
Summary
Introduction
Bioequivalence between a reference and a generic drug is based on the hypothesis that a ± 20% change in blood exposure (or ± 10% for drugs with narrow therapeutic index, NTI) following the generic/reference switch will not have any therapeutic consequences. However, the individual exposure ratio between generic and reference can be higher than 1.20 (or 1.10). This study aims to analyse the different parameters influencing the individual exposure ratio, hence the conditions for reference/generic interchangeability.
Methods
Bioequivalence studies with a double cross-over design for a virtual drug were simulated using 100 random sets of 12, 24, 48 or 100 pairs of areas under the curve (AUC), varying the generic/reference AUC geometric mean ratios between 0.80 and 1.25 and the within-subject exposure variance of the reference and the generic formulations.
Results
The proportion of subjects with an exposure generic/reference ratio outside the ± 10% or ± 20% acceptance intervals increases when (1) the reference within-subject variance increases; (2) the ratio of the generic within-subject variance on the reference within-subject variance increases; and (3) the generic/reference mean AUC ratio diverges from 1.0. When only considering replicated administrations of the reference, the individual exposure ratio increases with the within-subject variance, yielding values outside the usually accepted individual exposure ratio range of 0.5 to 2 for drugs with narrow therapeutic index as soon as the within-subject variance standard deviation is ≥ 0.25 (equivalent to within-patient CV% > 25%).
Conclusions
Interchangeability between reference and generic formulations, especially for drugs with narrow therapeutic index can only be assumed if, the within-subject variance of generic is less or equal to the within-subject variance of reference or, if this is not the case, if the distribution of the generic/generic individual exposure ratios is included within the therapeutic margins of the reference drug.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Therapeutic imbalance has been reported after switching patients from generic (G) to reference (R) drugs, especially for those with a narrow therapeutic index such as some anti-epileptics [1] and l-thyroxin [2]. However, the mechanism of therapeutic imbalance remains unclear and very likely multifactorial. Therefore, certain regulatory authorities do not recommend any replacement during treatment for a list of medicines (no-switch list), especially those with narrow therapeutic index. It means that the criteria of Average Bioequivalence required for marketing authorisation of generic drugs are insufficient to guarantee clinical interchangeability at the individual level i.e. the absence of a therapeutic impact of switching between bioequivalent formulations in all patients. Regulatory agencies have proposed adaptating the Average Bioequivalence methodology by expanding the acceptation limits for drugs with large within-subject variance (CV > 30%) and narrowing them for drugs with narrow therapeutic index [3,4,5]. The acceptance limits of the European Medicine Agency (EMA) for drugs with narrow therapeutic index were narrowed to 0.9–1.11 for the geometric mean ratio of generic/reference while the Food and Drug Administration (FDA) has proposed adaptative acceptance limits according to the reference-scaled average bioequivalence methodology (RSABE) [5,6,7,8].
The Average Bioequivalence methodology and reference-scaled average bioequivalence criteria
In the usual Average Bioequivalence approach, bioequivalence is accepted if the difference between the logarithmic means of exposure (generic-reference) falls between preset regulatory limits: ln (0.8) ≤ µT - µR ≤ ln (1.25) where µT and µR are the population average exposure of the log transformed measure for the generic and reference formulations (ln (0.8) = −0.224 and ln (1.25) = 0.224), corresponding to the limits of the geometric mean ratio between 0.8 and 1.25.
With the reference-scaled average bioequivalence methodology adopted by the FDA, the acceptable limits of µT - µR are calculated as follows:
where σW0 is a predetermined constant set by the regulatory agency and σWR the standard deviation (SD) of the WS variance of reference.
According to the equation:
-
(1)
\({\sigma }_{\mathrm{WR}}={\sigma }_{\mathrm{W}0}\) indicates the implied limits are equal to the standard Average Bioequivalence,
-
(2)
\({\sigma }_{\mathrm{WR}}>{\sigma }_{\mathrm{W}0}\) indicates the implied limits of bioequivalence acceptance are wider than the standard limits (0.8–1.25 for geometric mean ratio),
-
(3)
\({\sigma }_{\mathrm{WR}}<{\sigma }_{\mathrm{W}0}\) indicates the implied limits are narrower than the standard limits.
The FDA has set σW0 at 0.25 [5, 7] and this approach is applied for drugs with high within-subject variance (CV > 30%), which allows enlargement of bioequivalence acceptance limits for such drugs. For drugs with narrow therapeutic index, σW0 is set at 0.10, corresponding to bioequivalence limits of 0.9–1.11 for geometric mean ratio of generic/reference exposure. If σWR is < 0.10, the bioequivalence acceptance limits will then be narrower.
The alternative EMA strategy has been to consider a maximal reduction of Average Bioequivalence limits to 0.9–1.11 for the geometric mean ratio for drugs with narrow therapeutic index.
In addition, for drugs with narrow therapeutic index, the FDA (and not the EMA) has proposed that the within-subject variance of generic should not be higher than that of reference to ensure switchability [6]. However, such calculations require the full replication of the cross-over design of bioequivalence studies, with two periods of exposure for reference and two also for generic.
In order to determine if the the standard deviation (σ) values of the within-subject variance of generic (test) and reference are comparable, the upper limit of the σWT/σWR ratio has to be evaluated. The FDA requires that the upper limit of the 90% equal-tails confidence interval for σWT/σWR is ≤ 2.5 [6].
The (1 − α)100% CI for \(\frac{{\sigma }_{WT}}{{\sigma }_{WR}}\) is given by
According to the equation:
-
SWT is the estimate of σWT with v1 as the degree of freedom
-
SWR is the estimate of σWR with v2 as the degree of freedom
-
Fα/2,v1,v2 is the value of the F-distribution with v1 (numerator) and v2 (denominator) degrees of freedom that has probablity of α/2 to its right.
-
F1−α/2,v1,v2 is the value of the F-distribution with v1 (numerator) and v2 (denominator) degrees of freedom that has probablity of 1 − α/2 to its right.
-
Here, α = 0.1.
For the EMA, the reference-scaled average bioequivalence criterion applies only to Cmax, not to the AUC [3]: if the replicated-reference within-subject CV of lnCmax > 30% (highly variable drugs) then bioequivalence limits are widened using scaled-average bioequivalence. Under scaled-average-bioequivalence, [U, L] = exp [± k·SWR], where U and L are the upper and lower limits of the acceptance range, k the regulatory constant set to 0.760 and SWR the within-subject standard deviation of the reference lnCmax.
If replicated-reference within-subject CV of lnCmax. ≥ 50%, then Cmax. acceptance limits are widened to a maximum of [69.84–143.19%].
For drugs with narrow therapeutic index, EMA has reduced the Average Bioequivalence limits, etc.
acceptance limits for 90% CI of geometric mean ratio of both AUC and Cmax. to [90–111%] but does not require any within-subject variance comparison between reference and generic.
Study rationale
The accepted bioequivalence methodology for marketing authorisation of generic drugs is based on the principle that differences in exposure within ± 20% (or 10% for drugs with narrow therapeutic index) between generic and reference should have no therapeutic impact. This is a generally accepted clinical pharmacological principle at the individual level. However, it has been used for comparison of exposure means between generics and reference drugs, which cannot guarantee the absence of therapeutic consequence in all, or most, individuals after a switch between formulations. Indeed, the individual exposure ratio can be outside the interval (80–120%) (or [90–110%] for drugs with narrow therapeutic index) in a rather “significant” proportion of subjects even if the Average Bioequivalence criteria are met [9].
We investigated the determinants of the generic/reference individual exposure ratio to propose additional criteria that could guarantee the absence of therapeutic impact of the switch of one reference to one generic during chronic treatment in individuals.
Methods
We successively calculated the individual exposure ratios derived from two available EMA data sets and simulated in silico bioequivalence studies following different scenarios by varying the parameters affecting bioequivalence.
Distribution of individual exposure ratios in two EMA datasets of bioequivalence studies
We studied the two EMA individual datasets of bioequivalence studies provided through the EMA Q&A document on bioequivalence [4]. The first (EMA dataset 2) corresponds to a three-period bioequivalence study with replicated administration of the reference drug to 24 healthy subjects. The second (EMA dataset 1) corresponds to a replicated cross-over design with 4 exposure periods in 78 subjects (69 subjects with complete data), two periods with the reference and two with the generic formulation. For each subject, we calculated and plotted the individual exposure ratios (IER) of the reference versus itself and of the generic versus the reference. In both bioequivalence studies, the usual criteria of bioequivalence acceptance for the generic compared to the reference were met with the 90% CI of the geometric mean ratio totally included within the standard interval of 80 to 125%.
In silico studies
We performed 100 sets of random lognormal simulations (µ and SD values) to mimic a 2 × 4 cross-over design (reference 1, reference 2, generic 1 and generic 2) for a virtual drug and an increasing number of subjects of 12, 24, 48 and 100 (script of simulation provided as supplemental data).
For comparison of reference 1 to reference 2: the simulation consisted in log normal simulation for reference 1 with a mean AUC fixed to 100 µg × h/L and simulation of the IER R1/R2 with a mean ratio = 1. For R1 and R2, different levels of WSv were investigated using SD values from 0.05 to 0.40 (same for references 1 and 2 but with different seed for simulations). Finally, the proportion of patients with reference 1/reference 2 IER outside the 10% or 20% acceptance limits was calculated.
Similar calculations were made to compare reference and generic: (i) in log normal simulation for reference 1 with a mean AUC fixed to 100 µg × h/L and simulation of the individual exposure ratio R1/G1 with variation of the ratio between 80 and 125%; (ii) values of the standard deviation SD of the within-subject variance were varied from 0.05 to 0.80 for the generic and from 0.1 to 0.3 for the reference; (iii) the proportions of patients with generic/reference IER outside the 10% or 20% ABE acceptance limits were calculated.
The mean and SD of the proportions of individuals outside the 10% and 20% acceptance ranges, the geometric mean ratios and their corresponding 90% CI limits across the 100 sets of simulations were calculated.
All calculations were performed using log-transformed simulated data. We assumed subject-by-formulation interaction to be negligible. All the analyses were performed in R version 4.2.1 using the Tidyverse [10] frameworks.
Results
Description of the individual exposure ratios (IERs) in the EMA datasets
Individual exposure ratios were explored in two steps: The first one corresponds to the classical bioequivalence study comparing exposure of a generic to a reference with a two-period cross-over design. This situation is meant to ensure interchangeability between a reference formulation and a generic in patients during chronic treatment, with the limitation that only single doses are administered.
The second step investigates individual exposure ratios after replicated single-dose administration of the reference, which may be seen as more representative of a chronic treatment. However, here again, only two periods of exposure are compared, with a wash-out period between them.
Individual exposure ratios between the reference and generic drugs
The objective was to evaluate the dispersion of individual values of generic/reference individual exposure ratio when the Average Bioequivalence criteria are met. Since Average Bioequivalence acceptance is based on a variation of ± 20% of the mean exposure ratio (or ± 10% for drugs with narrow therapeutic index), we calculated (from raw exposure data) the percentage of patients with individual exposure ratio outside the ranges of ± 20 and ± 10%.
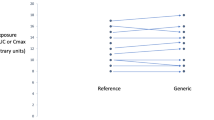
Figure 1 shows the generic/reference individual exposure ratios in the EMA dataset bioequivalence Q&A document [4], corresponding to a bioequivalence study performed in 24 subjects. The generic/reference geometric mean AUC ratio (× 100) was 102.26% with 90% CI = 97.32%–107.46%. Then the Average Bioequivalence criterion was met, even with respect to the more drastic acceptance range for a medicine with narrow therapeutic index (90–111.11% for GMR following the EMA, 90–112% following the Canadian Agency). However, as shown in Fig. 1, six subjects (25%) were out of the 20% acceptance range and 14 subjects (58%) out of the 10% acceptance range. Figure 2 depicts the same individual exposure data corresponding to the EMA dataset from a fully replicated cross-over bioequivalence study in 69 subjects. The Average Bioequivalence criteria are borderline but met, with a geometric mean AUC ratio (× 100) = 115.66% and 90% CI = 107.11%–124.89%. When only considering data from the first dosing periods of the reference and generic, 48 out of the 69 subjects with complete data (69%) are outside the ± 20% acceptance range.

Individual generic/reference exposure ratio in 24 subjects (EMA dataset 2). Geometric mean ratio = 102%; IC 90% = 97%–107%; 6/24 individual exposure ratios (25%) are outside the ± 20% acceptance interval (dashed red lines); 14/24 (58%) are outside the ± 10% interval (dashed blue lines)

Individual generic/reference exposure ratio in 69 subjects (EMA dataset 1). Geometric mean ratio = 115%, IC 90% = (107%–124%); 48/69 subjects (69%) are outside the ± 20% acceptance interval
Individual exposure ratios after repeated administration of the reference drug
Further examination of the EMA datasets [4] shows that AUC (and Cmax) values of the active substance vary with time according to the within-subject variance of exposure to the reference drug (Figs. 3 and 4).

Reference/reference individual exposure ratios in 24 subjects (EMA dataset 2). Geometric mean ratio = 100%, 90% CI = (96%–107%). CV (%) of within-subject variability derived from the ANOVA residual variance = 11.5%; 4/24 subjects (16.6%) are outside the ± 20% interval (dashed red lines) and 11/24 (45%) outside the ± 10% acceptance interval (blue lines)

Reference/reference individual exposure ratios in 69 subjects (EMA dataset 1); geometric mean ratio = 110%, 90% CI = (97%–125%), WSv CV (%) = 47%; 41/69 subjets (59%) are outside the 20% interval
In 24 subjects with two periods of administration of the reference and one of the generic, the individual AUC ratio between the two administrations of the reference is outside the ± 20% range for 4 of them (16.6%) and outside the ± 10% range for 9 (37%) (Fig. 3). As expected, the average bioequivalence criterion is met, with (reference 1/reference 2) mean AUC ratio = 102.20% and 90% CI = 96.72%–107.10%. The corresponding within-subject geometric coefficient of variation (derived from the SD of the ANOVA residual variance) provided in the EMA document is 11.14%, which can be regarded as rather low variability, compatible with a drug with narrow therapeutic index. For coefficient of variation (CV) values < 40%, the SD of within-subject variance (SDWSv) is very close to the CV value (× 100). For higher CV values, SDWSv is slighltly lower than the CV. Indeed CV and SDWSv are related by the formula:
In the BE study performed on 69 subjects (Fig. 4), the same comparison provides a similar result: 41/69 subjects (59%) were outside the ± 20% range and 50/69 (72%) outside the ± 10% range. Actually, the Average Bioequivalence criteria of reference versus itself are hardly met: GMR = 110.64% 90% CI = 97.74%–125.24%. Consistently, the within-subject CV% = 44.64% (also issued from the ANOVA residual variance) can be regarded as high.
Simulation of different scenarios
The determinants of individual exposure ratios are further studied using simulations, following the same two scenarios: the first of a reference formulation versus itself (replicated single dose, as a proxy of a chronic treatment); and the second of a reference versus a generic, as a proxy of switching formulations during a chronic treatment, to assess interchangeability.
Influence of within-subject variance of the reference
By simulating replicate single-dose administrations of a given (reference) formulation, we studied the influence of within-subject variance of exposure on the distribution of individual exposure ratios, on geometric mean ratios and on bioequivalence acceptance criteria. Indeed, in this situation, the geometric mean ratio (GMR) of exposure tends to 1 by definition and the distribution of individual exposure ratios is intrinsically linked to the within-subject variance of the active substance.
Distribution of individual exposure ratios
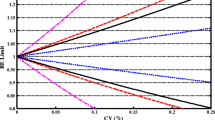
As shown on Fig. 5, the distribution of (reference/reference) individual exposure ratios enlarges when SDWSv increases. Up to 95% of individual exposure ratios are strictly between 0.5 and 2 when SDWSv < 0.25 (CV < 25%), but with higher values of SDWSv, individual exposure ratios may be < 0.5 in some individuals and loss of efficacy is possible, while in others individual exposure ratio may be > 2 with an incidence of adverse effects that may increase. The higher the SDWSv of the reference formulation, the higher are the risks. Clinical outcome will depend on the therapeutic margin of the active substance in terms of individual exposure fluctuations.

Distribution of individual exposure ratios (reference versus reference) (Y axis) across 24 simulated subjects according to different values of SDWSV of the reference (X axis). The reference SD value on the X axis is 100 × standard deviation of within-subject variance of the reference formulation
The limits of the boxes indicate the 25 and 75% percentiles, the limits of the vertical lines indicate the 2.5 and 97.5% values. Points indicate the “outliers” and correspond to 2.5% of the individual exposure ratio values.
The horizontal red lines at 0.5 and 2 IER materialize the limits for efficacy and toxicity generally condidered for drugs with narrow therapeutic index, respectively.
Percentage of subjects with reference/reference IER outside the ± 20% or ± 10% acceptance ranges
The percentage of subjects outside the ± 20% range increases when SDWSv of the reference formulation increases, following a sigmoid relationship (Fig. 6). Almost no subjects present with individual exposure ratio outside the ± 20% range when SDWSv < 0.10 (CV < 10%). With high values of SDWSv (> 0.40), up to 75% of IERs are outside ± 20%. This relationship is not significantly modified when the number of subjects is increased, which is not surprising since the true value of WSv is independent from the number of subjects.

Proportion of patients (and its standard deviation) with a reference/reference individual exposure ratio out of the ± 20% or ± 10% acceptance range, according to the reference SDWSv
As expected, for a same value of SDWSv, the % IERs outside the ± 10% range is higher than that for the ± 20% range (Fig. 6).
SD value for reference on X axis = 100 × standard deviation of within-subject variance of the reference formulation.
Average Bioequivalence acceptance following replicate administration of the reference formulation
Figure 7 and Table 1 illustrate the relationship between the number of subjects to be included in the bioequivalence study of the reference versus itself to remain within the acceptance limits of Average Bioequivalence when SDWSv of the reference formulation increases. In this situation, the geometric mean ratio is close to 1 by definition. Without surprise, when SDWSv increases, more subjects have to be enrolled in order to remain within the acceptance limits of Average Bioequivalence, especially for drugs with narrow therapeutic index. For SDWSv > 0.50, demonstrating Average Bioequivalence for a drug with narrow therapeutic index requires more than 48 subjects. With 100 subjects, the Average Bioequivalence acceptance limits are met even with SDWSv values up to 0.70.

Number of subjects and acceptance reference/reference bioequivalence limits according to the within-subject variance of the reference formulation. In this case, geometric mean ratio is close to 100% by definition. The total width (× 100) of 90% CI of the geometric mean ratio of exposure (and its standard deviation) according to the reference SDWSv (X axis) and to the number of subjects enrolled (n = 12 to 100). Since the whole 90%CI of the GMR should be included within (80–125%) for bioequivalence acceptance, the total width of this interval has to be less than 45% (dashed red line). For drugs with narrow therapeutic index, the width of the 90% CI acceptance interval (90–111%) is 21% (dashed blue line). See also Table 1
In Table 1, the results of ABE of reference vs reference when SDWSv increases are provided for the ± 20% and ± 10% acceptance ranges. With the latter, it is even more obvious that reaching ABE requires to increase the number of subjects: when SDWSv = 35 and n = 12 subjects, this ± 10% acceptance range is not reached (90% CI limits outside the 90.0–111.11% range), whereas it is with 100 subjects.
When SDWSv = 35 and n = 12 subjects, the ± 10% acceptance range is not reached (90% CI not totally included in [90.0–111.11%]), whereas it is with 100 subjects.
Simulation of different generic-versus-reference scenarios
In this situation there are two exposure within-subject variance (WSv) values, one for the reference and one for the generic. We investigated the impact of the difference between the two within-subject variances on the distribution of generic/reference individual exposure ratios and on bioequivalence acceptance according to geometric mean ratios values and to the number of enrolled subjects.
Influence on the generic/reference individual exposure ratios for a Geometric Mean Ratio GMR close to 1
For a value of GMR close to 1 and a given within-subject variance of the reference, the % of generic/reference individual exposure ratios outside the ± 20% range increases when the generic within-subject variance increases (Fig. 8). Symmetrically, for a given generic within-subject variance, the distribution of generic/reference individual exposure ratios also widens when the reference within-subject variance increases. Similar results were obtained for the ± 10% range. It is worth noting that when the generic SDWSv < reference SDWSv, the % of individual exposure ratios IERs outside the 20% range is less than that of the reference versus itself (comparison of Figs. 6 and 8).

Proportion of patients (with its SD values) with generic/reference individual exposure ratio out of the ± 20% acceptance range (Y axis) depending on the generic SDWSv (X axis) and on the reference SDWSv (10, 20 or 30: colored curves)
Generic SD value on the X axis = 100 × standard deviation of within-subject variance of the generic.
Combined influence on the generic/reference individual exposure ratios of the generic/reference ratio of SDWSv, SDWSv of the reference, number of subjects and GMR values
Figure 9 shows that the proportion of generic/reference individual exposure ratios outside the ± 20% acceptance interval increases when (1) the generic/reference SDWSv ratio increases, (2) SDWSv of the reference increases and (3) geometric mean ratio greatly diverges from 100% (GMR values within [90–110%] had very little influence).

Proportion of patients with generic/reference individual exposure ratio outside the 20% acceptance range depending on: the generic/reference SDWSv ratio, the reference SDWSv, the GMR and the number of subjects evaluated
For a fixed value of generic/reference SDWSv ratio = 2.5 as proposed by the FDA (6) (materialized by red lines on Fig. 9), the proportions of generic/reference individual exposure ratios outside the ± 20% range are 45%, 70% and 80% for respective 100 × SDWSv values of 10, 20 and 30 and geometric mean ratio between 90 and 110%. In addition, the number of subjects does not influence this relationship.
Similar results are obtained with the ± 10% acceptance range, however with higher numbers of individuals outside the range (Supplemental Fig. 1s).
Columns represent reference SDWSv × 100 = 10, 20 and 30.
Lines represent different numbers of subjects evaluated: 12, 24, 48, 100.
X axis = ratio between the generic and reference SDWSv values.
Y axis = proportion of patients with generic/reference individual exposure ratio outside the ± 20% acceptance range.
Colored curves according to GMR values: results are very similar with GMR values of 1, 0.9 and 1.1.
For a given generic/reference SDWSv ratio (X axis), the proportion of subjects with a generic/reference individual exposure ratio outside the ± 20% acceptance range increases when reference SDWSv increases (columns from left to right) and when geometric mean ratio values are < 90% or > 110%, but are independent from the number of subjects evaluated.
Average bioequivalence acceptance
We studied the combined influence on average bioequivalence acceptance (90% CI limits of geometric mean ratio within [80–125%], or [90–111%] for drugs with narrow therapeutic index) of the generic/reference ratio of SDWSv, SDWSv of the reference, number of subjects and percentage of patients with IER outside ± 20%.
For a fixed value of generic/reference SDWSv ratio (e.g. 2.5): (i) the proportion of generic/reference individual exposure ratios outside ± 20% increases when within-subject variance of the reference increases, regardless of the number of subjects (Fig. 10); (ii) in contrast, BE acceptance depends on both the reference within-subject variance and the number of subjects. Bioequivalence may not be reached if within-subject variance is high and the number of subjects is not sufficient (Fig. 10). But, when the generic/reference SDWSv ratio = 2.5 and reference SDWSv is < 0.20, bioequivalence is obtained whatever the number of patients.

Influence on bioequivalence acceptance of the proportion of generic/reference individual exposure ratios outside the ± 20% acceptance range (Y axis), according to the generic/reference SDWSv ratio (X axis), for reference SDWSv = 10, 20, 30 (columns) and number of subjects = 12, 24, 48 and 100 (lines)
Similar results are obtained for bioequivalence acceptance of generics of drugs with narrow therapeutic index when reference within-subject variance increases or when the number of subjects decreases (Supplemental Fig. 2), however with a lower probability of bioequivalence acceptance for a given generic/reference SDWSv ratio.
Whatever the situations, the proportion of generic/reference individual exposure ratios outside the ± 20% acceptance range increases when the generic/reference SDWSv ratio increases. Consequences on bioequivalence acceptance (standard 90% CI limits of GMR included within the 80 to 125% interval): blue curves represent situations where bioequivalence is accepted (1), and red curves where it is not (0). For an upper limit of 2.5 for the generic/reference SDWSv ratio, bioequivalence is accepted when the number of patients increases and/or when the reference SDWSv decreases.
Discussion
In this study based on actual data and simulations, we observed that even when the Average Bioequivalence criteria are met between a generic and its corresponding reference formulation, a significant proportion of individuals exhibit a generic/reference exposure ratio outside the assumed acceptance limits. This observation is in apparent contradiction with the basic principles of Average bioequivalence that any difference of exposure < 20% (or 10% for NTI drugs) should not have any therapeutic impact, assuming the therapeutic effects of the active substance are linked to its blood/plasma concentrations. However, it simply illustrates the basic mathematical principle that the individual fluctuations of exposure between generic and reference may be higher than the geometric mean ratio estimated in simple cross-over bioequivalence studies.
Actually, for any active substance and formulation, individuals exhibit a wide range of within-subject variability of exposure. The ± 20% bioequivalence acceptance range proposed for the IC90% uncertainty interval of the mean of the individual geometric mean exposure ratios might better be applied to the individual reference-to-generic exposure ratios themselves (90% of the sample distribution).
The mean within-subject exposure variability (within-subject variance) is derived from the bioequivalence study ANOVA. Our calculations show that the individual exposure ratio of a reference drug versus itself (regarded as a proxy of repeated administration of the same drug dose to patients) increases when the standard deviation of within-subject variance (SDWSv) increases (Fig. 5). The relationship between SDWSv and the proportion of reference/reference individual exposure ratios outside ± 20% or ± 10% is almost linear, up to SDWSv values of 0.30 and reaches for higher values a plateau of approx. 75% and 85%, respectively (Fig. 6), independent of the number of subjects.
Our simulation study demonstrates that when the reference drug SDWSv increases, the number of subjects has to be increased so that the reference drug versus itself remains within the acceptance range limits of Average Bioequivalence (Fig. 7). It is for this reason that for highly variable drugs (CV > 30%), the acceptance limits of ABE for generic formulations compared to their reference have been enlarged by the EMA and FDA, using the Reference Scaled Average Bioequivalence (RSABE) principles [3, 5, 9]. This is acceptable since their therapeutic margin is generally large enough to allow such enlargement of bioequivalence limits [9].
Indeed, for any drug (active substance) with a marketing authorisation, whatever its level, the actual within-subject variance of individual exposure at steady-state is associated with a favourable benefit/risk balance i.e. the within-subject variance is included within the therapeutic margin defined by a minimal and a maximal value of exposure. It is interesting to note however that within-subject variance of replicated single doses may be higher than that measured at steady-state of a chronic treatment, due to the dampening effect of drug accumulation on AUC and Cmax variability. However, for a similar mean exposure, one can anticipate that no change in the individual benefit-risk ratio will be observed when a generic replaces a reference drug if (i) the generic exposure within-subject variance is less than that of the reference; or (ii) the range of individual exposure ratios with the generic is narrower than the therapeutic range of the active substance. This fact is illustrated in Supplemental Figs. 3s and 4s adapted from a previous paper by Lechat [9]. However, the acceptable distance between the minimal and maximal blood exposures of the active substance and fluctuations around the mean for a given subject are generally not known. It is generally accepted that for drugs with narrow therapeutic index the ratio between the minimal toxic and minimal therapeutic plasma (or blood) concentrations is small, in the order of 2 to 3.
Given such a potential impact for drugs with narrow therapeutic index of the difference in within-subject variance between the reference and generic formulations, the FDA has proposed to include an upper limit of 2.5 for the 90% CI of the SDWSv ratio between generic and reference [6], in order to ensure interchangeability [6, 8]. This comes in addition to the reference-scaled average bioequivalence (RSABE) (cf “The Average Bioequivalence methodology and reference-scaled average bioequivalence criteria” section) in the Average Bioequivalence acceptance criteria. It is worth nothing that the reported range of SDWSv for drugs with narrow therapeutic index is 0.05–0.22 [8]. This value of 2.5 might be too high to guarantee in all cases the absence of therapeutic impact of switching between a generic and its reference i.e. interchangeability. It depends on the “distance” between this upper limit and the therapeutic margin of the reference. Our calculations show that the proportion of subjects with individual exposure ratio outside the ± 10% acceptance range for drugs with narrow therapeutic index is quite high when this SD ratio reaches a value of 2.5 (Fig. 8 and Supplemental Fig. 1s). For example, with SDWSv = 0.25 for the generic and 0.10 for the reference (with a generic/reference ratio of 2.5), 45% of patients have a generic/reference exposure ratio out of the ± 20% range (Fig. 8), and 75% out of the ± 10% range (Supplemental Fig. 1s). This situation may result in increased incidence of inefficacy or adverse reactions following conversion across formulations.
We propose to use the diagram of Fig. 5 applicable to all active substances to help solve this issue. It displays the 95% CI interval of individual exposure ratios after replicate administration of a same formulation, for each level of SDWSv of this formulation. It helps to estimate the maximal SDWSv value of let us say a generic formulation (X axis) corresponding to the maximal or minimal acceptable generic/generic individual exposure ratio (Y axis) according to the therapeutic margin of the reference formulation. For SDWSv < 0.25, all individual exposure ratio values are between 0.5 and 2.5 (including outliers), suggesting that no therapeutic impact of the conversion from reference to generic (or vice versa) is expected if their mean bioequivalence is close to 100%.
Generics with higher SDWSv than the reference may not be clinically equivalent i.e. interchangeable. The clinical effects of the switch will depend on the therapeutic margin of the active substance. For reference SDWSv ≥ 0.25, the acceptable values of within-subject variance of the generic will depend on the acceptable individual exposure ratio values for the reference. In all cases however, when the generic SDWSv is equal to or less than that of reference, no therapeutic impact of switching between formulations is to be expected.
The multitude of adverse reactions reported following substitution of the old reference by a new reference formulation of levothyrox [11, 12] in France, despite bioequivalence had been demonstrated using the usual methodology, could be partly explained by the (unexplored) difference of within-subject variance between the two formulations. Indeed, within-subject variance cannot be calculated for the two formulations separately, because of the two-period cross-over design of the bioequivalence trial. Only a fully replicated cross-over design with two periods of exposure for each formulation would have allowed calculing and comparing formulation-specific within-subject variances [11].
Another hypothesis proposed by Concordet et al. [12] to explain this dramatically increased incidence of adverse reactions following the switch from the old to the new formulation of levothyrox was a possible subject-by-formulation interaction, which has to be differentiated from the fluctuations of individual exposure ratioconsidered in the present study. Such a statistical interaction has been discussed in different articles on individual bioequivalence [7, 13, 14]. It refers to the difference between subjects of the generic/reference exposure ratio during a bioequivalence study. Our simulation study clearly demonstrated that individual exposure ratio varies across subjects according to the usual fluctuations of any biological process, including oral bioavailability of a formulated active substance. The true subject-by-formulation interaction occurs when the difference of exposure level between subjects remains constant with time when subjects are shifted from one reference to a generic drug [9]. However, such a true subject-by-formulation interaction is rather theoretical and is rarely observed in practice, consistent with the fact that no pharmacological explanation can be proposed to this phenomenon. There are no convincing reports of any clinical impact of such subject-by-formulation interaction in the literature. While reviewing applications of generic drugs with modified-release formulations using the individual bioequivalence design they proposed, the FDA failed to detect the presence of clinically significant subject-by-formulation interactions [7, 14]. Such an interaction was reported with methylphenidate but was in fact related to different within-subject variances between formulations [15]. In general, when explored in replicated cross-over bioequivalence studies, the variance associated with the subject-by-formulation interaction is negligible as compared to the within-subject variance [16, 17]. In the study by Yu et al. [16], they concluded that only the within-subject variability (and not the subject-by-formulation interaction) seems to play a crucial and decisive role in the variations of drug exposure. In addition, with in silico calculations, we could show that similar distribution of individual exposure ratio could be obtained in the study of Concordet et al. [12], without any subject by formulation interaction [18]. For these reasons, we did not include in our statistical models for simulations any subject-by-formulation interaction term, which can be considered as a limitation of our study. Another limitation is that in this simulation study, we assumed that deviations are randomly distributed while in practice, there may be individuals with deviating absorption or metabolism. But such deviations will similarly occur with both reference and generic formulations.
Conclusions
In addition to the usual Average Bioequivalence criteria applicable to bioequivalence studies, the within-subject variance of the reference and of any test formulations (including generics) should be assessed and compared in order to improve the prediction of no therapeutic impact of the switch, especially for drugs with narrow therapeutic index. This requires bioequivalence studies with fully replicated designs. If their geometric mean exposure ratio is close to 100% and the within-subject variance of the generic is equal to or less than that of the reference, no therapeutic consequences are expected. If the generic within-subject variance is higher than that of reference, interchangeability can only be assumed if the 95% CI limits of the generic/generic individual exposure ratios are included within the therapeutic margins of the reference formulation.
Data availability
No data are available since this is not really appropriate but the R program for the simulation studies is provided as associated available document.
References
Berg MJ, Gross RA, Tomaszewski KJ, Zingaro WM, Haskins LS (2008) Generic substitution in the treatment of epilepsy: case evidence of breakthrough seizures. Neurology 71:525–530
Dernières données issues de l'enquête de pharmacovigilance de l'ANSM sur les médicaments à base de lévothyroxine. https://ansm.sante.fr/actualites/medicaments-a-base-de-levothyroxine-lansm-publie-les-dernieres-donnees-issues-de-lenquete-de-pharmacovigilance. Accessed 30 Mar 2023
EMA guideline on the investigation of bioequivalence: Guideline o the Investigation of Bioequivalence (europa.eu). Accessed 30 Mar 2023
EMA: Questions and answers on bioequivalence studies. https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/clinical-pharmacology-pharmacokinetics/clinical-pharmacology-pharmacokinetics-questions-answers. Accessed 30 Mar 2023
FDA Guidance for Industry Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs — General Considerations. https://www.fda.gov/files/drugs/published/Bioavailability-and-Bioequivalence-Studies-Submitted-in-NDAs-or-INDs-%E2%80%94-General-Considerations.pdf. Accessed 30 Mar 2023
FDA Draft Guidance on Warfarin Sodium (2012). https://www.accessdata.fda.gov/drugsatfda_docs/psg/Warfarin_Sodium_tab_09218_RC12-12.pdf. Accessed 30 Mar 2023
Davit BM, Chen ML, Conner DP et al (2012) Implementation of a reference-scaled average bioequivalence approach for high variable generic products by the US Food and Drug Administration. AAPS J 14:915–924
Yu LX, Jiang W, Zhang X et al (2015) Novel bioequivalence approach for narrow therapeutic index drugs. Clin Pharmacol Ther 97:286–291
Lechat P (2022) Interchangeability between generic and reference products: limits of average bioequivalence methodology. Eur J Drug Metab Pharmacokinet 47:777–787
Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R et al (2019) Welcome to the Tidyverse. J Open Source Softw 4:1686
Gottwald-Hostalek U, Uhl W, Wolna P, Kahaly GJ (2017) New levothyroxine formulation meeting 95–105% specification over the whole shelf-life: results from two pharmacokinetic trials. Curr Med Res Opin 33:169–174
Concordet D, Gandia P, Montastruc JL et al (2019) Levothyrox® new and old formulations: are they switchable for millions of patients? Clin Pharmacokinet 58:827–833
Chen ML, Patnaik R, Hauck WW et al (2000) An individual bioequivalence criterion: regulatory considerations. Stat Med 19:2821–2842
Food and Drug Administration (2000) Guidance for industry on Bioavailability and bioequivalence Studies for orally administered Drug products – General considerations. Availability. Food and Drug Administration. Fed Regist 65:64449–64450
Meyer MC, Straughn AB, Jarvi EJ et al (2000) Bioequivalence of methylphenidate immediate-release tablets using a replicated study design to characterize intrasubject variability. Pharm Res 17:381–384
Yu Y, Teerenstra S, Neef C et al (2015) A comparison of the intrasubject variation in drug exposure between generic and brand-name drugs: a retrospective analysis of replicate design trials. Br J Clin Pharmacol 81:667–678
Van Lancker G, Van Bortel L, Delafontaine B et al (2019) Switchability of gabapentin formulations : a randomized trial to assess bioequivalence between neurontin and gabasandoz on the individual subject level. Clin Pharmacol Ther 106:195–203
Lechat P, Ropers J, Barre J. Mouly S (2019) Comment on: “Levothyrox® New and Old Formulations: Are they Switchable for Millions of Patients?” Clin Pharmacokinet 10:1351–1352
Author information
Authors and Affiliations
Contributions
Each author has contributed to the conduct of the study and reviewed the manuscript. Jean Baptiste Woillard and Fatma Kir specifically set up the programm of the different simulation studies with R coding.
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest is declared fo Philippe Lechat, Fatma Ikr and Jean Baptiste Woillard. Pierre Marquet declares a conflict of interest with Sandoz laboratories.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lechat, P., Kir, F., Marquet, P. et al. Within-subject pharmacokinetic variability has a strong influence on individual exposure ratios in bioequivalence studies, hence on drug formulation interchangeability. Eur J Clin Pharmacol 79, 1565–1578 (2023). https://doi.org/10.1007/s00228-023-03565-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-023-03565-6