT-5b, and STAT-6, are phosphorylated by the receptor-associated JAKs. Phosphorylated STATs dimerise with each other, and translocate to the cell nucleus to act as transcription factors to regulate gene transcription (Shuai and Liu 2003; Lundquist et al. 2014), Fig. 1.
Under normal conditions, this pathway plays an obvious role in cell growth, survival, and differentiation and in regulating protein expression involved in angiogenesis, extracellular matrix composition, inflammation, apoptosis, and cellular signaling (Al-Rasheed et al. 2016), Fig. 1.
In RA, many immune cells infiltrate the synovium in response to pro-inflammatory cytokines and chemokines, leading to inflammation and tissue destruction (O’Sullivan et al. 2007; Pesu et al. 2008). Binding of a cytokine to its receptor activates the receptor-associated JAKs leading to activation, dimerization, and translocation of STAT into the nucleus causing an increase in the transcription of more and more inflammatory cytokines, which continues the loop of inflammatory signaling. Inhibiting this loop of inflammatory signaling may be mediated by inhibiting the JAK pathways (McInnes and Liew 2005).
Tofacitinib was the first jakinib to be approved for the treatment of moderate-to-severe conditions of RA patients who had an inadequate response to methotrexate. Other JAK inhibitors with variable selectivity, such as barcitinib, upadacitinib, decernotinib, filgotinib, and peficitinib, possess multiple adverse effects. The suppressors of cytokine signaling (SOCS) are family of intracellular proteins that are considered to be negative regulators of JAK/STAT pathway. The expression of SOCS is altered in RA leading to deregulation of JAK/STAT pathway. Thus, future therapies for RA is to discover novel targets that will improve the expression of SOCS to restore normal JAK/STAT signaling (Singh and Singh 2020). Also, assessment of the anti-arthritic efficacy of STAT inhibitors used in cancer studies may be considered as new target for treatment of RA (Bose et al. 2020).
TLR/NF-κB pathway
The NF-κB is a transcription factor that plays an important role in immunity and inflammation. The NF-κB is formed of an inactive complex of three subunits inhibitor kappa B (IκB), P65, and P50. Activation of NF-κB can be mediated via inhibitor kappa B kinase (IKK) which is capable of phosphorylating the IκB, leading to its dissociation and translocation of P50 and P65 into the nucleus and increase in the transcription of pro-inflammatory genes and anti-apoptotic proteins (Joosten et al. 2016; Mori et al. 2011), Fig. 1.
Additionally, NF-κB promotes T helper 17 (Th17) differentiation, by both direct and indirect ways as well as activates B cells, increasing the production of auto-antibodies which emphasis its important role in the pathogenesis of RA (Liu et al. 2017). One study illustrated that NF-κB has a role in pain sensation, as it was found that increased NF-κB activity in astrocytes decreases transcription of catechol-o-methyltransferase (COMT), causing an increase in the level of catecholamines that are responsible for pain sensation (Hartung et al. 2015).
The TLRs are transmembrane receptors, which play a key role in both innate and adaptive immune responses. Under normal conditions, the inflammatory response mediated by the innate immune system acts together with the adaptive immune response, to eliminate the invading pathogen, resulting in mitigation of the inflammatory response (Huang and Pope 2009).
In contrast, in RA, all aspects of the innate and adaptive immune response persist, so the inflammation becomes chronic, with persistence production of pro-inflammatory mediators. This occurs due to activation of TLRs by various ligands, subsequently leading to activation of the NF-κB enhancing the expression of pro-inflammatory cytokines causing further activation of several TLRs (Huang and Pope 2009, Ospelt et al. 2008).
The TLR-4 is important in the pathogenesis of RA, as shown by enhanced expression in the synovial tissue of patients with RA and the protection from experimental models of RA in TLR-4 knockout mice (Aksoy et al. 2012; Duffy and O’Reilly 2016).
Activation of TLR-4 pathway induces the myeloid differentiation primary response protein (MyD88) and the tumor necrosis factor receptor-associated factor 6 (TRAF6), and finally leads to translocation of NF-κB into the cell nucleus and the up-regulation of the expression of many pro-inflammatory cytokines, such as [interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), etc.] (Joosten et al. 2016), Fig. 1.
The TLR signaling can induce inflammation via an indirect mechanism through the activation of the protein kinase inositol-requiring enzyme 1α (IRE1α). Active IRE1α splices X box-binding protein (XBP1). Interestingly, XBP1 is considered as a transcription factor involved in the further expression of pro-inflammatory cytokines, such as TNF-α and IL-6 (Duffy and O’Reilly 2016).
Antagonizing TLR-4, inhibition of NF-κB activation or targeting other signaling molecules in the pathway may be considered as new targets for mitigating RA inflammatory condition. As highlighted previously that the combination therapy of sitagliptin and tofacitinib offers anti-inflammatory actions through inhibiting both JAK/STAT and TLR-4/NF-κB pathways in adjuvant-induced model of arthritis in rats (Ibrahim et al. 2020).
PI-3K/AKT/mTOR pathway
Firstly, the PI-3K was illustrated to be an oncogene, which is classified into three classes according to their structure and function. However, later on, this pathway was found to be implicated in the pathogenesis of many diseases such as psoriatic arthritis and RA (Malemud 2013). In RA, when this pathway is activated by tumor necrosis-related apoptosis inducing ligand (TRAIL) and IL-15, PI-3K phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) to produce phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which subsequently activates protein kinase B (PKB or AKT) and mTOR through a cascade of signal transduction, and this can result in promoting aggressive immune cell infiltration, synoviocyte proliferation, production of IL-17 by CD4+T cells, and resistance of RA synoviocytes to Fas (CD95)-induced apoptosis (Malemud 2013), Fig. 1. In one study, it has been illustrated that microRNA-125 (miR-125) can directly inhibit the expression of Poly [ADP-ribose] polymerase 2 (PARP2), which subsequently inhibits the activity of PI-3K/AKT/mTOR signaling pathway in experimental-induced RA model in rats (Liu et al. 2019). Another study stated that inhibition of this pathway mitigated the production of pro-inflammatory cytokines in macrophages and monocytes (Xie et al. 2014).
SAPK/MAPK pathway
Mitogen-activated protein kinase (MAPK) cascades are signaling components activated by a variety of cytokines and TLRs. This pathway starts with MAPK kinase kinases (also known as MKKKs or MAP3Ks) at the top, and MAPK kinases (also known as MKKs, MEKs, or MAP2Ks) are found in the middle, and ends with MAPKs at the bottom, leading to activation of many transcription factors such as NF-κB, and this may lead to matrix metalloproteinase (MMP) gene expression, cell survival, and resistance to apoptosis (Martínez-Limón et al. 2020), Fig. 1.
The MAPK include extracellular signal-regulated kinases (ERK) and the p38 kinases (p38). Growth factors and certain cytokines can activate ERK 1 and 2, finally leading to activation of transcription factors such as Elk-1 and c-Myc. Toll-like receptors, oxidative stress, and inflammatory cytokines can activate the p38 kinases. Since the p38 pathways are activated mostly by environmental and genotoxic stresses, they are also known as stress activated protein kinases or SAPKs (Fang et al. 2020).
It was thought that p38 is a useful target in the treatment of RA. The use of p38 α inhibitors, such as VX-702 and pamapimod, in animal models of RA gave promising results; however, in RA clinical trials, they were not effective and their use was associated with side effect (Malemud 2013). In RA therapy, targeting other components of SAPK/MAPK signaling may be assessed.
SYK pathway
The SYK is a non-receptor tyrosine kinase that is up-regulated in the synovium of RA patients. Upon its activation, it can activate downstream MAPKs and PI-3K leading to mast cell activation, macrophage phagocytosis, cytokine secretion (IL-6), and fibroblast-like synoviocytes matrix metalloproteinase (FLS MMP) synthesis, among other functions. Also, SYK has a role in B-cell receptor signaling and antigen presentation, as well as osteoclast differentiation (Okamoto and Kobayashi 2011), Fig. 1.
The SYK was considered to be a promising target in the treatment of RA as fostamatinib, a selective inhibitor of SYK, mitigated the severity of arthritis in murine model of collagen-induced arthritis (CIA), as well as in clinical arthritis trials. However, in phase 2 clinical trials, adverse events including diarrhea, hypertension, neutropenia, and infections were reported (Deng et al. 2016).
Notch signaling pathway
The Notch signaling pathway regulates various cellular processes including proliferation and differentiation during development as well as adult tissue homeostasis. Having inflammatory disorders, like RA, the binding of Notch’s ligands leads to the proteolytic cleavage of Notch1 by γ-secretase and the generation of Notch1 intracellular domain (NICD1), which is able to translocate to the nucleus and regulate transcription of its downstream genes that accelerate production of pro-inflammatory cytokines leading to modulation of disease activity (Shang et al. 2016; Choi et al. 2018), Fig. 1. It has been shown that Notch signaling stimulates synoviocytes and accelerates their production of pro-inflammatory cytokines in CIA or Notch antisense transgenic mice by immunization with chicken type II collagen (CII) (Park et al. 2015). Another study showed that Notch1 targeting via small interfering RNA delivery nanoparticles (siRNA-NPs) can suppress Notch1 pathway, resulting in retardation of the progression of inflammation, bone erosion, and cartilage damage in CIA mice (Kim et al. 2015). In addition, the pharmacological and genetic inhibition of Notch1 signaling suppresses the progression of inflammatory arthritis through modulating the Treg cells in CIA and collagen antibody-induced arthritis (CAIA) (Choi et al. 2018).
Furthermore, Notch signaling is implicated in M1/M2 imbalance in RA. It has been illustrated previously that the activation of Notch signaling in BM-derived macrophages in RA synovial tissue enhanced M1 polarization. While the use of Notch inhibitor (thapsigargin) attenuated inflammation by switching M1 to M2 macrophages. We could conclude that targeting Notch signaling may represent a therapeutic target for RA by controlling the balance of M1 and M2 macrophage polarization (Sun et al. 2017).
The purinergic 2X7 receptor
The P2XRs are seven subtypes of mammalian ATP-gated receptors. They are cation-selective channels, which have equal permeability to Na+ and K+ and with significant Ca2+ permeability. The binding of three molecules of ATP to a P2XR induces the opening of a channel selective for small cations and finally leading to the release of inflammatory cytokines (Burnstock and Knight 2018), Fig. 1. It had been previously illustrated that P2X7R signaling is involved in regulating differentiation of Th17 cells in type II CIA in mice (Fan et al. 2016). Another study stated that P2X7R is highly expressed in human FLS and the P2X7R antagonist A804598 decreased the production of ROS (Liu et al. 2020).
NLRP-3 or inflammasome
The NLRP-3 is a cytoplasmic multiprotein complex, which is considered to be a milestone in innate immunity. Several endogenous or exogenous stimuli can lead to NLRP3 activation leading to maturation and secretion of IL-1β and IL-18 (Choulaki et al. 2015; Ruscitti et al. 2015).
The activation of the NLRP-3 or inflammasome occurs in two steps: priming and activation. In the priming step, activation of TLRs, P2X7R, and cytokine receptors result in subsequent activation of the NF-κB signaling pathway and up-regulation of proteins forming NLRP-3, pro-IL-1β, and pro-IL-18. During the activation step, NLRP-3 assembly occurs via the NACHT domain, followed by recruitment of ASC, resulting in recruitment and activation of pro-caspase-1. The assembly of these three proteins forms the NLRP-3 inflammasome. Activated caspase-1 activates both pro-IL-1β and pro-IL-18 and induces inflammation (Li et al. 2020), Fig. 1.
Recent studies have demonstrated that NLRP-3 is highly expressed in both synovia from RA patients and CIA in mice, whereas NLRP-3 inhibition ameliorated arthritic symptoms effectively and other inflammatory insults (Guo et al. 2018; Fusco et al. 2020).

Multiple signaling pathways involved in pathogenesis of rheumatoid arthritis. JAK/STAT Janus kinase/signal transducer and activator of transcription, TLR-4 Toll-like receptor-4, MYD88 myeloid differentiation primary response protein, IRAK-1/4 Interleukin-1 receptor-associated kinase 1/4, TRAF-6 tumor necrosis factor receptor-associated factor 6, TAK-1 transforming growth factor beta-activated kinase 1, IκB inhibitor kappa B, NF-κB nuclear factor kappa B, SYK spleen tyrosine kinase, RAF rapidly accelerated fibrosarcoma, MAPK mitogen-activated protein kinase, PI-3K phosphatidylinositide-3Kinase, AKT protein kinase B, mTOR mammalian target of rapamycin, NICD notch intracellular domain, NLRP-3 nucleotide binding oligomerization domain-like receptor family pyrin domain containing 3 or inflammasome
Illustration for the role of various Crosstalks between different signaling in extending the functions of individual pathways, resulting in a more complex regulatory network and aggrevating the pathogenesis of RA
Crosstalk between NF-κB and JAK/STAT pathways
A crosstalk between NF-κB and STAT-3 was observed in many inflammatory conditions and cancers. NF-κB family members interact with STAT-3 resulting in either synergy or repression of transcription of regulated genes, and many mechanisms for this have been suggested (Ambili and Janam 2017). First, the dephosphorylated STAT-3 may bind to NF-κB/IκB complex enhancing NF-κB activation. Second, STAT-3 may acetylate p65 in the nucleus increasing the time of its retention inside the nucleus and ensure constitutive NF-κB activation. Finally, NF-κB activation results in increased production of IL-6 which, in turn, will activate STAT-3. Simultaneous activation may be beneficial to mediate effective inflammatory response in many diseases (Ambili and Janam 2017), Fig. 2.
Crosstalk between SYK, TLR/NF-κB and MAPK pathways
Spleen tyrosine kinase (SYK) activation leads to coupling to the caspase recruitment domain family member 9/mucosa-associated lymphoid tissue lymphoma translocation protein 1/B-cell lymphoma 10 (CARD9 /Malt1/Bcl10) complex (Lowell 2011). This directly leads to activation of IKK, which in turn phosphorylated IκB and activates NF-κB pathways. Another mechanism has been suggested which is the ability of the aforementioned complex to activate the MAPK pathway or also facilitate the ability of the TLRs to activate the downstream MAPK pathway (Lowell 2011), Fig. 2.

Crosstalk between NF-κB and JAK/STAT as well as between SYK,TLR/NF-κB and MAPK pathways. JAK/STAT Janus kinase/signal transducer and activator of transcription, TLR-4 Toll-like receptor-4, MYD88 myeloid differentiation primary response protein, TRAF-6 tumor necrosis factor receptor-associated factor 6, IκB inhibitor kappa B, NF-κB nuclear factor kappa B, SYK spleen tyrosine kinase, MAPK mitogen-activated protein kinase, CARD9 caspase recruitment domain family member 9, MALT1 mucosa-associated lymphoid tissue lymphoma translocation protein 1, BCL10 B-cell lymphoma 10
Crosstalk between JAK/STAT, MAPK and PI-3K /AKT/mTOR pathways
Dimerization of IL-6-type cytokine receptors does not only activate the JAK/STAT signaling pathway, but also induce the MAPK cascade through recruitment of the SHP2 (SH2-domain-containing tyrosine phosphatase) to tyrosine-phosphorylated gp130 and its phosphorylation in a JAK1-dependent manner. The phosphorylated SHP2 binds to the Grb2–SOS (growth factor receptor-bound protein/Son of Sevenless) complex leads to the activation of the Ras–Raf–MAPK pathway (Heinrich et al. 2003), Fig. 3.

Crosstalk between JAK/STAT, MAPK, and PI-3K/AKT/mTOR pathways. JAK/STAT Janus kinase/signal transducer and activator of transcription, PI-3K phosphatidylinositide-3Kinase, AKT protein kinase B, mTOR mammalian target of rapamycin, SHP2 SH2-domain-containing tyrosine phosphatase, Grb2–SOS growth factor receptor-bound protein/Son of Sevenless, RAF rapidly accelerated fibrosarcoma, MAPK mitogen-activated protein kinase
Also, activation of JAKs leads to phosphorylation of the tyrosine residues of the attached receptors. Proteins containing SH2 domains (such as STATs and PI-3K) are able to bind the phosphotyrosines and carry out their function. Therefore, activation of the JAK–STAT pathway can also activate PI-3K/AKT/mTOR signaling (Rawlings et al. 2004).
Crosstalk between TLR/NF-κB, SYK/NLRP-3 and Notch Pathways
The TLRs activation can cause activation of Notch signaling directly and/or indirectly, which, in turn, induces expression of pro-inflammatory cytokines such as TNF-α and IL-6. The increase in the level of the cytokines can induce the expression of Notch ligands and this can sustain the inflammatory response in RA (Keewan and Naser 2020), Fig. 4. Additionally, the injection of complete Freund’s adjuvant caused an excessive genetic expression of SYK and subsequently its downstream molecule NLRP-3 in the untreated arthritic animals. Previous studies showed that stimulation of NLRP-3 inflammasome is one of quintessential sparks that upregulate cellular synthesis of several pro-inflammatory cytokines that in turn prompt propagation of inflammation in arthritis (Fioravanti et al. 2019). Also, it has been found in other study that Notch 3 activation, which enhances the activation of p38 MAPK, which facilitates phosphorylation of p65 on Ser276, which in turn increases the transcriptional activity of NF-κB (López-López et al. 2020).

Crosstalk between TLR/NF-κB, SYK/NLRP-3, and Notch Pathways. TLR toll-like receptor, SYK spleen tyrosine kinase, JUN Jun-N-terminal kinases, IκB inhibitor kappa B, NF-κB nuclear factor kappa B, NICD notch intracellular domain, NLRP-3 nucleotide binding oligomerization domain-like receptor family pyrin domain containing 3 or inflammasome
Crosstalk between P2X7R/NLRP-3/NF-κB pathways
The P2X7R belongs to one of the P2X ion channel receptors family members that have been suggested that they can mediate pain and inflammation in arthritis (Zeng et al. 2019). In addition, other studies illustrated that activation of P2X7R by elevated levels of ATP mediated by pannexin channels can lead to activation of NF-κB and subsequent release of IL-1β, IL-6, TNF-α, MMP13, and PGE2 (Zeng et al. 2019).
The P2X7R activation can decrease the intracellular K+ concentration which permits NLRP-3 or inflammasome assembly and release of IL‐1β and IL‐18 (Li et al. 2020), Fig. 5.

Crosstalk between P2X7R/NLRP-3/NF-κB pathways. P2X7R purinergic 2X7 receptor, NLRP-3 nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 or inflammasome, ATP adenosine triphosphate, NF-κB nuclear factor kappa
Conclusion
The biological agents are widely used in the management of RA patients, but they are of high cost and can induce many adverse events. Therefore, new strategies to reach better efficacy and safety are needed. In this review, we illustrated more details about the pathogenesis of RA. It was concluded that the pro-inflammatory cytokines can be considered the mysterio of orchestrated crosstalking intracellular signaling pathways involved in the pathogenesis and the state of sustained inflammation in RA. Also, this review highlights new targets suggested for the development of new therapeutic approaches.
References
Aksoy E, Taboubi S, Torres D, Delbauve S, Hachani A, Whitehead MA et al (2012) The p110delta isoform of the kinase PI-3K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat Immunol 13:1045–1054. https://doi.org/10.1038/ni.2426
Al-Rasheed NM, Al-Rasheed NM, Hasan IH, Al-Amin MA, Al-Ajmi HN, Mahmoud AM (2016) Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats. Drug Des Dev Ther 10:2095–2107. https://doi.org/10.2147/DDDT.S109287
Ambili R, Janam P (2017) A critique on nuclear factor-kappa B and signal transducer and activator of transcription 3: the key transcription factors in periodontal pathogenesis. J Indian Soc Periodontol 21:350–356. https://doi.org/10.4103/jisp.jisp_301_16
Bose S, Banerjee S, Mondal A, Chakraborty U, Pumarol J, Croley C (2020) Targeting the JAK/STAT signaling pathway using phytocompounds for cancer prevention and therapy. Cells 9:1451. https://doi.org/10.3390/cells9061451
Branimir A, Miroslav M (2014) Pathogenesis of rheumatoid arthritis. Reumatizam 61:19–23
Burnstock G, Knight GE (2018) The potential of P2X7 receptors as a therapeutic target, including inflammation and tumor progression. Purinergic Signal 14:1–18. https://doi.org/10.1007/s11302-017-9593-0
Choi BY, Choi Y, Park JS, Kang LJ, Baek SH, Park JS et al (2018) Inhibition of Notch1 induces population and suppressive activity of regulatory T cell in inflammatory arthritis. Theranostics 8:231–243. https://doi.org/10.7150/thno.26093
Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K et al (2015) Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther 17:257. https://doi.org/10.1186/s13075-015-0775-2
Choy E (2012) Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 51:v3–v11. https://doi.org/10.1093/rheumatology/kes113
Deng GM, Kyttaris VC, Tsokos GC (2016) Targeting SYK in autoimmune rheumatic diseases. Front Immunol 7:78. https://doi.org/10.3389/fimmu.2016.00078
Duffy L, O’Reilly SC (2016) Toll-like receptors in the pathogenesis of autoimmune diseases: recent and emerging translational developments. Immunotargets Ther 5:69–80. https://doi.org/10.2147/ITT.S89795
Fan ZD, Zhang YY, Guo YH, Huang N, Ma HH, Huang H et al (2016) Involvement of P2X7 receptor signaling on regulating the differentiation of Th17 cells and type II collagen-induced arthritis in mice. Sci Rep 6:35804. https://doi.org/10.1038/srep35804
Fang Q, Zhou C, Nandakumar KS (2020) Molecular and cellular pathways contributing to joint damage in rheumatoid arthritis. Mediators Inflamm 2020:3830212. https://doi.org/10.1155/2020/3830212
Fioravanti A, Tenti S, McAllister M, Chemaly M, Eakin A, McLaughlin J et al (2019) Exploring the involvement of NLRP3 and IL-1β in osteoarthritis of the hand: results from a pilot study. Mediators Inflamm 2019:1–11. https://doi.org/10.1155/2019/2363460
Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R (2020) Focus on the role of NLRP3 inflammasome in diseases. Int J Mol Sci 21:4223. https://doi.org/10.3390/ijms21124223
Guo H, Sun L, Ling S, Xu JW (2018) Levistilide A ameliorates NLRP3 EXPRESSION INVOLVING the SYK-p38/JNK pathway and peripheral obliterans in rats. Mediators Inflamm 12:1–11. https://doi.org/10.1155/2018/7304096
Hartung JE, Eskew O, Wong T, Tchivileva IE, Oladosu FA, O’Buckley SC et al (2015) Nuclear factor-kappa B regulates pain and COMT expression in a rodent model of inflammation. Brain Behav Immun 50:196–202. https://doi.org/10.1016/j.bbi.2015.07.014
Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F (2003) Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J 374:1–20. https://doi.org/10.1042/BJ20030407
Huang QQ, Pope RM (2009) The role of toll-like receptors in rheumatoid arthritis. Curr Rheumatol Rep 11:357–364. https://doi.org/10.1007/s11926-009-0051-z
Ibrahim S, Salama M, Selima E, Refaat R (2020) Sitagliptin and tofacitinib ameliorate adjuvant induced arthritis via modulating the cross talk between JAK/STAT and TLR-4/NF-κB signaling pathways. Life Sci 2020:118261. https://doi.org/10.1016/j.lfs.2020.118261
Joosten LA, Abdollahi-Roodsaz S, Dinarello CA, O’Neill L, Netea MG (2016) Toll-like receptors and chronic inflammation in rheumatic diseases: new developments. Nat Rev Rheumatol 12:344–357. https://doi.org/10.1038/nrrheum.2016.61
Keewan E, Naser SA (2020) The role of notch signaling in macrophages during inflammation and infection: implication in rheumatoid arthritis? Cells 9:111. https://doi.org/10.3390/cells9010111
Kim MJ, Park JS, Lee SJ, Jang J, Park JS, Back SH et al (2015) Notch1 targeting siRNA delivery nanoparticles for rheumatoid arthritis therapy. J Control Release 216:140–148. https://doi.org/10.1016/j.jconrel.2015.08.025
Li Z, Guo J, Bi L (2020) Role of the NLRP3 inflammasome in autoimmune diseases. Biomed Pharmacother 130:110542. https://doi.org/10.1016/j.biopha.2020.110542
Liu T, Zhang L, Joo D, Sun SC (2017) NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2:17023. https://doi.org/10.1038/sigtrans.2017.23
Liu K, Zhang Y, Liu L, Yuan Q (2019) miR-125 regulates PI-3K /Akt/mTOR signaling pathway in rheumatoid arthritis rats via PARP2. Biosci Rep. 39:BSR20180890. https://doi.org/10.1042/BSR20180890
Liu Y, Wu Y, Gu S, Yin Q, Li H, Wang J et al (2020) The P2X7 receptor (P2X7R)-specific antagonist A804598 inhibits inflammatory reaction in human fibroblast-like synoviocytes. Am J Transl Res 12:45–53
López-López S, Monsalve EM, Romero de Ávila MJ et al (2020) NOTCH3 signaling is essential for NF-κB activation in TLR-activated macrophages. Sci Rep 10:14839. https://doi.org/10.1038/s41598-020-71810-4
Lowell CA (2011) Src-family and SYK kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Perspect Biol 3:a002352. https://doi.org/10.1101/cshperspect.a002352
Lundquist LM, Cole SW, Sikes ML (2014) Efficacy and safety of tofacitinib for treatment of rheumatoid arthritis. World J Orthop 5:504–511. https://doi.org/10.5312/wjo.v5.i4.504
Malemud CJ (2013) Intracellular signaling pathways in rheumatoid arthritis. J Clin Cell Immunol 4:160. https://doi.org/10.4172/2155-9899.1000160
Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E (2020) The p38 pathway: from biology to cancer therapy. Int J Mol Sci 21:1913. https://doi.org/10.3390/ijms21061913
McInnes IB, Liew FY (2005) Cytokine networks–towards new therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol 1:31–39. https://doi.org/10.1038/ncprheum0020
Mori T, Miyamoto T, Yoshida H, Asakawa M, Kawasumi M, Kobayashi T et al (2011) IL-1beta and TNFalpha-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol 23:701–712. https://doi.org/10.1093/intimm/dxr077
Okamoto H, Kobayashi A (2011) Tyrosine kinases in rheumatoid arthritis. J Inflamm (Lond Engl) 8:21. https://doi.org/10.1186/1476-9255-8-21
Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP et al (2008) Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum 58:3684–3692. https://doi.org/10.1002/art.24140
O’Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC (2007) Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol 44:2497–2506. https://doi.org/10.1016/j.molimm.2006.11.025
Park JS, Kim SH, Kim K, Jin CH, Choi KY, Jang J et al (2015) Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann Rheum Dis 74:267–274. https://doi.org/10.1136/annrheumdis-2013-203467
Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O’Shea JJ (2008) Therapeutic targeting of Janus kinases. Immunol Rev 223:132–142. https://doi.org/10.1111/j.1600-065X.2008.00644.x
Rawlings JS, Rosler KM, Harrison DA (2004) The JAK/STAT signaling pathway. J Cell Sci 117:1281–1283. https://doi.org/10.1242/jcs.00963
Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F et al (2015) Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1β via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3 (NLRP3)-inflammasome activation: a possible implication for therapeutic decision in these patients. Clin Exp Immunol 182:35–44. https://doi.org/10.1111/cei.12667
Shang Y, Smith S, Hu X (2016) Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell 7:159–174. https://doi.org/10.1007/s13238-016-0250-0
Shuai K, Liu B (2003) Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 3:900–911. https://doi.org/10.1038/nri1226
Singh S, Singh S (2020) JAK-STAT inhibitors: Immersing therapeutic approach for management of rheumatoid arthritis. Int Immunopharmacol 86:106731. https://doi.org/10.1016/j.intimp.2020.10673
Sun W, Zhang H, Wang H, Chiu YG, Wang M, Ritchlin CT et al (2017) Targeting notch-activated M1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J Bone Miner Res 32:1469–1480. https://doi.org/10.1002/jbmr.3117
Xie S, Chen M, Yan B, He X, Chen X, Li D (2014) Identification of a role for the PI-3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE 9:e94496. https://doi.org/10.1371/journal.pone.0094496
Zeng D, Yao P, Zhao H (2019) P2X7, a critical regulator and potential target for bone and joint diseases. J Cell Physiol 234:2095–2103
Funding
No funding provide.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ibrahim, S.S.A., Huttunen, K.M. Orchestrated modulation of rheumatoid arthritis via crosstalking intracellular signaling pathways. Inflammopharmacol 29, 965–974 (2021). https://doi.org/10.1007/s10787-021-00800-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-021-00800-3