
Summary
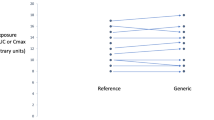
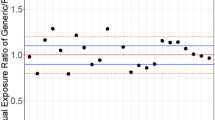
A generic drug product (T) in order to be approved for marketing authorization a bioequivalence trial is required. In the trial the generic product is compared to the innovator product (R) in terms of the pharmacokinetic parameters AUC and Cmax. The regulatory requirement for bioequivalence is that the 90% confidence intervals for the ratio (T/R) of the generic to innovator product pharmacokinetic parameter averages lies within the limits (80%, 125%). The design of the trial is usually a two-period crossover. This design has the limitation that if the statistical analysis reveal significant sequence effect then the bioequivalence results may be biased and their interpretation is difficult. The sequence effect is confounding with the unequal residual effect and with the formulation by period interaction. Since the existence of the sequence effect questions the quality of the trial, the applicant should provide possible explanations and information on the subjects, the trial conditions, the clinical settings and the assay methodology. An additional statistical analysis on the data from the first period of the trial may support the bioequivalence. If it is proven that the sequence effect is a true effect then the generic may be approved for marketing authorization.
Article PDF
Similar content being viewed by others

Avoid common mistakes on your manuscript.
References
Chow S.C., Liu J.P. (1992): Design and analysis of bioavailability and bioequivalence studies. New York: Marcel Dekker.
Zintzaras E., Bouka P. (1999): Bioequivalence studies: biometrical concepts of alternative designs and pooled analysis. Eur. J. Drug Metab. Pharmacokinet., 24, pp 225–232.
CPMP: NOte for guidance on the investigation of bioavailability and bioequivalence. In: the Rules Governing Medicinal Products for Human Use in the Member States of the European Community, Volume III, Addendum No 2, Guidelines and the Quality, Safety & Efficacy of Medicinal Products for Human Use, pp 149–168.
Bolton S. (1997): Pharmaceutical Statistics. New York: Marcel Dekker.
Schuirmann D.J. (1990): Statistical evaluation of Bioequivalence Studies. In K.N. Sharma, K.K. Sharma & P. Sen. Eds. Generics Drugs, Bioequivalence and Pharmacokinetics: Delhi: UCMS.
Sauter R., Steinijans V.W., Diletti E., Bohm A., Schulz H.U. (1992): Presentation of results from bioequivalence studies. Int. J. Clin. Pharmacol. Ther. Toxicol., 30, pp 233–256.
Hauschke D., Steinijans V. (1997): Cross-over trials for bioequivalence assessment. In Vollmar J. Eds:. Biometrics in the Pharmaceutical Industry. Stuttgard: Gustav Fischer.
Mead R. (1988): The design of experiments. Cambridge: Cambridge University Press.
Jones B., Kenward M.G. (1995): Design and Analysis of Cross-Over Trials. London: Chapman and Hall.
Ki F.Y., Liu J.P., Wang W., Chow S.C. (1995): The impact of outlying subjects on decision of bioequivalence. J Biopharm Stat, 5, pp 71–94.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Zintzaras, E. The existence of sequence effect in cross-over Bioequivalence trials. Eur. J. Drug Metab. Pharacokinet. 25, 241–244 (2000). https://doi.org/10.1007/BF03192321
Received:
Issue Date:
DOI: https://doi.org/10.1007/BF03192321