
Abstract
Marketing authorisation of generic drugs is based on a demonstration of the “average” bioequivalence (ABE), with acceptance limits of 0.8–1.25 for the 90% confidence interval (CI) of the ratio (generic versus reference) of geometric means of exposure (whole blood, serum or plasma drug concentration). However, when interchangeability of reference by one generic is considered during treatment of a given patient, such methodology cannot guarantee the lack of therapeutic impact especially for drugs with narrow therapeutic index. This review article describes the basis and limits of ABE methodology, and the adaptations that have been proposed by regulatory agencies. For highly variable drugs, given their large therapeutic margin, regulatory agencies even allow widening of the bioequivalence acceptance limits. For drugs with a narrow therapeutic index, the average bioequivalence methodology has been amended differently by regulatory agencies. The European Medicine Agency only requires the narrowing of the ABE acceptance limits to the 0.9–1.10 range. The US Food and Drug Administration (FDA) has proposed to narrow the ABE acceptance limits according to the reference within-subject variance. The FDA requires a fully replicate cross-over study (with four periods) which allows one to compare the within-subject variance between generic and reference drug, and to detect any subject-by-formulation interaction. Indeed, any within-subject variance difference or subject by formulation interaction is an obstacle to interchangeability at the individual level. These amendments for the ABE do not fundamentally change the fact that individual ratios of exposure (generic/reference) will vary to a larger extent than the ratio of their means. For these reasons, since true individual bioequivalence studies cannot be performed for practical reasons and statistical issues, one can suggest that, in addition to the usual average bioequivalence criteria, the limits of the 95% confidence interval of the individual generic/reference exposure ratios could be used to allow interchangeability during treatment (at least for narrow therapeutic index drugs). Limit values of such CI for interchangeability acceptance should be scaled to the therapeutic margin of the reference drug. Regulatory agencies could conduct calculations based on real datasets of bioequivalence studies to determine if such criteria could be acceptable to allow interchangeability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Marketing authorisation of generic drugs requires demonstration of bioequivalence which is based on an average bioequivalence methodology (ABE). |
This does not guarantee absence of therapeutic impact when a reference drug is switched to the corresponding generic during treatment in a given patient. |
Adaptation of ABE has been proposed by regulatory agencies for drugs with large within-subject variability and for drugs with narrow therapeutic index. |
It is suggested for such narrow therapeutic index drugs that an additional criterion allowing interchangeability could be proposed on the basis of 95% confidence interval of individual exposure ratio of generic versus reference. |
1 Introduction
The “average bioequivalence” (ABE) methodology has been used for years for marketing authorisation of generic drugs. The ABE is based on the determination of confidence intervals (CI) of the ratios of the geometric means of exposure (whole blood, serum or plasma drug concentration) between the generic and corresponding reference product. However, regulatory agencies do not indicate in the different marketing authorisation documents of generic drugs that replacement (interchangeability or switching) during treatment for a given patient of one reference by its generic is associated with lack of any therapeutic impact.
This article reviews the basis and limits of the average bioequivalence methodology and the adaptations that have been adopted by different regulatory agencies for drugs with large within-subject variability and for drugs with narrow therapeutic index. We suggest that, to allow interchangeability at the individual level, an additional criterion could be investigated especially for drugs with a narrow therapeutic index. In addition to the CI of the ratio of means, the CI of the mean of individual ratios of exposure (generic/reference) could be considered.
2 Average Bioequivalence Methodology
2.1 Principles of the “Average Bioequivalence” Methodology
Bioequivalence studies for marketing authorisation of generic drugs are based on a methodology that has been accepted for many years by different regulatory agencies [1,2,3]. It is based on the comparison of the means of exposure (whole blood, serum or plasma drug concentration) of the active substance of a generic drug with those of the corresponding reference product. We will simplify and consider the most frequent case of an oral route of administration for the two products and the use of plasma drug concentration for exposure. The two parameters of exposure that are studied are the area under the plasma concentration–time curve (AUC) and the peak plasma concentration (Cmax). The general principle of bioequivalence confirmation is that one accepts bioequivalence if the mean exposure of the active substance released by the generic product does not differ by more than 20% from that of the reference product. The use of this 20% criterion is based on a decision by medical experts of the US Food and drug administration (FDA), who found that, for most drugs, a 20% difference in the concentration of the active ingredient in the blood would not be clinically significant [4, 5].
This means that one hypothesises that a mean 20% difference does not impact either the therapeutic efficacy or the benefit/risk balance of the active substance at the individual level. The pharmacological basis of this hypothesis is that the pharmacological effect (and the therapeutic result) is closely linked to and correlated with the kinetics of the blood exposure of any active substance [6, 7]. Indeed, the drug effects depend on the appropriate concentration at the sites of action of the drug, and this concentration is a function of blood exposure, which itself is a result of the extent and rate of absorption, distribution, biotransformation and excretion of the substance.
Within the ABE methodology, one then accepts without difficulty that substitution of one reference drug by its corresponding generic can be proposed at the instauration of a new treatment for a given patient. One generally extrapolates such a substitution from the onset of treatment to the period during treatment. However, interchangeability (or switching) during treatment, i.e. the lack of any therapeutic impact at the individual level, cannot be guaranteed by the average-based bioequivalence demonstration.
Bioequivalence studies are generally performed on healthy subjects with a cross-over design spanning two successive periods, one with generic administration and the other with administration of the reference (Fig. 1) with two sequences of administration. Bioequivalence studies may be performed with a parallel group design if the elimination half-life of the active substance lasts several days or weeks. They are generally single-dose studies with the appropriate choice of dose according to the linear or nonlinear feature of the pharmacokinetics according to US FDA or European Medicine Agency (EMA) recommendations [1,2,3]. Such a condition is considered the most sensitive situation for detecting differences between formulations. These studies can very rarely be performed on patients with repeated doses [1,2,3]. Bioequivalence studies for generic drugs require only a demonstration of bioequivalence on pharmacokinetic parameters and not on additional pharmacodynamic parameters, as is the case for biosimilar medicines [8].

Bioequivalence study: comparison of plasmatic exposure of one active substance issued from the reference and the corresponding generic drug in ten healthy subjects. The study design is usually a cross-over with two periods (and two sequences) of exposure. The mean difference between the generic and reference exposures must be within ± 20% of the reference exposure (see Sect. 2.1). The slope of the lines joining the exposure values between generic and reference is not different between subjects (no subject-by-formulation interaction, and the within-subject variability of exposure is not different between generic and reference). AUC area under the plasma concentration-time curve, Cmax peak plasma concentration
Bioequivalence is then established if the difference in exposure between the generic and reference substance (for the AUC and Cmax) is within −0.2× reference exposure and +0.2× reference exposure, which also means that the generic/reference exposure ratio has to be between 0.8 and 1.20 (Table 1, Fig. 1) [4, 5].
Therefore, equivalence tests must be performed for the following hypotheses: 0.8 < AUC generic/AUC reference and 0.8 < AUC reference/AUC generic; the same tests can be used for Cmax. When both hypotheses are considered together, this gives 0.8 < AUC generic/AUC reference < 1.25 (since if 0.8 < AUC reference/AUC generic, then, mathematically, AUC generic/AUC reference is < 1/0.8 = 1.25; this explains the 1.25 value for the upper limit (and not 1.20) since 0.8 is the reciprocal value of 1.25. This is the so-called two one-sided test procedure for demonstrating bioequivalence as initially proposed by Schuirman [9].
For mathematical and statistical reasons according to regulatory agencies, the comparison of exposure means between generic and reference substances and calculation of the confidence intervals are based on an analysis of variance (linear mixed model) of the log-transformed exposure data [4, 10]. Indeed, analysis of variance assumes an additive model, equality of variances between compared groups, and normally distributed data. The logarithmic transformation of data complies with such requirements [10]. Under such conditions, the ± 20% interval for the initial non-log-transformed data, which is centred around zero, also becomes centred around zero since the log value of 0.8 is −0.22 and the log value of 1.25 is equal to +0.22. On the logarithmic scale, the acceptance of bioequivalence follows Eq. 1:
According to the FDA, bioequivalence must be demonstrated using the ratio of the geometric means of the exposure data [3]. The 90% confidence interval (CI) of the geometric means ratio must fall within the 0.8–1.25 range, with an overall alpha risk of 5% (Table 1). There is indeed a mathematical link between the difference between the means on a logarithmic scale and the ratio of the geometric means (on a linear non-logarithmic scale) [10]: the arithmetic mean of the logarithmic values of a series Xi equals the logarithmic value of the geometric mean of Xi.
Therefore, the assessment of bioequivalence is based upon the 90% confidence intervals of the ratio of the population geometric means (test/reference) for the parameters under consideration. This method is equivalent to two one-sided tests with the null hypothesis of bioinequivalence at the 5% significance level [1, 2, 9].
In the special case of a two-period, two-sequence cross-over design, when the number of individuals per sequence is the same (N/2), the limits of the 90% confidence interval for the logarithm of the geometric means ratio are calculated as follows (Eq. 2):
S2res being the residual variance from variance analysis (performed on a logarithmic scale), N being the number of included subjects in the cross-over design and XT and XR being the logarithm of the geometric means observed in the study for the tested and reference formulations, respectively.
Then, in the ABE, according to Eqs. 2 and 3, the 90% confidence interval limits of the ratio of geometric means (generic/reference) are proportional to the square root of the residual variance (which is the within-subject variance) of plasmatic exposure and inversely proportional to the square root of the number of included subjects.
The FDA has published a statistical analysis of 2070 ABE studies that were submitted to the US agency from 1996 to 2007 [4]: The mean differences of exposure between generic drugs and references were 4.3% for Cmax and 3.5% for AUC. For 98% of these studies, the mean generic AUC differed by less than 10% from that of the reference.
2.2 Limits of the Average Bioequivalence Methodology
2.2.1 Subject-by-Formulation Interaction and Within-Subject Variance Difference between Generic and Reference
In bioequivalence studies with a cross-over design and two periods of exposure (generic/reference) for each included subject, the statistical analysis of variance assumes an equality of within-subject variances of exposure between the generic and reference substances and cannot explore any interaction between subjects and formulation. Indeed, the two sources of variability, the difference in the within-subject variance of exposure between the generic and reference substances and the subject-by-formulation interaction, are included in the residual variance of the analysis of variance of the cross-over study. They cannot be distinguished or analysed separately when the design incorporates only two periods of exposure, one with the generic and the other with the reference substance.
Any difference in within-subject variance between generic and reference and any subject-by-formulation interaction can only be separately analysed when the cross-over is replicated with at least four periods of exposure (at least two with the generic and two with the reference).
The subject-by-formulation interaction (Fig. 2) reflects the observation that the exposure can vary differently from one subject to another between generic and reference. It may increase or decrease. This could be the consequence of a clinical characteristic of certain subgroups of subjects that differentially impact the bioavailability of the active substance between the generic and reference. It is rather theoretical in healthy subjects but could be possible in patients. In such cases, if the within-subject variance remains the same between generic and reference, one could solve the issue by dose adaptation in such subjects if necessary in cases of any therapeutic impact of this interaction. Indeed, such subject-by-formulation interactions should remain of the same amplitude during repeated administrations.

Bioequivalence study: Illustration of subject-by-formulation interaction in relation to the different behaviours of six subjects (without difference in within-subject variability between generic and reference). Hypothetical results of ten exposure periods. Each individual receives the reference formulation during the first five periods and the generic formulation during the last five periods. For the purpose of the demonstration, the within-subject variability for both the reference formulation and the generic formulation is of small extent (i.e. the exposure for a given subject does not importantly vary with the reference or with the generic formulation). There is an interaction between subject and formulation: when the formulation changes, the exposure of one subject changes and the magnitude of these changes is not the same for all patients. AUC24h area under the plasma concentration-time curve from 0 to 24 h
In contrast, any difference in the within-subject variance of exposure between generic and reference formulations (Fig. 3) can be the result of different disintegration and dissolution processes between the generic and reference substances that may impact the bioavailability of the active substance. Indeed, this is a quality issue for the finished product that depends on its excipients, which may differ between generic and reference. This issue induces a different within-subject variability (between generic and reference) for active substance availability from one intake to another (day-to-day variability in the case of one daily intake), as observed, for instance, with methylphenidate [11]. Such phenomena can then induce higher or lower bioavailability of the active substance during treatment for the same subject and may have a therapeutic impact on some patients if the amplitude is important in particularly sensitive patients. Importantly, the random nature of this phenomenon prevents any correction by dose adaptation.

Bioequivalence study: illustration of difference of within-subject variability between generic and reference formulations. Hypothetical situation with six individuals receiving the reference formulation during the first five periods and the generic formulation during the last five periods. For the purpose of the demonstration, the within-subject variability with the reference formulation is small. In contrast, the within-subject variability is higher with the generic formulation (i.e. the exposure of a subject changes randomly with the generic formulation). There is no interaction between the formulation and the subject, i.e., for each subject, the average exposures (over periods) remain the same for both formulations. In the classical two-period cross-over (periods 5 and 6), the exposure values are the same between the situation in both Figs. 2 and 3. Such two-period cross-over therefore cannot distinguish a subject by formulation interaction from a difference of within-subject variability (within-subject variance). They are both included in the residual variance of the analysis of variance (ANOVA). To assess such difference, at least, a replication of the cross-over is needed with at least four periods (two with the reference and two with the test). AUC24h area under the plasma concentration-time curve from 0 to 24 h
In general, bioequivalence studies with cross-over replication have shown that differences in within-subject variances between generic and reference are of small magnitude and that subject-by-formulation interactions are negligible or absent compared with the within-subject variance of the reference [1, 2]. These subject-by-formulation interactions and differences between within-subject variances were raised by Concordet et al. as a way to interpret the observed adverse reactions following the replacement of an old Levothyrox® formulation with a newer one [12].
Therefore, the hypothesis of equality of within-subject variance (between generic and reference) and the lack of investigation of the subject-by-formulation interaction with a usual two-period cross-over design makes ABE methodology acceptable for substitution on initiation of treatment but represents a first set of arguments against the guarantee of interchangeability during treatment at the individual level, especially when differences in the within-subject variances are present, since dose adaptation cannot correct such differences, as is the case for subject-by-formulation interactions.
2.2.2 Limits of the ABE Related to the Distribution between Subjects around the Mean of the Difference of Plasmatic Exposure between Generic and Reference
The ABE is based on the 90% CI limits of the ratio of the geometric means of the generic and reference exposures. The width of this CI, as mentioned in paragraph 2.1, is inversely proportional to the square root of the number of included subjects. Thus, any increase in the number of included subjects will reduce the width of this CI. Conversely, the distribution of the individual ratios of the generic/reference exposure is larger than the calculated CI of the ratio of their means. This is in accordance with the general relationship between sample distribution and standard error of the mean value of such a sample.
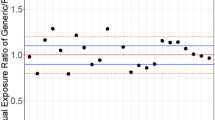
This is illustrated in Fig. 4 and is the consequence of the biological variability between subjects of the impact of excipients on the bioavailability of one active substance.

Individual differences in exposure (AUC) between two different formulations of theophylline (plotted from individual data reported by Rasheed and Siddiqui [10] in 18 subjects). The limits of average bioequivalence are ± 20% of reference exposure which means here ± 46.6 for the absolute values of reference exposure (dashed horizontal lines). Five of 18 subjects (28%) were outside this interval, although the average bioequivalence is demonstrated with the 90% CI of the mean difference (test – reference) = from −16.7 to +15.6 (horizontal continuous lines); 12 of 18 subjects (66%) are outside the interval of the 90% CI of the mean difference. 90% CI of geometric means ratio = 0.925–1.085 (inside 0.8–1.25) with a point estimate of = 0.998. AUC0-inf area under the plasma concentration-time curve from time 0 to infinity
Thus, even when bioequivalence can be demonstrated in terms of the ratio of means, including the hypothesis of the lack of difference between within-subject variances and the lack of any subject-by-formulation interaction, the between-subject fluctuations within the sample of included subjects will show that for some patients, the difference in exposure between generic and reference will be larger than ± 20% as illustrated in Fig. 4. Individual data were extracted from the publication by Rasheed and Siddiqui [10] on a bioequivalence study of two formulations of theophylline. The ABE is demonstrated with 90% CI limits of the ratio of means of 0.92–1.8. However, 5 out of the 18 included subjects (27%) had individual ratios of generic/reference exposure out of this range.
This means that the ABE methodology cannot guarantee interchangeability at the individual level since the difference in exposure between generic and reference will be larger than the accepted ± 20% range, when simultaneously, the difference in means will indeed well be within this interval.
The therapeutic impact of such a result depends on the therapeutic margin of the reference. If this therapeutic margin is large, and much larger than the 20% difference, any therapeutic impact cannot be expected in the case of switching the generic with the reference formulation during treatment. In contrast, if the therapeutic margin is narrow, close to the range of the acceptance limits of 20%, individual fluctuations can potentially lead to therapeutic changes of efficacy and of benefit/risk balance.
To solve these different limits of ABE, regulatory agencies and scientists have explored other bioequivalence methodologies and adjustments of ABE that we describe in the next paragraph.
3 Methodologies That Have Been Discussed by Regulatory Agencies to Solve the Limits of ABE
3.1 Individual Bioequivalence
For the last 20 years, statisticians and regulatory agencies have debated whether a methodology other than the ABE could be elaborated for bioequivalence that could allow interchangeability between the generic and reference during treatment at the individual level [13]. The idea of “individual” bioequivalence methodologies has emerged, but with rather confusing results. In fact, to establish bioequivalence at the individual level, one should repeat administration of the generic and reference substances several times on the same subject. This could allow determination of the mean value of exposure of the generic and reference substances and the within-subject variance for the generic and reference substances for each subject. One intuitively understands that the individual bioequivalence could be accepted if the observed differences between generic and reference exposure during repeated administrations was of the same (or even lower) magnitude than the difference in exposure between different administrations of the reference (and between different batches of the reference). In other words, as stated by Chen et al. [13], the motivation of such individual bioequivalence methodologies is to compare for each individual the difference in bioavailability of the test and reference formulation (T–R) with that of the reference against itself (R–R).
Such individual bioequivalence methodologies with repeated administrations of generic and reference formulations on the same subject are in practice almost impossible to perform. They also assume that the clinical status of the subject will remain perfectly stable with time. Such individual bioequivalence methodologies could be of more value for patients, but in such cases, the stability of the clinical status is even more difficult to obtain than in healthy subjects.
Statistical issues have also been raised for such individual bioequivalence studies, related to the number of periods, the limits of acceptance, the power of the test, etc.
For both reasons, such individual bioequivalence studies have not been incorporated into the usual practice.
3.2 Propositions Adopted by Regulatory Agencies for Adaptation of the ABE for Drugs with Large Within-Subject Variability and for Drugs with Narrow Therapeutic Indexes
3.2.1 Drugs with Large Within-Subject Variability
Some medicines contain active substances that present with large within-subject variability in bioavailability (and blood exposure) during repeated administrations of the same posology to a given patient. In general, such substances have low hydrosolubility, low lipophilia and low bioavailability and are subjected to active first-pass hepatic metabolism. The majority of these substances belong to class 4 of the international BCS classification (biochemistry classification of substances), which possess low hydrosolubility and low transmembrane permeability [14].
For medicines containing such a substance, given this large within-subject variability, it can become difficult to demonstrate bioequivalence versus itself and even more in comparison with a generic drug. In such a situation, the number of patients to be included in bioequivalence studies must be greatly enhanced [15,16,17].
To solve this issue for medicines with large within-subject variability and to reduce the number of subjects to be included in bioequivalence studies, regulatory agencies (such as the FDA and EMA) have decided to expand the limits of bioequivalence acceptance by adjusting them to the within-subject variability. This within-subject variability is measured by the within-subject variance, which is the residual variance of the ANOVA of the cross-over. The coefficient of variation (CV) is derived from the residual variance and has a value close to the square root of the residual variance [2, 18]. Adjustment of the BE limits is proposed for a CV > 30% by the EMA [1, 2] and for a CV > 25% by the FDA [3]. This method is referred to as reference-scaled average bioequivalence (RSABE) or average bioequivalence with expanding limits (ABEL). The FDA then proposes expanding the bioequivalence acceptation limits for drugs with a CV > 25% and applies it to both Cmax and AUC. The EMA proposes expanding the limits for drugs with a CV > 30% but applies it only to Cmax. Indeed, Cmax is considered more susceptible to variability than AUC. In addition, the EMA proposes not expanding these limits further for drugs with a CV > 50% [2].
However, such medicines with large within-subject variability generally have a large therapeutic margin, as otherwise they could not be used or obtain marketing authorisation. Indeed, their therapeutic effect remains at the same level despite the presence of large fluctuations in their plasmatic exposure (more than 20% between two successive administrations of the same dose). For this reason, the interchangeability between the generic and its reference (with demonstrated average bioequivalence) is of little concern since the therapeutic efficacy and the benefit/risk ratio remain the same within a large range of blood exposure (Fig. 5) of the active substance. Additionally, for these reasons, the expansion of the limits of ABE can be accepted to allow interchangeability.

Within-subject daily fluctuations of plasmatic exposure in the same subject following administration of the reference (continuous lines) and of the generic formulation (dashed lines) of a medicine with a large therapeutic margin. Even with some within-subject variance difference between generic and reference, exposure of the active substance remains within the therapeutic margin limits. In this case, interchangeability will not have any therapeutic impact. AUC24h area under the plasma concentration-time curve from 0 to 24 h
3.2.2 Medicines with a Narrow Therapeutic Index
Some medicines and their corresponding active substances are considered to have a narrow therapeutic index. However, there is no internationally accepted list of such active substances or common criteria for their definition. For such drugs, adverse reactions may occur with doses close to and just higher than the therapeutic ones, have abrupt dose–effect relationships and often require therapeutic drug monitoring, i.e. determination of plasma concentration for dose adaptation for each patient. These medicines include certain anti-epileptic drugs, immunosuppressants (tacrolimus, cyclosporine, mycophenolate), lithium, digoxin, vitamin K antagonist anticoagulants and l-thyroxine.
In general, medicines with a narrow therapeutic index have low within-subject variability (CV < 30%). If this was not the case, they could not be used. Indeed, the probability of induction of an adverse drug reaction or the risk of therapeutic failure appears important if a narrow therapeutic window faces a large variability of exposure from one administration to another in the same patient.
Drugs with a narrow therapeutic index are in most cases used as chronic treatments, during which one reference drug may be replaced by one corresponding generic drug and for which the issue of the therapeutic impact of interchangeability is raised (Fig. 6). It is indeed in this category of drugs that modification of therapeutic efficacy or changes in the benefit/risk ratio have been reported. This is especially the case with anti-epileptic drugs and thyroxin [12, 19], for which the usual criteria of average bioequivalence acceptance within the ± 20% range of plasmatic exposure have been challenged.

Same situation as in Fig. 5 but with an active substance with a narrow therapeutic index. Mean exposure is very similar for the same subject between the test and reference. However, the within-subject variance for the generic (dashed line) is larger than that of the reference (continuous line), and thus, on some days, generic exposure may be outside the range of the therapeutic margin. In this case, tests/reference interchangeability may have a therapeutic impact. AUC24h area under the plasma concentration-time curve from 0 to 24 h
To solve this difficulty, regulatory agencies have proposed different strategies for adapting the average bioequivalence criteria for drugs with a narrow therapeutic index. The EMA proposes maintaining the usual two-period cross-over design and restricting the acceptance limits of the 90% CI of the generic/reference ratio of the geometric means to 0.9–1.11 instead of the usual 0.80–1.25 limits [1, 2].
The FDA proposes reducing the average bioequivalence acceptance limits to the within-subject variability of the active substance (RSABE), with a maximum reduction to the 0.9–1.11 interval. In addition, the FDA requires comparisons (F test) of within-subject variances between generic and reference by realising a fully replicate cross-over design (four periods for each subject, two with the reference and two with the generic) [3, 20, 21]. The generic drugs will then be accepted by the FDA, as the within-subject variances of generic drugs will not significantly differ from those of reference drugs. This methodology has been specifically presented in detail for warfarin [22] and for l-thyroxin [23]. The replication of the cross-over design also allows testing of the subject-by-formulation interaction (cf. paragraph 2.2.1). However, as mentioned in paragraph 2.2.1, the subject-by-formulation interaction can be solved by individual dose adaptation following the generic/reference switch. In contrast, if the within-subject variance of the generic formulation is higher than that of the reference, dose adaptation will not solve the issue. This justifies the FDA position requiring a comparison of within-subject variances and rejection of a generic with a higher within-subject variance than the reference.
3.2.3 Therapeutic Impact of Interchangeability for Drugs with a Narrow Therapeutic Index
Adaptation of the criteria of average bioequivalence acceptance, however, does not solve the question of interchangeability at the individual level. Indeed, as previously mentioned in the introduction, regulatory agencies do not claim that average bioequivalence allows interchangeability; they merely avoid the issue.
A good illustration of the question is given by the study of Van Lancker et al. [19] on gabapentin. With this anti-epileptic medicine, reports of therapeutic impact and adverse reactions have been reported following the switch between the reference and corresponding generics [19]. Then, they performed a bioequivalence study complying with FDA requirements with a fully replicate cross-over design comparing the reference with one generic drug (Sandoz generic). They confirmed the average bioequivalence criteria (geometric mean ratio included in the 08–1.25 interval), but more interestingly, they also demonstrated the lack of a difference between within-subject variance (between generic and reference) and the lack of any subject-by-formulation interaction. This study showed that a therapeutic impact may occur (with occurrence of reported adverse reactions) when the formulations are switched even in the absence of a within-subject variance difference or a subject-by-formulation interaction.
The explanation arises partly from the fact that the distribution (95% confidence interval) of one sample of values of individual ratios of exposure (generic/reference) is larger than the 90% confidence interval of the ratio of the means. This is illustrated in Fig. 4. It is also illustrated in Fig. 3 of the publication of Van Lancker [19], which shows that the majority of individual exposure ratios (AUCs) are outside the 0.8–1.25 range. In the study of Concordet et al. [12] comparing both old and new formulations of Levothyrox (l-thyroxine), they also found (by calculations) that, for more than 50% of patients, the individual values of the ratio of thyroxin (T4) exposure were outside the narrowed 0.9–1.11 range, illustrating the fact that the average bioequivalence does not guarantee bioequivalence at the individual level. Such a large range of individual exposure ratios between both Levothyrox formulations is sufficient to explain the high rate of adverse reactions reported following the switch between both formulations.
The fact that the switch between both Levothyrox formulations was performed on a very large scale (more than two million of patients) and during a rather very short period of time (around 3–4 months) explains that such a pharmacovigilance signal could be detected. Such a generic/reference switch is never performed on such a large scale and within such a short period of time. Then, the very small proportion of patients who presented with therapeutic unbalance of their thyroid status following switch between old and new formulation represented quite an important absolute number of cases and could be detected by the pharmacovigilance organisation. Indeed, only 1.43% of the more than two million patients reported such adverse reactions following Levothyrox switch.
Therefore, several hypotheses may explain the occurrence of therapeutic impact and adverse reactions following a switch of one reference to its generic: the individual distribution of exposure ratios between both formulations greater than that of ratio of means, any within-subject variance difference between generic and reference and, finally, a subject-by-formulation interaction. For the specific case of Levothyrox, since adverse reactions progressively disappeared following use of the new formulation, the hypothesis of a difference of within-subject variance is unlikely. However, the relatively large CV (23.7%) reported in the two-period cross-over of the bioequivalence study of Levothyrox [24], higher than the usually reported CV for levothyroxine, is compatible with a larger CV for the new formulation, then approaching the 30% limit for drugs with high within-subject variability. Such a situation, as previously mentioned, is not appropriate for drugs with narrow therapeutic index such as levothyroxine [23]. The subject-by-formulation interaction hypothesised by Concordet et al. [12] remains a possible explanation, but we could show by calculations based on simulations that a similar result of bioequivalence could be obtained without such interaction [25]. Another aggravating factor for the Levothyrox case is derived from the fact that the initial higher tablet thyroxin content authorised for the old Levothyrox formulation (to compensate the progressive decline of thyroxin content due to degradation by oxidation) was no longer permitted for the new formulation. This difference may have induced some plasma exposure differences that could be enough to induce some therapeutic impact in very sensitive patients [25] despite the fact that the average bioequivalence was demonstrated [24]. The best way to prevent such therapeutic impact in the case of Levothyrox would have been to inform patients that it could happen and possibly be solved by dose adjustment.
In the case of therapeutic imbalance reported after generic/reference switching for drugs with a narrow therapeutic index and given the limits of the average bioequivalence methodology in its application to interchangeability, some regulatory agencies recommend not switching patients treated with these drugs with a narrow therapeutic index. Some “no switch” lists have then been elaborated, particularly including some anti-epileptic drugs, some immunosuppressants and l-thyroxin. In the case of substitution with these medicines, a posology adaptation can always be performed according to the therapeutic response if the therapeutic impact is the consequence of a subject-by-formulation interaction. As mentioned earlier, if the therapeutic imbalance is the consequence of a within-subject variance difference, no dose adaptation will solve the problem (cf. paragraph 2.2.2). This position of regulatory agencies recommending “no switch” for drugs with a narrow therapeutic index may, however, discourage pharmaceutical companies from improving the quality of such marketed medicines with new formulations of generic drugs.
4 Proposal for Complementary Criteria for Interchangeability in Addition to the ABE Criteria
To address the possibility of interchangeability between generic and reference formulations at the individual level, we suggest that regulatory agencies add some complementary criteria to the standard average bioequivalence acceptance criteria for drugs with a narrow therapeutic index. Such criteria should explore the distribution of individual values of ratios of exposure between generic and reference. Then, in addition to the CI of the ratio of means, the CI of the mean of individual ratios of exposure (generic/reference) should be considered. One could propose that the 95% confidence interval of the individual values of the exposure ratios for AUC and Cmax should be within a priori limits that could be set and scaled according to the therapeutic margin of the reference. For instance, such 95% CI limits could be ± 20% in accordance with the general pharmacological basis of bioequivalence [4,5,6,7] which basically refers to the individual level, compared with the ± 10% for the 90% CI for the ratio of means required by the average bioequivalence for drugs with a narrow therapeutic index. We suggest that regulatory agencies should perform calculations and simulations based on real datasets of bioequivalence studies (that they have or may request from pharmaceutical companies) to test the feasibility of such proposals.
5 Conclusion
With the acceptance of marketing authorisation of generic drugs based on the usual average bioequivalence methodology (ABE), interchangeability between the generic and reference formulation at the individual level during treatment does not impact therapeutic efficacy or the benefit/risk balance when the therapeutic margin is much larger than the usual limits of acceptance of the average bioequivalence. However, for drugs with a narrow therapeutic index, the ABE methodology cannot guarantee the absence of a therapeutic impact when generic/reference interchangeability is considered at the individual level. For these reasons, a generic/reference switch for drugs with a narrow therapeutic index is not recommended by many regulatory agencies unless a posology adjustment can be performed according to the therapeutic response. In addition to the usual ABE criteria, supplementary criteria could be tested by regulatory agencies for drugs with a narrow therapeutic index to allow generic/reference interchangeability during treatment. Such criteria should be based on the individual 95% CI limits of the individual exposure ratios between generic and reference exposure, scaled to the therapeutic margin of the reference drug.
References
EMA guideline on the investigation of bioequivalence. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 24 June 2022.
EMA: Questions and answers on bioequivalence studies. https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/clinical-pharmacology-pharmacokinetics/clinical-pharmacology-pharmacokinetics-questions-answers. Accessed 24 June 2022.
FDA guideline on bioequivalence. Guidance for industry bioavailability and bioequivalence studies submitted in NDAs or INDs—general considerations. https://www.fda.gov/files/drugs/published/Bioavailability-and-Bioequivalence-Studies-Submitted-in-NDAs-or-INDs-%E2%80%94-General-Considerations.pdf. Accessed 24 June 2022.
Davit BM, Nwakama PE, Conner GP, et al. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the united states food and drug administration. Ann Pharmacother. 2009;43:1583–97.
Approved products with therapeutic equivalence evaluation. 29th ed. Washington DC: US Department of Health and Human Services, Public Health Service, Food and Drug Administration, Center for Drug Evaluation and Research, Office of pharmaceutical sciences, Office of generic drugs, 2009. https://www.fda.gov/media/71474/download. Accessed 24 June 2022.
Karalis V, Macheras P, Van Peer A, Shah VP. Bioavailability and bioequivalence: focus on physiological factors and variability. Pharm Res. 2008;25:1956–62.
Goodman and Gilman’s. The pharmacological basis of therapeutics. New York: McGraw-Hill.
Biosimilar in EU. Information guide for health professionals. 2017. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 24 June 2022.
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–80.
Rasheed A, Siddiqui AA. Use of geometric mean in bioequivalence trials. Int J Stat Med Res. 2015;4:114–20.
Meyer MC, Straughn AB, Jarvi EJ, et al. Bioequivalence of methyphenidate immediate-release tablets using a replicated study design to characterize intrasubject variability. Pharm Res. 2000;17:381–4.
Concordet D, Gandia P, Montastruc JL, et al. Levothyrox® new and old formulations: are they switchable for millions of patients? Clin Pharmacokinet. 2019;58:827–33.
Chen ML, Patnaik R, Hauck WW, et al. An individual bioequivalence criterion: regulatory considerations. Stat Med. 2000;19:2821–42.
Amidon GL, Lennernäs H, Shah VP, Crison JR. Theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20.
Endrenyi L, Tothfalusi L. Bioequivalence for highly variable drugs: drug regulatory agreements, disagreements, and harmonisation. J Pharmacokinet Pharmacodyn. 2019;46:117–26.
Shah VP, Yacobi A, Barr WH, et al. Evaluation of orally administered variable drugs and drug formulations. Pharm Res. 1996;13:1590–4.
Tothfalusi L, Endrenyi L, Midha KK, et al. Evaluation of the bioequivalence of highly-variable drugs and drug products. Pharm Res. 2001;18:728–33.
Chow SC, Choi HW. On sample size calculation in bioequivalence trials. J Pharmacokinet Pharmacodyn. 2002;28:155–69.
Van Lancker G, Van Bortel L, Delafontaine B, et al. Switchability of gabapentin formulations: a randomized trial to assess bioequivalence between neurontin and gabasandoz on the individual subject level. Clin Pharmacol Ther. 2019;106:195–203.
Jayachandran P, Okochi H, Frassetto LA, et al. Evaluating within-subject variability for narrow therapeutic index drugs. Clin Pharmacol Ther. 2019;105:411–6.
Jiang W, Makhouf F, Schuirmann DJ, et al. A bioequivalence approach for generic narrow therapeutic index drugs: evaluation of the reference-scaled approach and variability comparison criterion. AAPS J. 2015;17:891–901.
FDA draft guidance on warfarin sodium. 2012. https://www.accessdata.fda.gov/drugsatfda_docs/psg/Warfarin_Sodium_tab_09218_RC12-12.pdf. Accessed 24 June 2022.
FDA Draft guidance on Levothyroxine Sodium. 2014. Available from URL: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Levothyroxine_Sodium%20capsules_NDA%20021924_RC%20Oct%202018.pdf. Accessed 24 June 2022.
Gottwald-Hostalek U, Uhl W, Wolna P, Kahaly GJ. New levothyroxine formulation meeting 95–105% specification over the whole shelf-life: results from two pharmacokinetic trials. Curr Med Res Opin. 2017;33:169–74.
Lechat P, Ropers J, Barre J, Mouly S. Comment on: “Levothyrox® new and old formulations: are they switchable for millions of patients?” Clin Pharmacokinet. 2019;10:1351–2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding sources were used to prepare the review.
Conflict of interest
The author has no conflicts of interest to declare.
Ethic approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contribution
Philippe Lechat conceived and wrote the review.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lechat, P. Interchangeability between Generic and Reference Products: Limits of Average Bioequivalence Methodology. Eur J Drug Metab Pharmacokinet 47, 777–787 (2022). https://doi.org/10.1007/s13318-022-00785-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-022-00785-6