
Abstract
Platelet activation and aggregation are key components in the cascade of events causing thrombosis following plaque rupture. Antiplatelet therapy is essential in the treatment of patients with acute coronary syndromes (ACS) and for those requiring percutaneous coronary intervention (PCI). Aspirin (acetylsalicylic acid) is a well established antiplatelet therapy and is mandated for secondary prevention of cardiovascular events following ACS. In patients with ACS, the addition of clopidogrel to aspirin is more effective than aspirin alone. For patients undergoing PCI, dual antiplatelet therapy with aspirin and clopidogrel is warranted. Aspirin should be continued indefinitely after PCI. Pretreatment of patients with clopidogrel prior to PCI lowers the incidence of cardiovascular events, yet the optimum timing of drug administration and dose are still being investigated, as is the duration of therapy following PCI. Late-stent thrombosis with drug-eluting stents has pushed the recommendation for duration of clopidogrel therapy up to 1 year and perhaps beyond, in patients without risks for bleeding. The concepts of aspirin and clopidogrel resistance are important clinical questions. No uniform definition exists for aspirin or clopidogrel resistance. Measurements of resistance are often highly variable and do not necessarily correlate with clinical resistance. Noncompliance remains the most prominent mode of resistance. Screening of selected patient populations for resistance or pharmacologic intervention of those patients termed ‘resistant’ warrants further study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
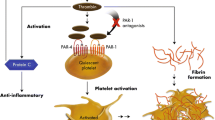
Thrombosis is the sentinel event leading to cardiovascular morbidity and mortality in acute coronary syndromes (ACS). ACS results from the complex interplay between atherosclerosis and platelet-mediated thrombosis and is influenced by multiple factors.[1] Platelet adhesion, activation, and aggregation are central to the development of a thrombus.[2] Plaque rupture exposes subendothelial von Willebrand Factor (vWF) and collagen to circulating blood and the platelets adhere to collagen and vWF via α2β1 integrin and glycoprotein (GP) VI and 1b receptors.[3,4] The activation of platelets is mediated by a number of substrates including collagen, vWF, thrombin, adenosine diphosphate (ADP), thromboxane A2 (TxA2), and epinephrine (adrenaline).[3] Platelet activation induces exocytosis of platelet granules, which propagates further platelet adhesion and activation.[4] Activated platelets increase arachidonic acid production via phospholipase A2.[4] Arachidonic acid is converted into prostaglandin (PG) G2/H2 by cyclo-oxygenase-1 (COX-1) and subsequently to TxA2 by thromboxane synthase.[5] TxA2 amplifies the thrombotic signal by promoting further platelet activation and acting as a potent vasoconstrictor.[6] Platelet aggregation results from the enhanced expression of the GP IIb/IIIa receptors, which bind adhesive proteins to form a stable thrombotic aggregate.[3] The steps and factors leading to platelet activation and aggregation are summarized in figure 1.

A graphical presentation of steps in platelet activation and aggregation and site of action of aspirin (acetylsalicylic acid) and adenosine diphosphate (ADP) antagonists. COX-1 = cyclo-oxygenase-1; GP = glycoprotein; TxA 2 = thromboxane A2; vWF = von Willebrand Factor.
The role of platelets in atherothrombosis is at the forefront of treatment of ACS.[2] Short- and long-term antiplatelet therapy are required to mitigate the deleterious effects of arterial thrombosis. Platelet activation in not just an acute event and has been shown to persist well after the onset of ACS.[7,8] Numerous heritable and environmental factors contribute further to the risk of enhanced platelet activity.[9] Worldwide, cardiovascular risk factors that contribute to atherothrombosis are undertreated.[10] Understanding the genomic influences on ACS has recently emerged as a new avenue towards individualized therapy, and the genetic variations in platelet biology may change the course of antiplatelet therapy in the future.[11] This review focuses on the importance of antiplatelet therapy in ACS and percutaneous coronary intervention (PCI).
1. Aspirin (Acetylsalicylic Acid)
Aspirin (acetylsalicylic acid) has been used for centuries as a medicinal agent and plays an essential role in the treatment of atherothrombosis. It specifically inhibits arachidonic acid metabolism by acetylation of a single serine residue (Ser529) on cyclo-oxygenase (COX), an enzyme that produces a precursor of TxA2.[12–14] COX is irreversibly inhibited by aspirin for the entire lifespan of a platelet (8.2 ± 2.2 days).[15] Inhibition of COX results in the decreased production of the potent platelet activator TxA2. Originally, aspirin was shown to decrease TxA2 levels in a dose-dependent manner as evidenced by a decrease in its stable serum metabolite thromboxane B2 (TxB2).[16] Once platelet COX is acetylated by aspirin, TxA2 cannot be synthesized until new platelets with unacetylated COX are produced by megakaryocytes. The ability to restore TxA2 production and, moreover, platelet function is directly related to platelet turnover.[17] COX exists as two isoforms; COX-1 is a constitutive form expressed in platelets, macrophages, and endothelial cells, while COX-2 is an inducible form that requires inflammatory stimuli for expression. Aspirin is primarily an irreversible COX-1 inhibitor, but can also inhibit COX-2 at very high doses.[18,19] The difference in activity of aspirin against the two COX isoforms can be inferred from differences in the their crystal structures.[20] More recently, aspirin has been shown in some studies to play a role in decreasing markers of inflammation. In a prospective, randomized, crossover study,[21] 40 patients with chronic stable angina had elevated inflammatory cytokine levels that were reduced following 6 weeks of therapy with aspirin versus placebo. Aspirin also reduces polymorphonuclear neutrophil (PMN) diapedesis in vivo.[22]
C-reactive protein (CRP) is an inflammatory marker of systemic inflammation and is a predictor of cardiovascular risk.[23] In the Physicians’ Health Study,[24] elevated baseline CRP levels were associated with a higher risk for myocardial infarction (MI) or ischemic stroke in men. When male patients were stratified into quartiles of baseline CRP levels, the relative risk reduction (RRR) for MI when randomized to aspirin versus placebo was 13.9% for the lowest quartile of baseline CRP levels and 55.7% for the highest quartile.[24] It remains controversial if aspirin has a direct effect on lowering CRP levels. Conflicting results have emerged as aspirin has been shown to both reduce and increase CRP levels.[21,25,26] In a 12-month prospective cohort study[27] of patients experiencing a non-Q-wave MI, higher maximum CRP concentrations at baseline were associated with a higher odds ratio (OR) to develop cardiovascular death or re-infarction. Patients treated with aspirin prior to the non-Q-wave MI had a reduction in cardiovascular death or nonfatal MI compared with those not treated with aspirin at 12 months (OR 0.98 vs 2.64; p = 0.04).[27] While the association between prior aspirin use and reduced rates of non-Q-wave MI is interesting, increased risk of recurrent cardiovascular events including death or MI has been shown in ACS patients receiving prior aspirin.[28] It is unknown whether aspirin has a direct role in modulating CRP levels or just mitigates inflammation in atherosclerosis. The correlation between decreasing markers of inflammation and risk reduction of cardiovascular events could represent a new paradigm in therapeutic regimens for the future.[23]
Despite the benefit of aspirin as an antiplatelet agent, bleeding is a potential risk that must be considered with each patient. The Antithrombotic Trialists’ Collaboration was a meta-analysis of 287 randomized controlled trials (RCTs) of antiplatelet therapy for the prevention of MI, stroke and death in high-risk patients.[29] Major extracranial bleeds occurred in 787 patients out of which 20% were found to be fatal.[29] The risk for a major extracranial bleed was increased 60% for patients receiving antiplatelet therapy versus controls (OR 1.6; 95% CI 1.4, 1.8).[29] Several other meta-analyses have been completed looking at aspirin and the risk for GI bleeding and intracranial hemorrhage. One meta-analysis of 28 RCTs found the risk of GI bleeding to be 2.47% in patients treated with aspirin compared with 1.42% in patients randomized to placebo (OR 1.68; 95% CI 1.51, 1.88).[30] This study also concluded that there was no increased risk of GI bleeding with lower doses of aspirin. In a review of epidemiologic studies from 1990–2001, patients who received aspirin at dosages <300 mg/day were still associated with roughly a 2-fold increase in risk for GI bleeding.[31] A later analysis of six RCTs for secondary prevention of cardiovascular or cerebrovascular events showed that with low-dose aspirin ≤325 mg/day patients were 2.5 times more likely versus placebo to experience GI bleeding (95% CI 1.4, 4.7; p = 0.001).[32] The analysis determined that one death was prevented for every 67 patients treated with aspirin, while one nonfatal GI bleed occurred per 100 patients treated.
The GI effects of aspirin are the result of a local toxic or systemic effect increasing the risk of GI bleeding. Enteric-coated aspirin was developed to reduce the toxic effects on the gastric mucosa and was shown to reduce gastric damage endoscopically in healthy subjects compared to the use of regular aspirin.[33] However, in one retrospective population study of 550 patients receiving aspirin and admitted for upper GI bleeding versus 1202 matched controls, the relative risk (RR) for upper GI bleeding was not significantly different for regular and enteric-coated aspirin.[34] Two meta-analyses of patients receiving enteric-coated or regular aspirin showed no significant differences in the risk for GI bleeding.[30,31] Several studies have determined that enteric-coated and regular aspirin were similar in their ability to inhibit platelet function as measured by various platelet function assays.[33,35–37] In one study, in 44% of patients with stable cardiovascular disease, enteric-coated aspirin was associated with persistent COX activity as measured by elevated serum TxB2.[38] However, the clinical significance of this laboratory finding remains unknown.
Aspirin is a potent antiplatelet agent with significant benefit as an antithrombotic agent, yet the use of aspirin nationally and worldwide is suboptimal. In one survey of outpatient physicians in 2003, only 32.8% of patients at high risk for cardiovascular disease were treated with aspirin.[39] The Reduction of Atherothrombosis for Continued Health (REACH) registry is an international, prospective, observational registry that evaluated 67 888 patients with risk factors for cardiovascular disease or known cardiovascular disease from 44 different countries followed over a 4-year timeperiod.[10] Data from large international registries such as REACH will assess the prevalence of aspirin use in the US and worldwide more accurately. The recent emphasis on guideline adherence has shown an increase in the appropriate use of aspirin in many registries.
2. Thienopyridines
Thienopyridines represent a second family of antiplatelet agents that inhibit platelet activation by a mechanism independent of the effect of aspirin on platelets. The most commonly used thienopyridine is clopidogrel. Ticlopidine is an alternative but used less frequently because of its adverse effect profile. Both agents are irreversible inhibitors of platelet activation lasting for the lifespan of the platelet.[40]
The active metabolites of thienopyridines bind to the ADP receptors on platelets, blocking the ability of ADP to activate platelets.[40] Specifically, the human ADP receptor, called P2, is comprised of three different subtypes, P2X1, P2Y1, and P2Y12.[41] ADP mediates the activation of platelets through the P2 receptor, where P2Y12 utilizes a G-coupled protein to inhibit the action of adenylyl cyclase and decrease cyclic adenosine monophosphate levels.[42] Antagonism of the P2Y12 receptor blocks ADP-induced platelet activation and aggregation.[41]
The first available thienopyridine that was used as an antiplatelet agent for ACS and PCI was ticlopidine.[43] This compound is now used as a second line thienopyridine because of its adverse effect profile; ticlopidine has been associated with neutropenia and thrombotic thrombocytopenic purpura (TTP). The incidence of neutropenia with ticlopidine is 0.8–2.3%. [44] Ticlopidine-induced neutropenia is reversible but there have been case reports of patients developing neutropenic fever.[44,45] In the ISAR[46] and STARS[47] trials there was no added risk of neutropenia with ticlopidine[40] (see table I for definitions of studies/trials used in this review). A more serious adverse effect of ticlopidine is TTP. In the STARS trial[47] patients undergoing coronary stenting did not show an increased risk of TTP after ticlopidine and aspirin therapy versus aspirin alone. A retrospective analysis of the EPISTENT trial[48] showed that the incidence of TTP with ticlopidine was 0.02% with a mortality rate of 20.1%. A case review of TTP in patients treated with ticlopidine reported that the incidence increases with a longer duration of therapy and was fatal in 60% of patients who did not undergo plasmapharesis.[49] Thus, a complete blood cell count every 2 weeks is recommended for any patient prior to initiating treatment with ticlopidine.

Definitions of studies/trials used in this review
3. Acute Coronary Syndrome
3.1 Aspirin
Aspirin is a powerful secondary preventive agent for ACS. The Canadian Multicenter Trial showed that aspirin initiated in the first 8 days following unstable angina reduced cardiac death and nonfatal MI by 51% compared with placebo over a mean follow-up of 18 months.[50] Subsequently, the ISIS-2 and RISC trials both found similar results for prevention of recurrent cardiac events with aspirin following ACS.[51–53] In the ISIS-2 trial, vascular mortality was reduced by 23% for patients randomized to aspirin 160 mg/day versus placebo at 5 weeks (9.4% vs 11.8%; 95% CI 15, 30; p < 0.00001).[51] The Antithrombotic Trialists’ Collaboration showed no significant differences between daily aspirin doses of 75–150, 160–325, or 500–1000 mg in preventing serious vascular events.[29] Doses of aspirin <75 mg were not statistically different compared with aspirin 75–150 mg for prevention of serious vascular events; however, only three clinical trials involving 3655 patients have so far investigated the effects of aspirin <75 mg on vascular events and thus this dose is less established.[29]
3.2 Clopidogrel versus Aspirin
In the CAPRIE trial,[54] clopidogrel 75 mg/day was compared with aspirin 325 mg/day for a mean of 1.91 years in 19 185 patients with recent MI, ischemic stroke, or symptomatic peripheral arterial disease (PAD). The trial showed an 8.7% risk reduction for clopidogrel versus aspirin for ischemic stroke, MI, or other vascular death (5.32% vs 5.83%; 95% CI 0.3, 16.5; p = 0.043).[54] Further analysis of the CAPRIE trial found that clopidogrel was an independent predictor for reducing ischemic stroke, MI, vascular death, or rehospitalization for ischemic events or bleeding.[55] The event rate and risk reduction was even more pronounced in high-risk patient populations, which included patients with previous coronary artery bypass grafting (CABG), history of previous ischemic events, involvement of multiple vascular beds, diabetes mellitus, or hypercholesterolemia.[56]
3.3 Dual Therapy with Aspirin and Thienopyridines
Dual antiplatelet therapy with aspirin and clopidogrel has been shown to synergistically inhibit platelet function in clinical ex vivo studies.[57] The CURE trial[58] studied 12 562 patients with ACS treated with clopidogrel in addition to aspirin. Patients were randomized within 24 hours of onset of ACS without ST-segment elevation to receive a loading dose of clopidogrel 300 mg followed by 75 mg/day or placebo. Patients in the clopidogrel arm showed a 20% reduction in cardiovascular death, MI, or stroke compared with placebo at 1 year (figure 2) but clopidogrel increased the risk of major bleeding versus placebo (3.7% vs 2.7%; RR 1.38; 95% CI 1.13, 1.67; p = 0.001).[58] In further analysis of the CURE trial, varying dosages of aspirin did not have any effect on lowering ischemic events, but doses of aspirin ≥200 mg were associated with a higher risk for major bleeding.[59] Thus, the addition of clopidogrel to aspirin in patients with ACS without ST-segment elevation is beneficial.

Cumulative hazard rates for the primary outcome of cardiovascular death, myocardial infarction or stroke in patients with acute coronary syndromes treated with clopidogrel versus placebo during a period of 12 months. The cumulative hazard rate for the primary outcome was significantly less (p < 0.001) in clopidogrel recipients compared with placebo recipients (reproduced from Yusuf et al.,[58] with permission).
The CURE trial did not include patients with ST-segment elevation MI (STEMI). The CLARITY-TIMI-28 trial[60] studied a loading dose of clopidogrel 300 mg followed by 75 mg/day in addition to aspirin in 3491 patients with STEMI. The 30-day combined outcome of cardiovascular death, MI, or recurrent ischemia leading to urgent revascularization was lower in the clopidogrel plus aspirin arm compared with aspirin alone arm (11.6% vs 14.1%; p = 0.03).[60] The larger COMMIT trial[61] randomized 45 582 patients with STEMI within 24 hours to receive clopidogrel 75 mg/day or placebo in addition to aspirin. All-cause mortality by hospital discharge was significantly lower in the clopidogrel compared with the aspirin alone group (7.5% vs 8.1%; p = 0.03) and did not increase the risk for fatal or intracerebral bleeds.[61] Despite the lack of a loading dose of clopidogrel in the COMMIT trial,[61] both CLARITY-TIMI-28[59] and COMMIT trials prove the benefit of combination therapy with clopidogrel and aspirin in the setting of STEMI.
The CHARISMA trial[62] analysed dual antiplatelet therapy with clopidogrel and aspirin for the primary and secondary prevention of atherothrombosis in high-risk patients. Patients were randomized to receive clopidogrel 75 mg or placebo in addition to aspirin for a mean follow-up of 2.3 years. Of the 15 603 patients, 77.9% patients had documented coronary, cerebrovascular, or symptomatic peripheral arterial disease and were termed the symptomatic group, while 21% of patients did not have any documented cardiovascular disease but rather had multiple risk factors for atherothrombosis (i.e. asymptomatic group).[62] There was no significant difference in the primary endpoint of cardiovascular death, MI, or stroke between the clopidogrel and placebo groups (6.8% vs 7.3%; RR 0.93; 95% CI 0.83, 1.05; p = 0.22).[62] Clopidogrel reduced the risk for the secondary endpoint of cardiovascular death, first occurrence of MI or stroke, hospitalization for unstable angina or transient ischemic attack, or any revascularization compared with placebo (16.7% vs 17.9%; RR 0.92; 95% CI 0.86, 0.995; p = 0.04).[62] In the symptomatic cohort of patients with clinically evident atherothrombosis, clopidogrel added to aspirin showed a significant benefit in the primary endpoint (6.9% vs 7.9% with placebo; RR 0.88; p = 0.046). This benefit was not seen in the asymptomatic cohort of patients with multiple risk factors for atherothrombotic events (6.6% for clopidogrel and 5.5% for placebo; RR 1.20; p = 0.20).[62] This analysis suggests that dual antiplatelet therapy may be beneficial for those patients with established cardiovascular disease but not for those patients at high risk for cardiovascular disease. In particular, patients with prior MI, ischemic stroke, or symptomatic PAD appeared to benefit from dual antiplatelet therapy as shown in figure 3.[63]

Kaplan-Meier curves for cardiovascular death, myocardial infarction (MI), or stroke for clopidogrel + aspirin (ASA; acetylsalicylic acid) versus placebo + aspririn in 9478 patients with prior MI, ischemic stroke, or symptomatic peripheral arterial disease. Dual antiplatelet therapy significantly reduced the primary outcome event rate of cardiovascular death, MI, or stroke in patients with a previous history of cardiovascular events compared with aspirin alone (8.8% vs 7.3%; relative risk reduction 17.1%; 95% CI 4.4, 28.1; p = 0.01) [reproduced from Bhatt et al.,[63] with permission].
4. Percutaneous Coronary Intervention
Percutaneous treatment with balloon angioplasty to dilate stenotic arteries was first mentioned in the literature in the late 1970s and has since evolved with the utilization of stent therapy.[64,65] Stent therapy in PCI has been proven to be clinically superior to percutaneous balloon angioplasty.[66–72] PCI has largely replaced CABG as treatment for ACS, except for patients with left main or three-vessel disease.[73] PCI can be associated with significant peri- and post-procedural complications that have been minimized by the use of appropriate antithrombotic therapy.[74] Prior to the use of antiplatelet therapy, early thrombosis was a common occurrence after stent placement.[75] Drug-eluting stents (DES) have become increasingly used in selected populations to decrease the rates of restenosis.[76] More recently, late-stent thrombosis with DES has been a controversial topic and thought to be associated with early discontinuation of antiplatelet therapy.[77,78]
4.1 Aspirin
Aspirin use before and after PCI is essential in the prevention of thrombosis. Early studies showed aspirin to be effective antiplatelet therapy when used with and without dipyridamole for the prevention of ischemia following angioplasty.[79–81] Pretreatment with aspirin prior to intervention reduced the angiographic evidence of thrombus before and after angioplasty.[79] In the MHEART II trial,[82] 752 patients were randomized to receive aspirin 325 mg, sulotroban (a selective TxA2 inhibitor), or placebo 1 hour prior to angioplasty and for a duration of 6 months. Death, MI, restenosis, or the need for revascularization was significantly reduced with aspirin compared with placebo (30% vs 41%; p = 0.046).[82]
The optimum dose of aspirin that needs to be administered prior to angioplasty or PCI has not been studied sufficiently. In early bare metal stent (BMS) trials, treatment regimens that included aspirin were found to reduce peri- and post-procedural complications.[83] Antiplatelet therapy with aspirin and ticlopidine reduced MI and the need for urgent revascularization compared with anticoagulant therapy including aspirin, unfractionated heparin, and phenprocoumon.[46] In early trials[47,84] in patients undergoing PCI, aspirin reduced mortality, MI, urgent revascularization, or stent thrombosis both with and without thienopyridines.
The most up to date guidelines by the American College of Cardiology (ACC)/American Heart Association (AHA)/Society of Cardiovascular Angiography and Interventions (SCAI) recommend a starting dose of aspirin 300–325 mg at least 2 hours prior to the procedure and preferably up to 24 hours prior to PCI.[85] Patients should continue treatment with aspirin for at least 1, 3, and 6 months, after PCI with BMS, sirolimus- or paclitaxel-eluting stents, respectively; treatment with low-dose aspirin should be continued indefinitely, thereafter.[85] The use of aspirin indefinitely following PCI is warranted.
4.2 Ticlopidine
Despite the risks of neutropenia and TTP, ticlopidine has been well established as an antiplatelet therapy for PCI. A trial by Hall et al.[84] randomized 226 patients, undergoing intravascular-ultrasound guided stent placement, to aspirin or aspirin plus ticlopidine and found a trend at 30 days that favored the addition of ticlopidine to aspirin in reducing death, stent thrombosis, MI, or need for revascularization (0.8% vs 3.9%; p = 0.10). The ISAR trial compared antiplatelet (aspirin and ticlopidine) and anticoagulation (unfractionated heparin, warfarin, and phenprocoumon) regimens after BMS deployment and found that the antiplatelet regimen reduced the risk of cardiac death, MI, or need for urgent revascularization (1.6% vs 6.2%; RR 0.25; 95% CI 0.06, 0.77; p = 0.01).[46] A later analysis of the ISAR trial determined at 6 months that there was no significant angiographic difference in restenosis between the two regimens.[86]
In STARS, 1653 patients undergoing PCI with BMS were randomized to one of three antithrombotic regimens, aspirin alone, aspirin and warfarin, or aspirin and ticlopidine to assess 30-day primary outcomes (death, MI, revascularization of target lesion, or angiographic evidence of thrombosis).[47] The primary endpoint occurred in 0.5% of patients receiving dual antiplatelet therapy versus 3.6% receiving aspirin alone and 2.7% receiving aspirin and warfarin (p = 0.001).[47] Hemorrhagic complications did increase from 1.8% with aspirin alone to 5.5% with aspirin and ticlopidine and 6.2% with aspirin and warfarin (p < 0.001) without any increased risk of neutropenia or thrombocytopenia.[47] Thus, dual antiplatelet therapy is beneficial compared with aspirin alone but has a higher risk for bleeding complications.
The FANTASTIC trial[87] randomized patients undergoing emergent or elective BMS implantation to ticlopidine and aspirin or oral anticoagulation and aspirin. The rate of stent occlusion, death, or MI favored antiplatelet therapy versus anticoagulation therapy, but did not reach statistical significance (5.6% vs 8.3%; p = 0.37).[87] A significantly higher risk of bleeding occurred in the anticoagulation group compared with the dual antiplatelet therapy group (21% vs 13.5%; OR 0.59; 95% CI 0.36, 0.98; p = 0.03), yet this difference was mainly due to an increase in local ecchymosis (16.5% vs 6.6%; p = 0.006) rather than any increase in blood transfusions (1.6% vs 2.6%; p = 0.45).[87]
MATTIS[88] studied the 30-day combined outcomes of cardiovascular death, MI, or urgent revascularization in 350 patients at high risk for stent thrombosis who were randomized to receive aspirin and ticlopidine or oral anticoagulation and aspirin. A trend that favored a reduction in the combined outcomes was seen with antiplatelet therapy compared with anticoagulation therapy (5.6 vs 11%; RR 1.94; 95% CI 0.93, 4.06; p = 0.07).[88] There was a significant reduction in the risk for major vascular or bleeding complications in patients treated with aspirin and ticlopidine versus oral anticoagulation and aspirin (1.7% vs 6.9%; p = 0.02).[88] Taken together, results from the Hall et al.[84] study, ISAR, STARS, FANTASTIC, and MATTIS trials show a reduction in ischemic events with dual antiplatelet therapy with aspirin and ticlopidine.
An observational study[89] in 175 consecutive patients undergoing PCI and receiving ticlopidine pretreatment found a strong correlation between reductions in periprocedural non-Q wave MI and increased duration of pre-treatment with ticlopidine. Pretreatment ≥3 days significantly reduced MI compared with treatment <3 days (OR 0.18; 95% CI 0.04, 0.78; p = 0.01).[89] The EPISTENT trial evaluated the potential benefit of ticlopidine pretreatment in stented patients randomized to abciximab or placebo and assessed outcomes through 1 year of follow-up.[90] All patients who underwent PCI received treatment with ticlopidine after the procedure and 58% of patients received ticlopidine therapy prior to PCI.[90] Among patients randomized to placebo, ticlopidine pretreatment compared with no pre-treatment was associated with a significant reduction in MI and death at 30 days, which remained present up to 1 year (11.2% vs 15.8%; p = 0.048).[90] Thus, pretreatment with thienopyridines is an important component of antiplatelet therapy for PCI.
4.3 Ticlopidine versus Clopidogrel
There has been a change in the standard antiplatelet regimen for PCI from ticlopidine to clopidogrel because of its quick onset of action when a loading dose is used, better tolerability, and once-daily dosing.[54,91] Ticlopidine 250 mg twice daily or clopidogrel 75 mg daily inhibits ADP-induced platelet aggregation by 40–60% after 3–5 days; a similar timeframe for recovery of platelet function has been noted following discontinuation of these agents.[92]
In the CLASSICS trial,[93] 1020 patients were randomized to three different 28-day therapeutic regimens following PCI: ticlopidine 250 mg twice daily; a loading dose of clopidogrel 300 mg followed by 75 mg/day; or clopidogrel 75 mg/day.[93] All regimens included aspirin 325 mg/day. Rates of cardiac death, MI, or revascularization was not significantly different between treatment groups.[93] Major or peripheral bleeding, neutropenia, thrombocytopenia, or early discontinuation secondary to noncardiac events was significantly lower in the combined clopidogrel group (4.6%) compared with the ticlopidine group (9.1%) [RR 0.50; 95% CI 0.31, 0.81; p = 0.005].[93] Thus, the results of CLASSICS trial showed a beneficial safety profile of clopidogrel compared with ticlopidine with similar efficacy; this study was not powered to examine differences in rates of stent thrombosis.
Studies have been conducted comparing the efficacies of clopidogrel and ticlopidine in PCI. A meta-analysis,[91] involving 13 955 patients from three RCTs and seven registries, compared the efficacy of ticlopidine with that of clopidogrel after PCI. Results of this analysis revealed that clopidogrel was associated with significant reductions at one month in major adverse cardiac events (OR 0.51; p = 0.001), in MI (OR 0.51; p = 0.001), and mortality (OR 0.44; p = 0.001).[91] In addition, reports of rare severe adverse affects (i.e. neutropenia and TTP) associated with ticlopidine and once-daily dosing with clopidogrel have led interventionalists to favor clopidogrel. In patients with mild allergies or intolerance to clopidogrel, which occurs rarely, ticlopidine may be substituted for clopidogrel. If ticlopidine is utilized, a 500-mg loading dose should be administered 6 hours prior to planned PCI or 24 hours if the patient is aspirin intolerant. Therapy should be continued for at least 2 weeks after the placement of a BMS.[94]
4.4 Clopidogrel
Pretreatment of patients undergoing PCI with clopidogrel is essential.[95] One of the first studies exploring the effects of clopidogrel pretreatment in patients undergoing PCI with GP IIb/IIIa inhibitors followed up 299 consecutive patients who were treated with a loading dose of clopidogrel 300 mg followed by 75 mg/day within 5 days prior to PCI or after PCI.[96] Pretreatment with clopidogrel reduced cardiovascular death, MI, or the need for urgent revascularization compared with no pretreatment (5.5% vs 14%; p = 0.03).[96] TARGET[97] randomized 4809 patients to tirofiban or abciximab prior to PCI.[98] In a post hoc analysis of TARGET, the effect of pretreatment with clopidogrel 300 mg was evaluated for the combined clinical endpoints of cardiovascular death, MI, or urgent revascularization at 30 days, 6 months, and 1 year. Patients who received clopidogrel pretreatment had improved combined clinical endpoints at 30 days and 6 months compared with no pretreatment (for 30 days, 6.6% vs 10.4%; hazard ratio [HR] 0.63; 95% CI 0.44, 0.89; p = 0.009; for 6 months, 14.6% vs 19.8%; HR 0.71; 95% CI 0.55, 0.92; p = 0.01).[97] There may have been selection bias in the group that was pretreated if this group included less urgent cases. There were no statistical differences between the two groups for bleeding risk at 30 days.[97] One-year mortality was significantly reduced with pretreatment compared with no pretreatment (1.7% vs 3.6%; HR 0.46; 95% CI 0.25, 0.85; p = 0.011).[97] These results suggest that pretreatment prior to PCI improves the short- and long-term clinical outcomes. In the TARGET trial, in an analysis of the timing of pre-treatment, patients who received pretreatment >6 hours before PCI had improved combined clinical endpoints compared with those who received pretreatment <6 hours prior to PCI at 30 days (4.9% vs 6.9%; p = 0.045).[97] This was primarily due to a reduced number of MI events (4.2% vs 6.3%; p = 0.028).[97] There was no significant differences between groups for the time intervals of 0–2 hours and 2–6 hours prior to the procedure (6.5% vs 7.7%; p = 0.198).[97]
Similar results were seen in the CREDO trial. CREDO[99] randomized 2116 patients undergoing elective PCI to placebo or a loading dose of clopidogrel 300 mg 3–24 hours prior to PCI. Thereafter all patients received clopidogrel 75 mg for 28 days. Patients in both groups received aspirin throughout the study.[99] Patients who received pre-treatment with clopidogrel 3 to <6 hours prior to PCI did not show any benefit in death, MI, or urgent revascularization compared with no pretreatment (RR −13.4%; 95% CI 29.8, −83.3; p = 0.60); patients treated with clopidogrel 6 to 24 hours prior to PCI showed a benefit in risk reduction for this endpoint (RR 28.6%; 95% CI −1.6, 62.9; p = 0.051).[99] Results from the TARGET and CREDO trials suggest that the optimum timing for pretreatment with clopidogrel 300 mg should be ≥6 hours prior to PCI. Potentially, longer intervals provide even greater benefit.[100]
Ex vivo data have suggested that clopidogrel 600 mg accelerates the inhibition of ADP stimulated platelet aggregation measured by light aggregometry as early as 2 hours after drug administration compared to less inhibition with clopidogrel 300 mg.[101] A post hoc analysis of the ISAR-REACT trial studied the effects of a pretreatment dose of clopidogrel 600 mg in 2159 patients randomized to abciximab versus placebo at various time intervals (2–3, 3–6, 6–12, and >12 hours) prior to PCI.[102] There were no significant differences between any of the time intervals for death, MI, or urgent revascularization at 30 days.[102] This data suggest that a higher dose of clopidogrel eliminates the need for a longer duration of pretreatment seen with a loading dose of clopidogrel 300 mg.
Several studies have been conducted to test the effect of various doses of clopidogrel on platelet aggregation in patient with ACS undergoing PCI. The ARMYDA-2 trial[103] randomized 255 patients undergoing PCI to clopidogrel 300 mg or 600 mg 4–8 hours prior to PCI with a 30-day follow-up period. On average, clopidogrel was administered 6 hours prior to PCI.[103] Death, MI, or target vessel revascularization was reduced with a higher loading dose compared with a lower dose (4% vs 12%; p = 0.041), primarily as a result of a 50% reduction in periprocedural MI (OR 0.48; 95% CI 0.15, 0.97; p = 0.044).[103]
The ISAR-CHOICE trial[104] randomized 60 patients with suspected or documented coronary artery disease (CAD) to 300, 600, or 900 mg loading doses of clopidogrel prior to coronary angiography. ADP-induced optical platelet aggregometry was measured before and 4 hours after the administration of clopidogrel.[104] No significant differences were found between clopidogrel 600 and 900 mg for inhibition of measured ADP-induced platelet aggregation (52.7% vs 49.7%; p = 0.59), yet increased inhibition occurred with clopidogrel 600 mg compared with clopidogrel 300 mg (52.7% vs 66.5%; p = 0.01).[104]
The ALBION[105] trial randomized 103 low to moderate risk patients with non-ST elevation MI to a loading dose of clopidogrel 300, 600, or 900 mg and measured various markers of platelet aggregation at different time intervals 24 hours after administration. Clopidogrel doses >300 mg inhibited platelet aggregation to a greater extent as early as 2 hours through 24 hours after administration, with no difference in bleeding risk.[105] Death, MI, unplanned PCI, or rehospitalization for angina at 30 ± 7 days did not differ significantly between the three loading doses of clopidogrel.[105] Taken together, ARMYDA-2, ISAR-CHOICE, and ALBION suggest a greater inhibition of platelet function at clopidogrel doses >300 mg.
The largest trial studying different loading doses of clopidogrel on platelet parameters randomized 292 consecutive high-risk non-ST elevation MI patients undergoing PCI to clopidogrel 300 mg or 600 mg at least 12 hours prior to PCI.[106] All patients received concurrent aspirin therapy and clopidogrel 75 mg/day for 1 month.[106] ADP-induced platelet aggregation prior to PCI was significantly lower in patients who received clopidogrel 600 mg compared with 300 mg (50 ± 19% vs 61 ± 16%; p < 0.0001).[106] At 1 month, cardiovascular death, acute, or subacute stent thrombosis, recurrent ACS, or stroke was significantly reduced in the higher loading dose group compared with the lower loading dose group (5% vs 12%; p = 0.035).[106] In a post hoc analysis, patients with high post-treatment platelet reactivity (HPPR), defined as platelet aggregation >70%, were examined.[106] HPPR was far less prevalent in the clopidogrel 600 mg group than in the 300 mg group (15% vs 25%; p = 0.04).[106] Of the patients who experienced a cardiovascular event, 67% of patients in the clopidogrel 300 mg group and 86% of patients in the 600 mg group were characterized as having HPPR.[106] Thus, patients who are unable to adequately suppress platelet aggregation are at an increased risk for cardiovascular events post-PCI. In the clopidogrel 600 mg group, patients with HPPR may represent a population of individuals who are nonresponders to or ‘resistant’ to treatment with clopidogrel.
Overall, pretreatment with clopidogrel 600 mg appears to be as safe as using a lower dose and seems to be more efficacious. The beneficial effects of pretreatment with clopidogrel may be due to maximal suppression of platelet aggregation prior to PCI or possibly in part due to a reduction in the levels of inflammatory markers associated with poorer cardiovascular outcomes.[107,108] The CURRENT/OASIS 7 trial will randomize 14 000 patients with unstable angina or non-STEMI (NSTEMI) undergoing early PCI to different doses of clopidogrel and aspirin with a 30-day follow-up to determine whether higher doses of antiplatelet agents results in reduced rates of ischemia.[109] Specifically, patients will be randomized to loading doses of either clopidogrel 300 mg or 600 mg followed by clopidogrel 150 or 75 mg/day for the first week, then 75 mg/day up to 30 days.[109] All patients will receive a loading dose of aspirin 300 mg and will be randomized to either aspirin 75–100 mg or 300–325 mg/day for the 30 days.[109] CURRENT/OASIS 7 may help to determine if higher doses of clopidogrel and/or aspirin improve efficacy outcomes.
The duration of clopidogrel therapy following PCI is currently a topic of much debate. A prespecified analysis of the CURE trial called PCI-CURE investigated the benefit of long-term clopidogrel in patients undergoing PCI. In PCI-CURE,[110] 2658 patients with NSTEMI undergoing PCI received placebo or pretreatment with a loading dose of clopidogrel 300 mg then 75 mg/day for a median duration of 10 days. All patients were receiving aspirin prior to PCI, with limited use of GP IIb/IIIa inhibitors.[110] Post-PCI, patients received open label thienopyridine for 4 weeks after which the original study drug was resumed in 80% of patients in both groups for a mean duration of 8 months.[110] From PCI to the end of follow-up (i.e. mean duration of 8 months from PCI), clopidogrel showed a significant reduction in cardiovascular death, MI, or urgent revascularization compared with placebo (18.3% vs 21.7%; RR 0.83; 95% CI 0.70, 0.99; p = 0.03), with the greatest effect occurring in the prevention of Q-wave MI for the clopidogrel group (1.5% vs 3.5%; RR 0.43; 95% CI 0.26, 0.73).[110]
The CREDO trial followed up patients for up to 1 year after PCI. Patients in this study were randomized to clopidogrel or placebo with both groups receiving concomitant aspirin.[99] At 1 year, the clopidogrel group had a 26.9% reduction in death, MI, or stroke compared with placebo (8.5 vs 11.5%; 95% CI 3.9, 44.4; p = 0.02).[99] Patients treated with clopidogrel had an increased risk for major bleeding compared with placebo but this result was not statistically significant (8.8% vs 6.7%; p = 0.07).[99] Results from PCI-CURE and CREDO trials indicate that a longer duration of treatment with clopidogrel and concomitant aspirin post-PCI was not only safe but improved the efficacy for the prevention of ischemic cardiovascular events up to 1 year.
Current ACC/AHA/SCAI guidelines for clopidogrel therapy for PCI recommend a loading dose of 300 mg administered 6 hours prior to the procedure and continuation of therapy for at least 1 month with BMS, 3 months with sirolimus-eluting stents, and 6 months with paclitaxel-eluting stents.[85] A recommendation to continue therapy up until 1 year is suggested for DES patients without a high risk of bleeding.[85] Loading doses of clopidogrel >300 mg have been utilized to achieve greater antiplatelet activity but the safety and efficacy of higher doses have not been fully established.
4.5 Thienopyridine Therapy and Drug-Eluting Stents
DES have been associated with a phenomenon called late-stent thrombosis.[111] The BASKET-LATE trial[112] randomized 743 patients undergoing PCI to BMS, paclitaxel-eluting, or sirolimus-eluting stents. Patients were treated with clopidogrel for 6 months and aspirin indefinitely, then followed up for 1 year after stopping clopidogrel.[112] Cardiac death or MI was decreased 3-fold in the BMS group compared with the DES group during the 1-year follow-up after discontinuation of clopidogrel (1.3 vs 4.9%; p = 0.01).[112] The median time for a clinical event was 116 days after discontinuation of clopidogrel with a range of 15 to 362 days during the period of 1 year.[112] The above results suggest that the duration of clopidogrel therapy was insufficient to prevent late thrombotic events in patients with DES.
A recent observational study in 4666 patients who underwent PCI with BMS (n = 3165) or DES (n = 1501) were followed up at 6, 12, and 24 months for death, MI, or revascularization. Patients with DES and event-free at 6 months had significant reductions in adjusted 24-month outcomes with clopidogrel therapy versus no therapy for both death (2.0% vs 5.3%; 95% CI −6.3, −0.3; p = 0.03) and death or MI (3.1% vs 7.2%; 95% CI −7.6, −0.6; p = 0.02).[78] There was no statistically significant difference between groups for nonfatal MI alone (1.3 vs 3.6%; p = 0.24).[78] Similar results were reported for adjusted 24-month outcomes in patients who were event-free at 12 months and receiving clopidogrel compared with no clopidogrel (death: 0.0% vs 3.5%; p = 0.04; nonfatal MI: 0.0% vs 1.0%; p = 0.047; and death or MI: 0.0% vs 4.5%; p < 0.001).[78] Among patients with BMS, no statistically significant differences in death and death or MI were observed at 24 months for patients receiving clopidogrel versus no clopidogrel.[78] This observational study suggests that a longer duration of treatment with clopidogrel is associated with a reduced risk for death and death or MI in patients with DES.
A meta-analysis of ten controlled trials involving 5030 patients randomized to BMS (n = 2428) or DES (n = 2602) did not find any significant increase in the risk of stent thrombosis between DES or BMS (0.58% for DES vs 0.51% for BMS; OR 1.05; 95% CI 0.51, 2.15; p = 1.000).[77] Over a period of 12 months, the incidence of stent thrombosis with DES was 0.20% and was significantly increased in patients receiving longer stents.[77] Data on antiplatelet therapy each patient received at the time of stent thrombosis were not available.[77] A second meta-analysis of 14 RCTs in 6675 patients undergoing PCI with follow-up at 12–48 months found the incidence of stent thrombosis to be 0.50% with DES and 0% with BMS (RR 5.02; 95% CI 1.29, 19.52; p = 0.02).[113] The risk of stent thrombosis in DES was increased 4- to 5-fold after 6 months compared with BMS and surprisingly this risk remained constant up to 48 months after stent placement.[113] The majority of trials in this meta-analysis used dual antiplatelet therapy for 2–6 months and none of the trials mandated clopidogrel use beyond 1 year.[113] Despite the incidence of stent thrombosis being very low, results of these meta-analyses suggest that early discontinuation of dual antiplatelet therapy in patients with DES is associated with a significant risk for thrombotic events. These results were confirmed by a subsequent meta-analysis of four RCTs in patients with paclitaxel-eluting stents, which found that the incidence of late stent thrombosis with paclitaxel-eluting stents was 0.50% compared with 0.06% in patients with BMS followed up for 36 months.[114] Ten patients with paclitaxel-eluting stents (n = 1718) developed stent thrombosis after 30 days, while eight patients had thrombotic events >180 days after stent placement.[114] At the time of thrombosis, treatment with clopidogrel had been discontinued in all patients and three patients were not receiving treatment with aspirin.[114] Overall, results of the three meta-analyses suggest that DES are associated with a very low but significant risk for late stent thrombosis compared with BMS. Taking into account the results of the BASKET-LATE trial, early discontinuation of thienopyridine therapy seems to be correlated with an increased risk for late stent thrombosis in patients with DES. However, subsequent meta-analyses of the RCT have found no excess death or MI associated with DES compared with BMS.[115–117]
The devastating nature of stent thrombosis and its association with DES has led practitioners to re-think their approach toward stent selection and subsequent antiplatelet therapy. The use of prolonged dual antiplatelet therapy must be weighed against risk for bleeding.[58] Selecting which patients are appropriate candidates for DES and long-term or lifetime dual antiplatelet therapy is essential.[111] A consensus statement by the AHA/ACC/SCAI addressing the issue of early discontinuation of thienopyridine therapy in DES was recently published.[118] DES should be avoided if possible in any patient who will undergo a surgical or invasive procedure within 12 months or in any individual who may not comply with 12 months of thienopyridine therapy for any reason.[118] Improved efforts to educate patients and healthcare providers who may want to discontinue thienopyridine therapy during invasive or surgical procedures are needed.[118] Any elective invasive or surgical procedure that may increase bleeding should be delayed until patients have received at least 12 months of dual antiplatelet therapy following DES placement.[118] If a patient needs to discontinue thienopyridine therapy for an invasive or surgical procedure, then aspirin should be continued and treatment with thienopyridine resumed as soon as possible.[118] Finally, there must be adequate communication between the physician and the patient’s interventional cardiologist prior to discontinuation of antiplatelet therapy in patients with stents.[118]
5. Newer Platelet Adenosine Diphosphate Receptor Antagonists
Several newer ADP receptor antagonists are currently being investigated.[119] Prasugrel is a newer potent thienopyridine that irreversibly inhibits platelet activation and aggregation via the P2Y12 receptor.[120,121] Ex vivo studies have shown that prasugrel is a more potent inhibitor of platelet aggregation than clopidogrel and ticlopidine.[120] In a phase I study, prasugrel had a faster onset of action and fewer nonresponders compared with clopidogrel.[122] The JUMBO-TIMI 26[123] trial was a phase II clinical trial of 904 patients undergoing elective or emergent PCI randomized to clopidogrel or one of three different dosing regimens of prasugrel. At 30 days, there was no significant differences in bleeding between patients receiving clopidogrel or pooled prasugrel (1.2% vs 1.7%; 95% CI 0.40, 5.08; p = 0.59).[123] Patients receiving prasugrel had a nonsignificant reduction in major cardiac adverse events compared to clopidogrel (7.2% vs 9.4%; 95% CI 0.46, 1.24; p = 0.26).[123] The TRITON-TIMI 38[124] randomized 13 608 patients with UA/NSTEMI or STEMI to prasugrel (60-mg loading dose then 10 mg/day) or clopidogrel (300-mg loading dose then 75 mg/day) for a median of 14.5 months. Cardiovascular death, nonfatal MI, or nonfatal stroke was significantly lower in the prasugrel group compared with the clopidogrel group (9.9 vs 12.1 %; HR 0.81; 95% CI 0.73, 0.90; p < 0.001), while major bleeding was significantly higher in the prasugrel group (2.4 vs 1.8%; HR 1.32; 95% CI 1.03, 1.68; p = 0.03).[124] The study suggests that the greater anti-ischemic benefit of prasugrel must be weighed carefully against its higher bleeding risk, and future subgroup analysis needs to determine who may benefit the most from prasugrel with minimal risks of bleeding.[125] PRINCIPLE-TIMI 44[126] trial was a randomized, double-blind, crossover study examining patients undergoing elective PCI. Patients were loaded prior to PCI with prasugrel 60 mg or clopidogrel 600 mg and had post-loading dose and post-PCI aggregometry and biomarker studies carried out.[126] Treatment with prasugrel 10 mg or clopidogrel 150 mg was maintained for 14 days and therapies were crossed over for a further 14 days.[126] Clinical outcomes, aggregometry, and biomarkers were assessed at 14 and 30 days.[126] Basically, prasugrel was more effective at inhibiting platelets even when compared to higher doses of clopidogrel.[126]
Ticagrelor (AZD 6140) is a reversible oral P2Y12 receptor antagonist from a class called cyclopentyltriazolopyrimidines.[127] The DISPERSE trial randomized 200 patients with atherosclerosis to clopidogrel 75 mg or to different dosages of ticagrelor and reported improved antiplatelet pharmacokinetics compared with clopidogrel.[128] The DISPERSE-2 trial[129] randomized 984 patients with NSTEMI receiving concurrent aspirin to a loading dose of ticagrelor 270 mg followed by 90 or 180 mg twice daily or a loading dose of clopidogrel 300 mg followed by 75 mg/day. No significant differences were found between ticagrelor and clopidogrel for the primary endpoint of death, MI, stroke, or recurrent ischemia at 12 weeks.[129] Ticagrelor had no increased risk for significant bleeding and reduced the risk of MI compared with clopidogrel (2.7 and 2.1%, respectively, for ticagrelor 90 mg and 180 mg vs 4.3% for clopidogrel).[129] These results suggest that ticagrelor has a similar safety profile and may be more efficacious than clopidogrel. In some of the clinical trials, an increased frequency of dyspnea has been noted with ticagrelor.[130] The PLATO study is an ongoing randomized phase III clinical trial evaluating the efficacy of ticagrelor in comparison with clopidogrel for the prevention of vascular events in 18 000 patients with ACS.[119,131,132]
In ex vivo studies, an intravenous P2Y12 receptor antagonist called cangrelor has shown greater inhibition of platelet activation than clopidogrel and does not require hepatic metabolism for activation unlike clopidogrel.[133,134] Cangrelor achieves steady state concentrations within a few minutes of administration, and the majority of patients regain platelet function within one hour of drug discontinuation.[41,135] This compound has been evaluated in phase II clinical studies and has shown similar efficacy and tolerability to clopidogrel.[41,127,136] Two phase III clinical trials are ongoing, called CHAMPION-PCI and -PLATFORM, to determine the efficacy of cangrelor in comparison with clopidogrel in patients undergoing PCI.[119,132,137,138]
6. Aspirin Resistance
Despite the widespread use of aspirin for primary or secondary prevention of ACS, a certain proportion of patients receiving aspirin will experience ischemic events related to atherothrombosis. This clinical failure has sometimes been called aspirin ‘resistance’ or nonresponse. The biologic mechanism responsible for aspirin resistance is most likely multifactorial. The topic of aspirin resistance has been extensively reviewed in the literature.[139–143] It is well known that aspirin has a variable effect on platelet reactivity.[144] The intra- and inter-individual variability of aspirin on platelet function has been clearly demonstrated by laboratory measurements.[145] It remains to be elucidated how this variability correlates with clinical resistance or nonresponse.[146]
The prevalence of aspirin resistance is not known. Numerous studies have been published with estimates of aspirin resistance ranging from 5% to 60%.[143,147] However, it is difficult to make any judgments about the true prevalence of aspirin resistance from these studies because of: (i) the small sample size; (ii) variabilities in testing and measurement between studies; (iii) the variability in definitions used for aspirin resistance; and (iv) the different patient populations used in the studies. Estimating the true prevalence is also hindered by the under use of aspirin by patients and physicians, even in patients with known CAD.[148] Thus, at present, little is known about the true prevalence of aspirin resistance and this remains an important question for future study.
6.1 Laboratory Tests for Measurement of Platelet Response to Aspirin
Numerous methods are currently available to investigate platelet activity.[149,150] Aspirin decreases the production of TxA2 thus inhibiting platelet activation and aggregation. The effect of aspirin on TxA2 can be measured by assessing stable metabolites of TxA2, mainly serum levels of TxB2 and urinary levels of 11-β-dehydrothromboxane B2.[147] Serum TxB2 is a stable breakdown metabolite of TxA2 that is dependent on COX-1 activity.[151] However, serum levels of TxA2 are not always solely dependent on platelets or COX-1 activity. Monocytes and macrophages can also produce TxA2, independent of COX-1 activity.[147] COX-2 is an inducible form of COX that can produce TxA2, whose expression and activity is increased in diseased vessels with atherosclerotic lesions.[152,153] TxA2 can be produced in endothelial cells, monocytes, and macrophages by transcellular metabolism involving platelet thromboxane synthase.[154] Megakaryopoiesis induces COX-2 expression which can be found in states of high platelet turnover.[155] Serum TxA2 and its stable metabolite TxB2 do depend on COX-1 activity, yet there are several other mechanisms and cells by which it can be produced.
A second stable metabolite of TxA2 found in the urine is 11-β-dehydrothromboxane B2 and it has been utilized as a measure of TxA2 production.[156] The limitations of measuring levels of this metabolite of TxA2 are similar to those of measuring serum TxB2 levels. However, higher levels of urinary 11-β-dehydrothromboxane B2 have been correlated with increased cardiovascular risk in aspirin-treated patients.[142] Measurements of metabolites of TxA2 can be directly affected by COX-1 activity. A number of mechanisms exist that can bypass COX-1 mediated production of TxA2 and still activate platelets, thus making the measurement of TxA2 metabolites somewhat unreliable for testing platelet activity.
Direct measurement of platelet function is an alternative method to measure aspirin resistance and its effects on overall platelet function. The majority of the tests rely upon measuring the level of agonist-induced (e.g. adrenaline [epinephrine], collagen, arachidonic acid, and ADP) platelet aggregation, which should correlate with TxA2 production in platelets.[150] The gold standard of platelet function tests is light or optical aggregometry, whereby the amount of agonist-induced platelet aggregation is correlated with light transmission measured spectrophotometrically. Limitations to this test are that it is highly labor intensive, results are variable between laboratories and operator dependent.[157]
The Platelet Function Analyzer-100 (PFA-100™)Footnote 1 is a point of care test that contains a biologically active membrane coated with adrenaline or ADP.[158] A sample of blood flows through a small aperture that creates an artificial vessel.[158] Blood samples experience high shear stress coupled with exposure to biologic stimuli on the coated membrane to induce platelet activation and aggregation. The time it takes for a stable plug to form at the aperture correlates with the platelet reactivity of the sample.[158] Numerous factors including platelet count, red blood cell count, and vWF can affect the closure time, thus results can be highly variable.[147] The VerifyNow™ is another point of care test that measures the ability of whole blood to induce fibrinogen-coated bead agglutination.[141] It measures light transmission and values are expressed as aspirin resistance units (ARU), where a value >550 is defined as aspirin resistant.[141]
Despite several ways to measure platelet function, studies have shown that there is poor agreement between various testing methods. 100 consecutive patients with a history of stroke or transient ischemic attack treated with aspirin for 1 month had blood samples drawn and tested with arachidonic acid-induced optical aggregometry, PFA-100™, and VerifyNow™.[159] The correlation between the tests for aspirin nonresponders was poor.[159] The incidence of aspirin resistance was 5% by optical aggregometry, 22% by PFA-100, and 17% by VerifyNow™; only 2% of patients were confirmed as nonresponders by all three tests.[159]
More importantly, does the absence of a clinical response to aspirin correlate with laboratory evidence of aspirin resistance? It is likely that a number of factors are responsible for this clinical failure, yet a certain population of patients may be truly resistant to aspirin.[160] A more logical approach to the debate may be to take cues from how blood pressure is currently analysed and to think of the laboratory evidence as a continuum of aspirin inhibition of platelet function.[140,141]
6.2 Potential Mechanisms of Aspirin Resistance
The most common cause for aspirin resistance is non-compliance. Numerous studies have correlated aspirin resistance to noncompliance.[161–163] In some small studies, higher doses of aspirin correlate with greater inhibition of platelet reactivity.[164,165] However, higher doses of aspirin do not show a greater clinical benefit compared with lower doses of aspirin and appear to confer a greater risk of both minor and major bleeding.[29,166] Younger age and increased body weight are correlated with decreased rates of COX-1 inhibition with low doses of aspirin but the clinical significance of this correlation remains unknown.[167]
Aspirin crosses the intestinal barrier as a weakly acidic lipophilic drug that can be metabolized to its inactive form, salicylic acid, by mucosal esterases.[168,169] It has been proposed that proton pump inhibitors can increase the metabolism of aspirin to its inactive form by esterases.[147] The concurrent use of NSAIDs by an individual receiving aspirin has been postulated as a cause for resistance.[170] NSAIDs can inhibit COX-1 activity directly independent of aspirin. NSAIDs and aspirin have the potential to interact pharmacokinetically, and NSAIDs could both actively or allosterically inhibit the binding of aspirin to COX-1.[171,172] Patients receiving NSAIDs, particularly ibuprofen, and aspirin have been shown to have an increased risk of MI.[173,174] Adequately powered trials have not been conducted to determine the true effect of NSAIDs as augmenting or accelerating inhibition of COX-1.[175] True pharmacokinetic failure to inhibit COX-1 with aspirin has been shown in selected patient populations. Some studies[176–178] have shown that patients undergoing CABG or carotid surgery have impaired COX-1 inhibition with aspirin, yet the mechanism and clinical significance of this failure are unknown.
COX-2 induction by various clinical states and/or increased production in monocytes, macrophages, and endothelial cells can provide a mechanism for TxA2 production independent of COX-1.[152,176,179] Other alternate pathways for platelet activation could occur through non-TxA2 molecules like collagen, ADP, or vWF.[180,181] Interestingly, patients who were found to be aspirin resistant by PFA-100™ were more sensitive to stimulation by ADP.[181]
Exercise causes an increase in circulating catecholamines which may be associated with increased platelet activation; aspirin cannot protect against exercise-induced platelet activation in certain individuals.[182] Isoprostanes are products of lipid peroxidation and may act as thromboxane receptor agonists. Certain risk factors (e.g. smoking, diabetes mellitus, obesity, hyperlipidemia, hyperglycemia, and hyperhomocysteinemia) associated with cardiovascular disease have been shown to increase isoprostane production and oxidative stress.[183–188] Patients with severe unstable angina have increased production of F2-isoprostane 8-isoprostaglandin, a byproduct of arachidonic acid peroxidation. This provides a possible biochemical link between increased oxidant stress and platelet activation in aspirin resistant patients.[189] The role of systemic inflammation on platelet reactivity may provide an alternative mechanism for TxA2 production.[190] Cardiovascular disease itself may activate platelets, as seen in congestive heart failure (CHF) and ACS.[191,192] Numerous mechanisms or confounding variables may contribute to impaired platelet response, and clinical studies must control for these variables in order to determine the true cause for aspirin resistance.
An exciting aspect of aspirin resistance is seen in patients who may have a genetic cause for altered response to aspirin. Various genetic polymorphisms have been identified and associated with aspirin resistance.[193] Polymorphisms of the COX-1 gene have been identified, which could be involved with altered responsiveness to aspirin.[193,194] COX-2 polymorphisms are associated with decreased risks for MI and stroke.[195] Other polymorphisms associated with platelet surface glycoproteins may be associated with clinical response to aspirin.[196] More specifically, polymorphism PLA1/A2 of the gene encoding GP IIIa has been associated with increased bleeding time and impaired response to platelet aggregation agonists.[197–200] Aspirin resistance has also been associated with individuals with polymorphisms in the ADP receptor gene P 2 Y 1 on platelets, vWF, and the collagen receptor.[196,201,202] As more data are accumulated on genetic polymorphisms associated with impaired pharmacologic response to aspirin, this subset of individuals represents a unique group where primary and secondary prevention of cardiovascular disease with alternative antiplatelet therapies could be utilized. Potential mechanisms of aspirin resistance are summarized in figure 4.

Possible mechanisms of aspirin resistance. ADP adenosine diphosphate; CABG = coronary artery bypass grafting; COX-1 = cyclo-oxygenase-1; COX-2 = cyclo-oxygenase-2; GP = glycoprotein; TxA 2 = thromboxane A2; vWF = von Willebrand Factor.
6.3 Clinical Outcomes in Patients with Aspirin Resistance
Clinical trials as far back as 1993 have shown poorer clinical prognosis in aspirin nonresponsive individuals.[203] A substudy of the HOPE trial[142] analysed 966 patients with known cardiovascular disease receiving aspirin once daily in a nested case-control design and measured urinary-11-β-dehydrothromboxane B2 with a median follow-up of 4.5 years. After adjusting for baseline differences, individuals with urinary-11-β-dehydrothromboxane B2 levels (i.e. higher TxA2 levels) in the highest quartile were 1.8 times more likely to have cardiovascular death, MI, or stroke compared with those in the lowest quartile (OR 1.8; 95% CI 1.2, 2.7; p = 0.009).[142] The trial had several limitations including single baseline measurements of urinary TxB2 concentrations and multiple confounders between cases and controls.[142] Overall, the above data suggest that failure of aspirin to inhibit platelet TxA2 production correlates with adverse clinical outcomes.
A more recent trial in 171 patients undergoing elective PCI and receiving aspirin for ≥1 week and loaded with clopidogrel 300 mg 12–24 hours prior to PCI measured baseline cardiac enzymes (CKMB and TnI) and platelet function with VerifyNow™.[204] Patients who received treatment with GP IIb-IIIa inhibitors, other antiplatelet drugs or NSAIDs within 2 weeks of PCI, or with preprocedural elevations in cardiac enzymes were excluded. This study found that 19.2% of patients were aspirin resistant and had higher levels of cardiac enzymes compared with patients who were aspirin sensitive, which made aspirin resistance an independent risk factor for myonecrosis (OR 2.8; 95% CI 1.3, 6.0; p = 0.007).[204] The sensitivity and specificity of the VerifyNow™ were 92% and 85%, respectively.[204] Study limitations included a predominantly Asian patient population, small sample size, and collection of a single-baseline blood sample for defining aspirin resistance.[204] Randomized clinical trials assessing the prognostic significance of aspirin resistance are needed that are powered adequately and control for confounding variables, especially compliance.
Another trial randomized 36 patients with known peripheral arterial disease to clopidogrel or placebo.[205] All patients were treated with aspirin 100 mg/day for a 3-week run-in period prior to randomization and continued on aspirin through the duration of the trial.[205] Patients were treated with clopidogrel or placebo for 3 weeks, followed by a 3-week washout, and then a 3-week crossover of the randomized drugs.[205] Platelet aggregation by optical aggregometry was analysed after each 3-week period.[205] The highest inhibition of collagen induced platelet aggregation was found in patients with the highest levels of arachidonic acid induced platelet aggregation (i.e. the least sensitive to aspirin).[205] Thus, clopidogrel showed greater inhibition of platelet function in patients who were least responsive to aspirin. The trial suggests that clopidogrel may be a useful adjunct in patients who have biochemical evidence of aspirin resistance.
So the question remains: what do we do about aspirin resistance? At this point, there are limited clinical data on when it is appropriate to test for aspirin resistance, or if someone is defined as aspirin resistant, how one should manage the antiplatelet regimen. The Antithrombotic Trialists’ Collaboration had shown that increased doses of aspirin appear to have little clinical benefit in a population of patients.[29] Higher aspirin doses were associated with increased bleeding risk in the CURE trial.[59] Clinical trials are needed to determine if higher aspirin doses can overcome aspirin resistance and potentially decrease cardiovascular events. A substudy of the CHARISMA trial will examine a cohort of patients to determine prospectively if clopidogrel added to aspirin will be beneficial for those patients with high levels of urinary-11-β-dehydrothromboxane B2. RCTs further analysing the question of aspirin resistance in large populations of patients with ACS are required.
7. Clopidogrel Resistance
As in the case with aspirin, the concept of ‘resistance’ or nonresponse to clopidogrel has recently emerged as a controversial topic.[141,206,207] Many of the concepts and controversies that surround aspirin resistance are shared with clopidogrel resistance. Current literature has estimated the prevalence of clopidogrel resistance at 4% to 68%.[206,207] A reduction in the prevalence of nonresponders has been reported with longer durations of treatment with clopidogrel.[208] Various methods exist to test platelet response to clopidogrel. No single test or definition is available to determine if a patient is resistant to clopidogrel. Light or optical aggregometry with ADP as an agonist will induce P2Y12-dependent platelet aggregation.[150,157] Results with optical aggregometry can vary significantly with the concentration of ADP used and the time chosen to test platelet function after the administration of clopidogrel.[101,209,210] Other methods used to determine platelet reactivity to clopidogrel are currently being tested and include disaggregation studies and measurement of monoclonal antibodies to GP IIb/IIIa or P-selectin.[211] Phosphorylated vasodilator-stimulated phosphoprotein (VASP) levels have been shown to correlate with clopidogrel resistance but exhibits a wider inter-individual variability.[212] VASP is an intraplatelet regulatory protein which is phosphorylated via a P2Y12-dependent mechanism; the phosphorylated form can be detected by flow cytometry.[212] Significant inter- and intra-test variability occurs with the various testing modalities and hampers the ability to define clopidogrel resistance based on laboratory measurements.[210,211]
7.1 Potential Mechanisms for Clopidogrel Resistance
Research is being conducted to determine what influences an individual’s response to clopidogrel; numerous mechanisms have been postulated. Noncompliance is a common cause for clopidogrel resistance. It still remains to be proven if platelet reactivity to clopidogrel is influenced by systemic inflammation.[190] Variations in pharmacokinetics and pharmacodynamics may influence an individual’s response to clopidogrel. Clopidogrel absorption varies between individuals, and there is significant interindividual variability in hepatic cytochrome P450 (CYP) 3A4 activity.[213,214] CYP3A4 metabolizes the prodrug clopidogrel into its active form, thus drug-drug interactions could occur with other molecules metabolized by CYP3A4. Atorvastatin is metabolized by CYP3A4 and it has been postulated that this drug could inhibit the platelet effects of clopidogrel. In one trial[215] of 44 consecutive patients undergoing PCI, atorvastatin attenuated the antiplatelet activity of clopidogrel in a dose-dependent manner. However, results from the CREDO, PRONTO, and INTERACTION studies showed that no statins including atorvastatin altered the clinical antiplatelet effect of clopidogrel.[216–218]
In 544 individuals who underwent PCI or had heart failure there was a wide distribution in ADP-induced platelet aggregation as measured by optical aggregometry.[219] In a trial of 96 patients undergoing elective PCI, ADP-induced platelet aggregation was measured by optical aggregometry at various timepoints from baseline to 30 days following PCI.[210] All patients were treated with a loading dose of aspirin and clopidogrel 300 mg followed by 75 mg/day.[210] There was a wide interindividual variability in the platelet inhibitory response from clopidogrel; however, patients with higher baseline platelet reactivity showed higher platelet reactivity in subsequent testing post-PCI.[210] Both studies show that there is a fair amount of variability in response to clopidogrel, but do not provide guidance regarding what, if anything, to do about it.[210,219]
It is postulated that differences in platelet reactivity with clopidogrel could occur as a result of genetic differences in the P2Y12 ADP receptor on platelets. The H2 haplotype of the P2Y12 ADP receptor has been associated with enhanced ADP-induced platelet aggregation and is more prevalent in patients with peripheral arterial disease compared with age-matched controls.[220,221] A study[222] in 416 patients undergoing elective PCI and treated with aspirin and clopidogrel 600 mg prior to PCI, had 2-hour post-administration blood samples analysed for ADP-induced optical platelet aggregation. Patients were evaluated for P2Y12 haplotypes (H1/H2) and P2Y12 C32T genotypes and no differences occurred between haplotype combinations or genotypes in their platelet reactivity.[222] Prospective RCTs will allow further insight into the mechanisms surrounding clopidogrel platelet reactivity.
7.2 Prognostic Significance of Clopidogrel Resistance
Very few clinical studies have assessed the prognostic significance of clopidogrel resistance in patients with cardiovascular disease. One study[209] in 105 patients with known CAD undergoing elective PCI for stable angina, measured ADP-induced optical platelet aggregation at baseline and 4 hours after clopidogrel administration. Clopidogrel nonresponse was defined as platelet aggregation of <10% from baseline.[209] Roughly 5–11% of individuals were found to be nonresponders, of whom two individuals in the nonresponder subgroup had subacute thrombosis 6–7 days following PCI.[209] In another study,[223] a population of 60 patients with STEMI undergoing PCI were administered aspirin and clopidogrel 300 mg after PCI followed by 75 mg/day for 3 months. ADP-induced optical platelet aggregation studies were done at baseline and daily for 5 days post-PCI.[223] Patients were followed up for 6 months and compliance was assessed monthly with telephone calls.[223] During the follow-up period, 40% of patients in the first quartile (i.e. patients with the highest mean platelet aggregation), 6.7% of patients in the second quartile, and zero patients in the third and fourth quartile had cardiovascular events (p = 0.007).[223] Both of these studies give a glimpse of how variability in platelet response may have prognostic significance.
So what should physicians do about clopidogrel resistance? There are no clinical guidelines currently for screening and management of patients who are resistant to clopidogrel. One could make an argument for screening selected populations who may be at higher risk for impaired platelet reactivity. One group that may be of interest are patients who will undergo complex PCI with DES and require longer dual antiplatelet therapy, yet how one would manage patients with impaired reactivity remains unknown. Current ACC/AHA/SCAI guidelines recommend platelet aggregation studies in patients at high risk for catastrophic or lethal subacute stenosis and to increase the dose of clopidogrel to 150 mg/day if <50% of platelet aggregation is inhibited.[85] Higher doses of clopidogrel accelerate inhibition of platelet aggregation as early as 2 hours after drug administration.[101] For patients undergoing PCI, increasing the loading doses of clopidogrel may prove to be beneficial in patients who are resistant to clopidogrel as higher doses have been shown to improve efficacy as evidenced from results from the ARMYDA-2, ALBION, and ISAR-CHOICE trials.[103–105] The newer ADP antagonists, prasugrel, cangrelor, and ticagrelor, may be alternative agents for use in patients who clearly display impaired platelet reactivity to clopidogrel or have clinical failure to clopidogrel despite adequate compliance. Future RCTs that can help define clopidogrel resistance, determine which patients should be screened, and provide insight into treatment options may provide a more individualized approach to therapy for patients with ACS and/or undergoing PCI.
8. Conclusion
Atherothrombosis is a complex cascade of events that ensues from the interplay of atherosclerotic plaque rupture leading to platelet activation and aggregation. Antiplatelet agents are essential for the primary and secondary prevention of ACS. For primary prevention, aspirin should be considered in selected patient populations, especially those with Framingham risk scores >10%. After ACS, dual antiplatelet therapy for at least 1 year has emerged as standard therapy for prevention of ischemic events. In patients undergoing PCI, aspirin should be continued indefinitely and clopidogrel for at least 1 year. Clopidogrel pretreatment is important to decrease the incidence of cardiovascular events. Dual antiplatelet therapy is even more important with the advent of DES and the rare phenomenon of late-stent thrombosis. Continuing dual antiplatelet therapy for periods that may be longer than current guidelines in patients with low risk for bleeding might be prudent. The concept of resistance to aspirin and clopidogrel is still an emerging and important clinical question. Advances in pharmacogenomics are on the horizon and may change the landscape of how ACS and PCI should be managed. Individualized antithrombotic regimens may include specific therapy based on patients’ platelet reactivity to that specific agent. Newer ADP antagonists may be promising alternatives to currently available antiplatelet therapies for the treatment of ACS and for patients undergoing PCI.
Notes
The use of trade names is for product identification purposes only and does not imply endorsement.
References
Fuster V, Moreno PR, Fayad ZA, et al. Atherothrombosis and high-risk plaque: part I. Evolving concepts. J Am Coll Cardiol 2005; 46(6): 937–54.
Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy: nature reviews. Nat Rev Drug Discov 2003; 2(1): 15–28.
Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8(11): 1227–34.
McNicol A, Israels SJ. Platelets and anti-platelet therapy. J Pharmacologic Sci 2003; 93(4): 381–96.
Roth GJ, Calverley DC. Aspirin, platelets, and thrombosis: theory and practice. Blood 1994; 83(4): 885–98.
Patrono C, Renda G. Platelet activation and inhibition in unstable coronary syndromes. Am J Cardiol 1997; 80(5A): 17E–20E.
Merlini PA, Bauer KA, Oltrona L, et al. Persistent activation of coagulation mechanism in unstable angina and myocardial infarction. Circulation 1994; 90(1): 61–8.
Ault KA, Cannon CP, Mitchell J, et al. Platelet activation in patients after an acute coronary syndrome: results from the TIMI-12 trial: Thrombolysis in Myocardial Infarction. J Am Coll Cardiol 1999; 33(3): 634–9.
O’Donnell CJ, Larson MG, Feng D, et al. Genetic and environmental contributions to platelet aggregation: the Framingham heart study. Circulation 2001; 103(25): 3051–6.
Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295(2): 180–9.
Bhatt DL, Topol EJ. Clopidogrel added to aspirin versus aspirin alone in secondary prevention and high-risk primary prevention: rationale and design of the Clopidogrel for High Atherothrombotic Risk and Ischemie Stabilization, Management, and Avoidance (CHARISMA) trial. Am Heart J 2004; 148(2): 263–8.
Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci U S A 1975; 72(8): 3073–6.
Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets: I. Acetylation of a particulate fraction protein. J Clin Invest 1975; 56(3): 624–32.
Patrono C, Bachmann F, Baigent C, et al. Expert consensus document on the use of antiplatelet agents: the task force on the use of antiplatelet agents in patients with atherosclerotic cardiovascular disease of the European society of cardiology. Eur Heart J 2004; 25(2): 166–81.
Burch JW, Stanford N, Majerus PW. Inhibition of platelet prostaglandin synthetase by oral aspirin. J Clin Invest 1978; 61(2): 314–9.
Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest 1982; 69(6): 1366–72.
Patrono C, Ciabattoni G, Patrignani P, et al. Clinical pharmacology of platelet cyclooxygenase inhibition. Circulation 1985; 72(6): 1177–84.
Mitchell JA, Akarasereenont P, Thiemermann C, et al. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A 1993; 90(24): 11693–7.
Cipollone F, Patrignani P, Greco A, et al. Differential suppression of thromboxane biosynthesis by indobufen and aspirin in patients with unstable angina. Circulation 1997; 96(4): 1109–16.
Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol 1995; 2(8): 637–43.
Ikonomidis I, Andreotti F, Economou E, et al. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation 1999; 100(8): 793–8.
Perretti M, Chiang N, La M, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med 2002; 8(11): 1296–302.
Bhatt DL, Topol EJ. Need to test the arterial inflammation hypothesis. Circulation 2002; 106(1): 136–40.
Ridker PM, Cushman M, Stampfer MJ, et al. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men [published erratum appears in N Engl J Med 1997; 337 (5): 356]. N Engl J Med 1997; 336(14): 973–9.
Feng D, Tracy RP, Lipinska I, et al. Effect of short-term aspirin use on C-reactive protein. J Thromb Thrombolysis 2000; 9(1): 37–41.
Feldman M, Jialal I, Devaraj S, et al. Effects of low-dose aspirin on serum C-reactive protein and thromboxane B2 concentrations: a placebo-controlled study using a highly sensitive C-reactive protein assay. J Am Coll Cardiol 2001; 37(8): 2036–41.
Kennon S, Price CP, Mills PG, et al. The effect of aspirin on C-reactive protein as a marker of risk in unstable angina. J Am Coll Cardiol 2001; 37(5): 1266–70.
Alexander JH, Harrington RA, Tuttle RH, et al. Prior aspirin use predicts worse outcomes in patients with non-ST-elevation acute coronary syndromes: PURSUIT Investigators. Platelet IIb/IIIa in Unstable angina: Receptor Suppression Using Integrilin Therapy. Am J Cardiol 1999; 83(8): 1147–51.
Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. [published erratum appears in BMJ 2002; 324 (7330): 141]. BMJ 2002; 324(7329): 71–86.
Derry S, Loke YK. Risk of gastrointestinal haemorrhage with long term use of aspirin: meta-analysis. BMJ 2000; 321(7270): 1183–7.
Garcia Rodriguez LA, Hernandez-Diaz S, de Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiologic studies. Br J Clin Pharmacol 2001; 52(5): 563–71.
Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162(19): 2197–202.
Hawthorne AB, Mahida YR, Cole AT, et al. Aspirin-induced gastric mucosal damage: prevention by enteric-coating and relation to prostaglandin synthesis. Br J Clin Pharmacol 1991; 32(1): 77–83.
Kelly JP, Kaufman DW, Jurgelon JM, et al. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348(9039): 1413–6.
Ridker PM, Hennekens CH, Tofler GH, et al. Anti-platelet effects of 100mg alternate day oral aspirin: a randomized, double-blind, placebo-controlled trial of regular and enteric coated formulations in men and women. J Cardiovasc Risk 1996; 3(2): 209–12.
Van Hecken A, Juliano ML, Depre M, et al. Effects of enteric-coated, low-dose aspirin on parameters of platelet function. Aliment Pharmacol Ther 2002; 16(9): 1683–8.
Karha J, Rajagopal V, Kottke-Marchant K, et al. Lack of effect of enteric coating on aspirin-induced inhibition of platelet aggregation in healthy volunteers. Am Heart J 2006; 151(5): 976.e7–e11.
Maree AO, Curtin RJ, Dooley M, et al. Platelet response to low-dose enteric-coated aspirin in patients with stable cardiovascular disease. J Am Coll Cardiol 2005; 46(7): 1258–63.
Stafford RS, Monti V, Ma J. Underutilization of aspirin persists in US ambulatory care for the secondary and primary prevention of cardiovascular disease. PLoS Med 2005; 2(12): e353.
Sharis PJ, Cannon CP, Loscalzo J. The antiplatelet effects of ticlopidine and clopidogrel. Ann Intern Med 1998; 129(5): 394–405.
Storey RF. The P2Y12 receptor as a therapeutic target in cardiovascular disease. Platelets 2001; 12(4): 197–209.
Hollopeter G, Jantzen HM, Vincent D, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001; 409(6817): 202–7.
Ito MK, Smith AR, Lee ML. Ticlopidine: a new platelet aggregation inhibitor. Clin Pharm 1992; 11(7): 603–17.
Haushofer A, Halbmayer WM, Prachar H. Neutropenia with ticlopidine plus aspirin. Lancet 1997; 349(9050): 474–5.
Neumann FJ, Hall D, Schomig A. Neutropenia with ticlopidine plus aspirin. Lancet 1997; 349(9064): 1552–3.
Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334(17): 1084–9.
Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting: Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339(23): 1665–71.
Steinhubl SR, Tan WA, Foody JM, et al. Incidence and clinical course of thrombotic thrombocytopenic purpura due to ticlopidine following coronary stenting: EPISTENT Investigators. Evaluation of Platelet Ilb/IIIa Inhibitor for Stenting. JAMA 1999; 281(9): 806–10.
Bennett CL, Davidson CJ, Raisch DW, et al. Thrombotic thrombocytopenic purpura associated with ticlopidine in the setting of coronary artery stents and stroke prevention. Arch Intern Med 1999; 159(21): 2524–8.
Cairns JA, Gent M, Singer J, et al. Aspirin, sulfinpyrazone, or both in unstable angina: Results of a Canadian multicenter trial. N Engl J Med 1985; 313(22): 1369–75.
Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Lancet 1988; 2 (8607): 349–60.
Risk of myocardial infarction and death during treatment with low dose aspirin and intravenous heparin in men with unstable coronary artery disease: the RISC Group. Lancet 1990; 336 (8719): 827–30.
Wallentin LC. Aspirin (75 mg/day) after an episode of unstable coronary artery disease: long-term effects on the risk for myocardial infarction, occurrence of severe angina and the need for revascularization. Research Group on Instability in Coronary Artery Disease in Southeast Sweden. J Am Coll Cardiol 1991; 18(7): 1587–93.
A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE): CAPRIE Steering Committee. Lancet 1996; 348 (9038): 1329–39.
Bhatt DL, Hirsch AT, Ringleb PA, et al. Reduction in the need for hospitalization for recurrent ischemie events and bleeding with clopidogrel instead of aspirin: CAPRIE investigators. Am Heart J 2000; 140(1): 67–73.
Hirsh J, Bhatt DL. Comparative benefits of clopidogrel and aspirin in high-risk patient populations: lessons from the CAPRIE and CURE studies. Arch Intern Med 2004; 164(19): 2106–10.
Moshfegh K, Redondo M, Julmy F, et al. Antiplatelet effects of clopidogrel compared with aspirin after myocardial infarction: enhanced inhibitory effects of combination therapy. J Am Coll Cardiol 2000; 36(3): 699–705.
Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345(7): 494–502.
Peters RJ, Mehta SR, Fox KA, et al. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation 2003; 108(14): 1682–7.
Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352(12): 1179–89.
Chen ZM, Jiang LX, Chen YP, et al. COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised place-bo-controlled trial. Lancet 2005; 366(9497): 1607–21.
Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354(16): 1706–17.
Bhatt DL, Fiather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49(19): 1982–8.
Gruntzig A. Transluminal dilatation of coronary-artery stenosis [letter]. Lancet 1978; 1(8058): 263.
Gruntzig AR, Senning A, Siegenthaler WE. Nonoperative dilatation of coronary-artery stenosis: percutaneous transluminal coronary angioplasty. N Engl J Med 1979; 301(2): 61–8.
Fischman DL, Leon MB, Baim DS, et al. A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. Stent Restenosis Study Investigators. N Engl J Med 1994; 331(8): 496–501.
Altmann DB, Racz M, Battleman DS, et al. Reduction in angioplasty complications after the introduction of coronary stents: results from a consecutive series of 2242 patients. Am Heart J 1996; 132(3): 503–7.
Savage MP, Douglas Jr JS, Fischman DL, et al. Stent placement compared with balloon angioplasty for obstructed coronary bypass grafts: Saphenous Vein De Novo Trial Investigators. N Engl J Med 1997; 337(11): 740–7.
Savage MP, Fischman DL, Rake R, et al. Efficacy of coronary stenting versus balloon angioplasty in small coronary arteries: Stent Restenosis Study (STRESS) Investigators. J Am Coll Cardiol 1998; 31(2): 307–11.
Erbel R, Haude M, Hopp HW, et al. Coronary-artery stenting compared with balloon angioplasty for restenosis after initial balloon angioplasty: Restenosis Stent Study Group. N Engl J Med 1998; 339(23): 1672–8.
Grines CL, Cox DA, Stone GW, et al. Coronary angioplasty with or without stent implantation for acute myocardial infarction: Stent Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med 1999; 341(26): 1949–56.
Kiemeneij F, Serruys PW, Macaya C, et al. Continued benefit of coronary stenting versus balloon angioplasty: five-year clinical follow-up of Benestent-I trial. J Am Coll Cardiol 2001; 37(6): 1598–603.
O’Neill WW, Grines CL. Surgery or stent? The gap continues to narrow. Lancet 2002; 360(9338): 961–2.
Popma JJ, Weitz J, Bittl JA, et al. Antithrombotic therapy in patients undergoing coronary angioplasty. Chest 1998; 114(5 Suppl.): 728S–41S.
Nath FC, Muller DW, Ellis SG, et al. Thrombosis of a flexible coil coronary stent: frequency, predictors and clinical outcome. J Am Coll Cardiol 1993; 21(3): 622–7.
O’Neill WW, Leon MB. Drug-eluting stents: costs versus clinical benefit. Circulation 2003; 107(24): 3008–11.
Moreno R, Fernandez C, Hernandez R, et al. Drug-eluting stent thrombosis: results from a pooled analysis including 10 randomized studies. J Am Coll Cardiol 2005; 45(6): 954–9.
Eisenstein EL, Anstrom KJ, Kong DF, et al. Clopidogrel use and long-term clinical outcomes after drug-eluting stent implantation. JAMA 2007; 297(2): 159–68.
Barnathan ES, Schwartz JS, Taylor L, et al. Aspirin and dipyridamole in the prevention of acute coronary thrombosis complicating coronary angioplasty. Circulation 1987; 76(1): 125–34.
Schwartz L, Bourassa MG, Lesperance J, et al. Aspirin and dipyridamole in the prevention of restenosis after percutaneous transluminal coronary angioplasty. N Engl J Med 1988; 318(26): 1714–9.
Lembo NJ, Black AJ, Roubin GS, et al. Effect of pretreatment with aspirin versus aspirin plus dipyridamole on frequency and type of acute complications of percutaneous transluminal coronary angioplasty. Am J Cardiol 1990; 65(7): 422–6.
Savage MP, Goldberg S, Bove AA, et al. Effect of thromboxane A2 blockade on clinical outcome and restenosis after successful coronary angioplasty: Multi-Hospital Eastern Atlantic Restenosis Trial (M-HEARTII). Circulation 1995; 92(11): 3194–200.
Schatz RA, Baim DS, Leon M, et al. Clinical experience with the Palmaz-Schatz coronary stent: initial results of a multicenter study. Circulation 1991; 83(1): 148–61.
Hall P, Nakamura S, Maiello L, et al. A randomized comparison of combined ticlopidine and aspirin therapy versus aspirin therapy alone after successful intravascular ultrasound-guided stent implantation. Circulation 1996; 93(2): 215–22.
Smith SC, Feldman TE, Hirshfeld JW, et al. ACC/AHA/SCAI 2005 Guideline Update For Percutaneous Coronary Intervention: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/SCAI Writing Committee to Update the 2001 Guidelines for Percutaneous Coronary Intervention). Circulation 2006; 113(1): 156–75.
Kastrati A, Schuhlen H, Hausleiter J, et al. Restenosis after coronary stent placement and randomization to a 4-week combined antiplatelet or anticoagulant therapy: six-month angiographic follow-up of the Intracoronary Stenting and Antithrombotic Regimen (ISAR) Trial. Circulation 1997; 96(2): 462–7.
Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting: the full anticoagulation versus aspirin and ticlopidine (FANTASTIC) study. Circulation 1998; 98(16): 1597–603.
Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the multicenter aspirin and ticlopidine trial after intracoronary stenting (MATTIS). Circulation 1998; 98(20): 2126–32.
Steinhubl SR, Lauer MS, Mukherjee DP, et al. The duration of pretreatment with ticlopidine prior to stenting is associated with the risk of procedure-related non-Q-wave myocardial infarctions. J Am Coll Cardiol 1998; 32(5): 1366–70.
Steinhubl SR, Ellis SG, Wolski K, et al. Ticlopidine pretreatment before coronary stenting is associated with sustained decrease in adverse cardiac events: data from the Evaluation of Platelet IIb/IIIa Inhibitor for Stenting (EPISTENT) Trial. Circulation 2001; 103(10): 1403–9.
Bhatt DL, Bertrand ME, Berger PB, et al. Meta-analysis of randomized and registry comparisons of ticlopidine with clopidogrel after stenting. J Am Coll Cardiol 2002; 39(1): 9–14.
Quinn MJ, Fitzgerald DJ. Ticlopidine and clopidogrel. Circulation 1999; 100(15): 1667–72.
Bertrand ME, Rupprecht HJ, Urban P, et al. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: the clopidogrel aspirin stent international cooperative study (CLASSICS). Circulation 2000; 102(6): 624–9.
Popma JJ, Berger P, Ohman EM, et al. Antithrombotic therapy during percutaneous coronary intervention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126(3 Suppl.): 576S–99S.
Chew DP, Bhatt DL, Robbins MA, et al. Effect of clopidogrel added to aspirin before percutaneous coronary intervention on the risk associated with C-reactive protein. Am J Cardiol 2001; 88(6): 672–4.
Assali AR, Salloum J, Sdringola S, et al. Effects of clopidogrel pretreatment before percutaneous coronary intervention in patients treated with glycoprotein IIb/IIIa inhibitors (abciximab or tirofiban). Am J Cardiol 2001; 88(8): 884–6.
Chan AW, Moliterno DJ, Berger PB, et al. Triple antiplatelet therapy during percutaneous coronary intervention is associated with improved outcomes including one-year survival: results from the Do Tirofiban and ReoProGive Similar Efficacy Outcome Trial (TARGET). J Am Coll Cardiol 2003; 42(7): 1188–95.
Topol EJ, Moliterno DJ, Herrmann HC, et al. Comparison of two platelet glycoprotein IIb/IIIa inhibitors, tirofiban and abciximab, for the prevention of ischemic events with percutaneous coronary revascularization. N Engl J Med 2001; 344(25): 1888–94.
Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled. JAMA 2002; 288(19): 2411–20.
Steinhubl SR, Berger PB, Brennan DM, et al. Optimal timing for the initiation of pre-treatment with 300mg clopidogrel before percutaneous coronary intervention. J Am Coll Cardiol 2006; 47(5): 939–43.
Muller I, Seyfarth M, Rudiger S, et al. Effect of a high loading dose of clopidogrel on platelet function in patients undergoing coronary stent placement. Heart 2001; 85(1): 92–3.
Kandzari DE, Berger PB, Kastrati A, et al. Influence of treatment duration with a 600-mg dose of clopidogrel before percutaneous coronary revascularization. J Am Coll Cardiol 2004; 44(11): 2133–6.
Patti G, Colonna G, Pasceri V, et al. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111(16): 2099–106.
von Beckerath N, Taubert D, Pogatsa-Murray G, et al. Absorption, metabolization, and antiplatelet effects of 300-, 600-, and 900-mg loading doses of clopidogrel: results of the ISAR-CHOICE (Intracoronary Stenting and Antithrombotic Regimen: Choose Between 3 High Oral Doses for Immediate Clopidogrel Effect) Trial. Circulation 2005; 112(19): 2946–50.
Montalescot G, Sideris G, Meuleman C, et al. A randomized comparison of high clopidogrel loading doses in patients with non-ST-segment elevation acute coronary syndromes: the ALBION (Assessment of the Best Loading Dose of Clopidogrel to Blunt Platelet Activation, Inflammation and Ongoing Necrosis) trial. J Am Coll Cardiol 2006; 48(5): 931–8.
Cuisset T, Frere C, Quilici J, et al. Benefit of a 600-mg loading dose of clopidogrel on platelet reactivity and clinical outcomes in patients with non-ST-segment elevation acute coronary syndrome undergoing coronary stenting. J Am Coll Cardiol 2006; 48(7): 1339–45.
Vivekananthan DP, Bhatt DL, Chew DP, et al. Effect of clopidogrel pretreatment on periprocedural rise in C-reactive protein after percutaneous coronary intervention. Am J Cardiol 2004; 94(3): 358–60.
Quinn MJ, Bhatt DL, Zidar F, et al. Effect of clopidogrel pretreatment on inflammatory marker expression in patients undergoing percutaneous coronary intervention. Am J Cardiol 2004; 93(6): 679–84.
CURRENT/OASIS 7: clopidogrel optimal loading dose usage to reduce recurrent events/optimal antiplatelet strategy for interventions [online]. Available from URL: http://clinicaltrials.gov/ct/show/NCT00335452 [Accessed 2007 Mar 30].
Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358(9281): 527–33.
Rabbat MG, Bavry AA, Bhatt DL, et al. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74(2): 129–36.
Pfisterer M, Brunner-La Rocca HP, Buser PT, et al. Late clinical events after clopidogrel discontinuation may limit the benefit of drug-eluting stents: an observational study of drug-eluting versus bare-metal stents. J Am Coll Cardiol 2006; 48(12): 2584–91.
Bavry AA, Kumbhani DJ, Helton TJ, et al. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119(12): 1056–61.
Ellis SG, Colombo A, Grube E, et al. Incidence, timing, and correlates of stent thrombosis with the polymeric paclitaxel drug-eluting stent: a TAXUS II, IV, V, and VI meta-analysis of 3,445 patients followed for up to 3 years. J Am Coll Cardiol 2007; 49(10): 1043–51.
Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356(10): 998–1008.
Spaulding C, Daemen J, Boersma E, et al. A pooled analysis of data comparing sirolimus-eluting stents with bare-metal stents. N Engl J Med 2007; 356(10): 989–97.
Kastrati A, Mehilli J, Pache J, et al. Analysis of 14 trials comparing sirolimus-eluting stents with bare-metal stents. N Engl J Med 2007; 356(10): 1030–9.
Grines CL, Bonow RO, Casey Jr DE, et al. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: a science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. Catheter Cardiovasc Interv 2007; 69(3): 334–40.
Helton TJ, Bhatt DL. Emerging options in antiplatelets: a review of novel therapies on the horizon for the treatment of acute coronary syndromes. Acute Coronary Syndromes 2006; 8(1): 2–8.
Sugidachi A, Asai F, Ogawa T, et al. The in vivo pharmacological profile of CS-747, a novel antiplatelet agent with platelet ADP receptor antagonist properties. Br J Pharmacol 2000; 129(7): 1439–46.
Niitsu Y, Jakubowski JA, Sugidachi A, et al. Pharmacology of CS-747 (prasugrel, LY640315), a novel, potent antiplatelet agent with in vivo P2Y12 receptor antagonist activity. Semin Thromb Hemost 2005; 31(2): 184–94.
Jernberg T, Payne CD, Winters KJ, et al. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J 2006; 27(10): 1166–73.
Wiviott SD, Antman EM, Winters KJ, et al. Randomized comparison of prasugrel (CS-747, LY640315), a novel thienopyridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention: results of the Joint Utilization of Medications to Block Platelets Optimally (JUMBO)-TIMI 26 trial. Circulation 2005; 111(25): 3366–73.
Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357(20): 2001–15.
Bhatt DL. Intensifying platelet inhibition: navigating between Scylla and Charybdis [editorial]. N Engl J Med 2007; 357(20): 2078–81.
Wiviott SD, Trenk D, Frelinger AL, et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007; 116(25): 2923–32.
van Giezen JJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Semin Thromb Hemost 2005; 31(2): 195–204.
Husted S, Emanuelsson H, Heptinstall S, et al. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin. Eur Heart J 2006; 27(9): 1038–47.
The DISPERSE 2 trial: safety, tolerability, and preliminary efficacy of AZD6140, the first oral reversible ADP receptor antagonist compared with clopidogrel in patients with non-ST elevation acute coronary syndrome [abstract no. 2906]. Presented at the 78th Annual Scientific Sessions of the American Heart Association; 2005 Nov 13–16; Dallas (TX).
Serebruany VL. Dyspnoea after AZD6140: safety first? [letter]. Eur Heart J 2006; 27(12): 1505.
ClinicalTrials.gov. A randomised, double-blind, parallel group, phase 3, efficacy and safety study of AZD6140 compared with clopidogrel for prevention of vascular events in patients with non-st or st elevation acute coronary syndromes (ACS) [PLATO: a study of PLATelet Inhibition and Patient Outcomes] [online]. Available from URL: http://clinicaltrials.gov/ct/show/NCT00391872 [Accessed 2007 Mar 30].
Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100(9): 1261–75.
Storey RF, Wilcox RG, Heptinstall S. Comparison of the pharmacodynamic effects of the platelet ADP receptor antagonists clopidogrel and AR-C69931MX in patients with ischaemic heart disease. Platelets 2002; 13(7): 407–13.
Storey RF, Judge HM, Wilcox RG, et al. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb Haemost 2002; 88(3): 488–94.
Fugate SE, Cudd LA. Cangrelor for treatment of coronary thrombosis. Ann Pharmacother 2006; 40(5): 925–30.
Greenbaum AB, Grines CL, Bittl JA, et al. Initial experience with an intravenous P2Y12 platelet receptor antagonist in patients undergoing percutaneous coronary intervention: results from a 2-part, phase II, multicenter, randomized, placebo- and active-controlled trial. Am Heart J 2006; 151(3): 689.el–e10.
ClinicalTrials.gov. A clinical trial comparing cangrelor to clopidogrel in subjects who require percutaneous coronary intervention [online]. Available from URL: http://clinicaltrials.gov/ct/show/NCT00305162 [Accessed 2007 Mar 30].
ClinicalTrials.gov. A clinical trial comparing treatment with cangrelor (in combination with usual care) to usual care, in subjects who require percutaneous coronary intervention [online]. Available from URL: http://clinicaltrials.gov/ct/ show/NCT00385138 [Accessed 2007 Mar 30].
Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2(1): 15–28.
Bhatt DL. Aspirin resistance: more than just a laboratory curiosity. J Am Coll Cardiol 2004; 43(6): 1127–9.
Wang TH, Bhatt DL, Topol EJ. Aspirin and clopidogrel resistance: an emerging clinical entity. Eur Heart J 2006; 27(6): 647–54.
Eikelboom JW, Hirsh J, Weitz JI, et al. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002; 105(14): 1650–5.
Mason PJ, Jacobs AK, Freedman JE. Aspirin resistance and atherothrombotic disease. J Am Coll Cardiol 2005; 46(6): 986–93.
Quick AJ. Salicylates and bleeding: the aspirin tolerance test. Am J Med Sci 1966; 252(3): 265–9.
Homoncik M, Jilma B, Hergovich N, et al. Monitoring of aspirin (ASA) pharmaco-dynamics with the platelet function analyzer PFA-100. Thromb Haemost 2000; 83(2): 316–21.
Patrono C, Coller B, Fitzgerald GA, et al. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126(3 Suppl.): 234S–64S.
Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet 2006; 367(9510): 606–17.
Califf RM, DeLong ER, Ostbye T, et al. Underuse of aspirin in a referral population with documented coronary artery disease. Am J Cardiol 2002; 89(6): 653–61.
Rand ML, Leung R, Packham MA. Platelet function assays. Transfus Apheresis Sci 2003; 28(3): 307–17.
Michelson AD. Platelet function testing in cardiovascular diseases. Circulation 2004; 110(19): e489–93.
Patrono C. Aspirin resistance: definition, mechanisms and clinical read-outs. J Thromb Haemost 2003; 1(8): 1710–3.
Schonbeck U, Sukhova GK, Graber P, et al. Augmented expression of cyclooxy-genase-2 in human atherosclerotic lesions. Am J Pathol 1999; 155(4): 1281–91.
Maclouf J, Folco G, Patrono C. Eicosanoids and iso-eicosanoids: constitutive, inducible and transcellular biosynthesis in vascular disease. Thromb Haemost 1998; 79(4): 691–705.
Karim S, Habib A, Levy-Toledano S, et al. Cyclooxygenase-1 and -2 of endothelial cells utilize exogenous or endogenous arachidonic acid for transcellular production of thromboxane. J Biol Chem 1996; 271(20): 12042–8.
Rocca B, Secchiero P, Ciabattoni G, et al. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci USA 2002; 99(11): 7634–9.
Catella F, Healy D, Lawson JA, et al. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Natl Acad Sci USA 1986; 83(16): 5861–5.
Nicholson NS, Panzer-Knodle SG, Haas NF, et al. Assessment of platelet function assays. Am Heart J 1998; 135 (5 Pt 2 Su): S170–8.
Kundu SK, Heilmann EJ, Sio R, et al. Description of an in vitro platelet function analyzer: PFA-100. Semin Thromb Hemost 1995; 21 Suppl. 2: 106–12.
Harrison P, Segal H, Blasbery K, et al. Screening for aspirin responsiveness after transient ischemic attack and stroke: comparison of 2 point-of-care platelet function tests with optical aggregometry. Stroke 2005; 36(5): 1001–5.
Hennekens CH, Sacks FM, Tonkin A, et al. Additive benefits of pravastatin and aspirin to decrease risks of cardiovascular disease: randomized and observational comparisons of secondary prevention trials and their meta-analyses. Arch Intern Med 2004; 164(1): 40–4.
Carney RM, Freedland KE, Eisen SA, et al. Adherence to a prophylactic medication regimen in patients with symptomatic versus asymptomatic ischemic heart disease. Behav Med 1998; 24(1): 35–9.
Schwartz KA, Schwartz DE, Ghosheh K, et al. Compliance as a critical consideration in patients who appear to be resistant to aspirin after healing of myocardial infarction. Am J Cardiol 2005; 95(8): 973–5.
Tantry US, Bliden KP, Gurbel PA. Overestimation of platelet aspirin resistance detection by thrombelastograph platelet mapping and validation by conventional aggregometry using arachidonic acid stimulation. J Am Coll Cardiol 2005; 46(9): 1705–9.
Tohgi H, Konno S, Tamura K, et al. Effects of low-to-high doses of aspirin on platelet aggregability and metabolites of thromboxane A2 and prostacyclin. Stroke 1992; 23(10): 1400–3.
Cerletti C, Dell’Elba G, Manarini S, et al. Pharmacokinetic and pharmacodynamic differences between two low dosages of aspirin may affect therapeutic outcomes. Clin Pharmacokinet 2003; 42(12): 1059–70.
Serebruany VL, Steinhubl SR, Berger PB, et al. Analysis of risk of bleeding complications after different doses of aspirin in 192,036 patients enrolled in 31 randomized controlled trials. Am J Cardiol 2005; 95(10): 1218–22.
Maree AO, Curtin RJ, Dooley M, et al. Platelet response to low-dose enteric-coated aspirin in patients with stable cardiovascular disease. J Am Coll Cardiol 2005; 46(7): 1258–63.
Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet 1985; 10(2): 164–77.
Williams FM. Clinical significance of esterases in man. Clin Pharmacokinet 1985; 10(5): 392–403.
Curtis JP, Krumholz HM. The case for an adverse interaction between aspirin and non-steroidal anti-inflammatory drugs: is it time to believe the hype? J Am Coll Cardiol 2004; 43(6): 991–3.
Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med 2001; 345(25): 1809–17.
Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol 2005; 45(8): 1295–301.
Kimmel SE, Berlin JA, Reilly M, et al. The effects of nonselective non-aspirin non-steroidal anti-inflammatory medications on the risk of nonfatal myocardial infarction and their interaction with aspirin. J Am Coll Cardiol 2004; 43(6): 985–90.
Bhatt DL. NSAIDS and the risk of myocardial infarction: do they help or harm? Eur Heart J 2006; 27(14): 1635–6.
Steinhubl SR. The use of anti-inflammatory analgesics in the patient with cardiovascular disease: what a pain. J Am Coll Cardiol 2005; 45(8): 1302–3.
Zimmermann N, Wenk A, Kim U, et al. Functional and biochemical evaluation of platelet aspirin resistance after coronary artery bypass surgery. Circulation 2003; 108(5): 542–7.
Golaski J, Chlopicki S, Golaski R, et al. Resistance to aspirin in patients after coronary artery bypass grafting is transient: impact on the monitoring of aspirin antiplatelet therapy. Ther Drug Monit 2005; 27(4): 484–90.
Payne DA, Jones CI, Hayes PD, et al. Platelet inhibition by aspirin is diminished in patients during carotid surgery: a form of transient aspirin resistance? Thromb Haemost 2004; 92(1): 89–96.
Weber AA, Zimmermann KC, Meyer-Kirchrath J, et al. Cyclooxygenase-2 in human platelets as a possible factor in aspirin resistance [letter]. Lancet 1999; 353(9156): 900.
Kawasaki T, Ozeki Y, Igawa T, et al. Increased platelet sensitivity to collagen in individuals resistant to low-dose aspirin. Stroke 2000; 31(3): 591–5.
Macchi L, Christiaens L, Brabant S, et al. Resistance to aspirin in vitro is associated with increased platelet sensitivity to adenosine diphosphate. Thromb Res 2002; 107(1–2): 45–9.
Christiaens L, Macchi L, Herpin D, et al. Resistance to aspirin in vitro at rest and during exercise in patients with angiographically proven coronary artery disease. Thromb Res 2002; 108(2–3): 115–9.
Morrow JD, Frei B, Longmire AW, et al. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers: smoking as a cause of oxidative damage. N Engl J Med 1995; 332(18): 1198–203.
Davi G, Ciabattoni G, Consoli A, et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplemention. Circulation 1999; 99(2): 224–9.
Friend M, Vucenik I, Miller M. Research pointers: platelet responsiveness to aspirin in patients with hyperlipidaemia. BMJ 2003; 326(7380): 82–3.
Davi G, Guagnano MT, Ciabattoni G, et al. Platelet activation in obese women: role of inflammation and oxidant stress. JAMA 2002; 288(16): 2008–14.
Patrono C, Falco A, Davi G. Isoprostane formation and inhibition in atherothrombosis. Curr Opin Pharmacol 2005; 5(2): 198–203.
Csiszar A, Stef G, Pacher P, et al. Oxidative stress-induced isoprostane formation may contribute to aspirin resistance in platelets. Prostaglandins Leukot Essent Fatty Acids 2002; 66(5–6): 557–8.
Cipollone F, Ciabattoni G, Patrignani P, et al. Oxidant stress and aspirin-insensitive thromboxane biosynthesis in severe unstable angina. Circulation 2000; 102(9): 1007–13.
Ziegler S, Alt E, Brunner M, et al. Influence of systemic inflammation on efficiency of antiplatelet therapy in PAOD patients. Ann Hematol 2004; 83(2): 92–4.
Serebruany VL, Malinin AI, Jerome SD, et al. Effects of clopidogrel and aspirin combination versus aspirin alone on platelet aggregation and major receptor expression in patients with heart failure: the Plavix Use for Treatment Of Congestive Heart Failure (PLUTO-CHF) trial. Am Heart J 2003; 146(4): 713–20.
Borna C, Lazarowski E, van Heusden C, et al. Resistance to aspirin is increased by ST-elevation myocardial infarction and correlates with adenosine diphosphate levels. Thromb J 2005; 3: 10.
Cambria-Kiely JA, Gandhi PJ. Aspirin resistance and genetic polymorphisms. J Thromb Thrombolysis 2002; 14(1): 51–8.
Halushka MK, Walker LP, Halushka PV. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Therap 2003; 73(1): 122–30.
Cipollone F, Toniato E, Martinotti S, et al. A polymorphism in the cyclooxygenase 2 gene as an inherited protective factor against myocardial infarction and stroke. JAMA 2004; 291(18): 2221–8.
Quinn MJ, Topol EJ. Common variations in platelet glycoproteins: pharmacogenomic implications. Pharmacogenomics 2001; 2(4): 341–52.
Szczeklik A, Undas A, Sanak M, et al. Relationship between bleeding time, aspirin and the P1A1/A2 polymorphism of platelet glycoprotein IIIa. Br J Haematol 2000; 110(4): 965–7.
Andrioli G, Minuz P, Solero P, et al. Defective platelet response to arachidonic acid and thromboxane A(2) in subjects with P1(A2) polymorphism of beta(3) subunit (glycoprotein IIIa). Br J Haematol 2000; 110(4): 911–8.
Michelson AD, Furman MI, Goldschmidt-Clermont P, et al. Platelet GP IIIa P1(A) polymorphisms display different sensitivities to agonists. Circulation 2000; 101(9): 1013–8.
Macchi L, Christiaens L, Brabant S, et al. Resistance in vitro to low-dose aspirin is associated with platelet P1A1 (GP IIIa) polymorphism but not with C807T(GP Ia/IIa) and C-5T Kozak (GP Ibalpha) polymorphisms. J Am Coll Cardiol 2003; 42(6): 1115–9.
Jefferson BK, Foster JH, McCarthy JJ, et al. Aspirin resistance and a single gene. Am J Cardiol 2005; 95(6): 805–8.
Pontiggia L, Lassila R, Pederiva S, et al. Increased platelet-collagen interaction associated with double homozygosity for receptor polymorphisms of platelet GPIa and GPIIIa. Arterioscler Thromb Vasc Biol 2002; 22(12): 2093–8.
Grotemeyer KH, Scharafinski HW, Husstedt IW. Two-year follow-up of aspirin responder and aspirin non responder: a pilot-study including 180 post-stroke patients. Thromb Res 1993; 71(5): 397–403.
Chen WH, Lee PY, Ng W, et al. Aspirin resistance is associated with a high incidence of myonecrosis after non-urgent percutaneous coronary intervention despite clopidogrel pretreatment. J Am Coll Cardiol 2004; 43(6): 1122–6.
Eikelboom JW, Hankey GJ, Thorn J, et al. Enhanced antiplatelet effect of clopidogrel in patients whose platelets are least inhibited by aspirin: a randomized crossover trial. J Thrombosis Haemost 2005; 3(12): 2649–55.
Nguyen TA, Diodati JG, Pharand C. Resistance to clopidogrel: a review of the evidence. J Am Coll Cardiol 2005; 45(8): 1157–64.
Barsky AA, Arora RR. Clopidogrel resistance: myth or reality? J Cardiovasc Pharmacol Therap 2006; 11(1): 47–53.
Serebruany VL, Malinin AI, Atar D, et al. Consistent platelet inhibition during long-term maintenance-dose clopidogrel therapy among 359 compliant outpatients with documented vascular disease. Am Heart J 2007; 153(3): 371–7.
Muller I, Besta F, Schulz C, et al. Prevalence of clopidogrel non-responders among patients with stable angina pectoris scheduled for elective coronary stent placement. Thromb Haemost 2003; 89(5): 783–7.
Gurbel PA, Bliden KP, Hiatt BL, et al. Clopidogrel for coronary stenting: response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation 2003; 107(23): 2908–13.
Labarthe B, Theroux P, Angioi M, et al. Matching the evaluation of the clinical efficacy of clopidogrel to platelet function tests relevant to the biological properties of the drug. J Am Coll Cardiol 2005; 46(4): 638–45.
Aleil B, Ravanat C, Cazenave JP, et al. Flow cytometric analysis of intraplatelet VASP phosphorylation for the detection of clopidogrel resistance in patients with ischemic cardiovascular diseases. J Thromb Haemost 2005; 3(1): 85–92.
Taubert D, Kastrati A, Harlfinger S, et al. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost 2004; 92(2): 311–6.
Lau WC, Gurbel PA, Watkins PB, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004; 109(2): 166–71.
Lau WC, Waskell LA, Watkins PB, et al. Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation: a new drug-drug interaction. Circulation 2003; 107(1): 32–7.
Saw J, Steinhubl SR, Berger PB, et al. Lack of adverse clopidogrel-atorvastatin clinical interaction from secondary analysis of a randomized, placebo-controlled clopidogrel trial. Circulation 2003; 108(8): 921–4.
Serebruany VL, Midei MG, Malinin AI, et al. Absence of interaction between atorvastatin or other statins and clopidogrel: results from the interaction study. Arch Intern Med 2004; 164(18): 2051–7.
Serebruany VL, Malinin AI, Callahan KP, et al. Statins do not affect platelet inhibition with clopidogrel during coronary stenting. Atherosclerosis 2001; 159(1): 239–41.
Serebruany VL, Steinhubl SR, Berger PB, et al. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45(2): 246–51.
Fontana P, Dupont A, Gandrille S, et al. Adenosine diphosphate-induced platelet aggregation is associated with P2Y12 gene sequence variations in healthy subjects. Circulation 2003; 108(8): 989–95.
Fontana P, Gaussem P, Aiach M, et al. P2Y12 H2 haplotype is associated with peripheral arterial disease: a case-control study. Circulation 2003; 108(24): 2971–3.
von Beckerath N, von Beckerath O, Koch W, et al. P2Y12 gene H2 haplotype is not associated with increased adenosine diphosphate-induced platelet aggregation after initiation of clopidogrel therapy with a high loading dose. Blood Coagul Fibrinolysis 2005; 16(3): 199–204.
Matetzky S, Shenkman B, Guetta V, et al. Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation 2004; 109(25): 3171–5.
Acknowledgments
Dr Bhatt has received research grants from Bristol-Myers Squibb, Eisai, Ethicon, Sanofi-Aventis and The Medicines Company. He has received honoraria (over 2 years ago) from AstraZeneca, Bristol-Myers Squibb, Centocor, Daiichi-Sankyo, Eisai, Eli Lilly, GlaxoSmithKline, Millennium, Paringenix, PDL, Sanofi-Aventis, Schering-Plough, The Medicines Company, and TNS Healthcare (proceeds donated to non-profit organizations). Dr Bhatt has also been in the Speaker’s bureau of Bristol-Myers Squibb, Sanofi-Aventis, The Medicines Company and acted as a Consultant/Advisory Board for AstraZeneca, Bristol-Myers Squibb, Cardax, Centocor, Cogentus, Daiichi-Sankyo, Eisai, Eli Lilly, GlaxoSmithKline, Johnson & Johnson, McNeil, Medtronic, Millennium, Otsuka, Paringenix, PDL, Philips, Portola, Sanofi-Aventis, Schering-Plough, The Medicines Company, TNS Healthcare, and Vertex (honoraria donated to non-profit organizations for over 2 years). He has also given expert testimony regarding clopidogrel (compensation donated to a nonprofit organization).
The Cleveland Clinic Coordinating Center currently receives or has received research funding from Abraxis, Alexion Pharma, AstraZeneca, Atherogenics, Aventis, Biosense Webster, Biosite, Boehringer Ingelheim, Boston Scientific, Bristol-Myers Squibb, Cardionet, Centocor, Converge Medical Inc., Cordis, Dr Reddy’s, Edwards Lifesciences, Esperion, GE Medical, Genentech, Gilford, GlaxoSmithKline, Guidant, Johnson & Johnson, Kensey-Nash, Lilly, Medtronic, Merck, Mytogen, Novartis, Novo Nordisk, Orphan Therapeutics, P&G Pharma, Pfizer, Roche, Sankyo, Sanofi-Aventis, Schering-Plough, Scios, St Jude Medical, Takeda, The Medicines Company, VasoGenix, and Viacor.
Dr Depta has no conflicts of interest relevant to the content of this manuscript. No sources of funding were used to assist in the preparation of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Depta, J.P., Bhatt, D.L. Aspirin and Platelet Adenosine Diphosphate Receptor Antagonists in Acute Coronary Syndromes and Percutaneous Coronary Intervention. Am J Cardiovasc Drugs 8, 91–112 (2008). https://doi.org/10.1007/BF03256587
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03256587