
Abstract
Surface molecularly imprinted polymer of solanesol (SA-SMIP) was prepared by reversed phase suspension polymerization using modified titanium dioxide (TiO2) as carrier, and operation conditions were investigated and optimized. Structures of modified TiO2 and SA-SMIP obtained at optimal conditions were characterized by Fourier transform infrared spectrometer adopting original TiO2 and non-surface molecularly imprinted polymer as reference. The SA-SMIP synthesized under optimal conditions displayed an excellent recognition of SA from the mixture of SA and triacontanol. The maximum separation degree of SA was 2.90. Finally, the adsorption kinetics and isotherm were investigated and analyzed. Adsorption kinetics results indicated that the adsorption of SA-SMIP to SA was a pseudo-second order process, and the adsorption of beginning and later stages was controlled by homogeneous particle diffusion and adsorption reaction process, respectively. Adsorption isotherm results documented hereby were two sorts of bonding sites, complete imprinted cavities and defective imprinted cavities. The adsorption for two bonding sites could be well lined up with the Langmuir model.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Solanesol (SA), a nine polyisoprene primary alcohol with all-trans configuration, was reported as the starting material for the synthesis of coenzyme Q10, vitamin K2, and SDB, an anticancer synergist agent [1, 2]. Besides its use as a precursor to prepare above medicine, SA itself also possesses extensive pharmaceutical value, such as antibiosis and antiphlogosis, and used in cardiovascular disease treatment [3]. The demand for SA has been increased annually because of its above pharmaceutical activity [4]. Driven by the growing demand, many attempts had been made to extract pure SA from natural resources [5, 6]. However, the application of SA requires high purity, and the actual contents of SA in plants are small, which made the separation and purification of SA from natural products become a cost and complex process naturally. For example, reports on SA’s purification show that chromatographic technology was always used, with more than one time of chromatography steps [7, 8, 9], which meant the preparation of SA at present is very costly and could not be used for obtaining high purity SA. Thus, an effective separation and purification technology or material for SA is still needed urgently [10].
Molecular imprinted polymers (MIPs) had shown a promised prospective in separation and purification field [11, 12, 13, 14, 15, 16] because of its special features, such as structure predetermination, specific recognition, and extensive practicability. However, reports on the preparation of MIPs were always focused on bulk or solution polymerization, and some MIPs prepared by suspension polymerization [5, 14]. Because the products synthesized by bulk/solution polymerization always have the massive shape and need grinding and screening, which would result in the waste of raw materials and deform the imprinting cavities inevitably, the adsorption capacity and affinity of MIP to the template molecule would decrease naturally. Besides, MIP prepared by both solution/bulk polymerization and general suspension polymerization still faces certain challenges [17]. If MIPs prepared with the above methods, most imprinted cavities would be distributed in the deeper range of MIP matrix and quantities of imprinted cavities distributed on the outer surface would be small, which would decrease the efficiency and effectivity of MIP in the process of separating template molecular, because less specific binding cavities on the outer surface would decrease the adsorption capacity of MIP, while higher hindering rebinding of imprinted cavities in deeper range of MIP matrix would prolong the operation time [17, 18]. Comparatively, the imprinted cavities of surface molecular imprinted composite (SMIC) was mainly generated on the surface, and template molecular could access the imprinted cavities and be absorbed with less steric resistance and faster mass transfer speed [19, 20, 21] and had been evidenced as a promising strategy for enhancing the application of separation and purification. At present, there are many reports about the preparation and performance of SMIC [14, 21, 22, 23, 24, 25, 26, 27, 28, 29]. However, the preparation and selectivity of SMIC for SA are under active investigation.
Based on our previous research on MIP of SA [30], this report focused on the design and synthesis of SA-SMIC using titanium dioxide (TiO2) as carrier. To realize this strategy, TiO2 particles were modified with silane coupling agent KH570 to change them to hydrophobic and polymerizable composite monomers first and then hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) were copolymerized by suspension polymerization to prepare SA-SMIC. Focused on the adsorption capacity and separation degree of SA-SMIC to SA from the mixture of SA and triacontanol (TA) with the molar ratio of nSA/nTA = 1/3, the effect of the monomer, cross-linker, initiator, etc., was investigated and optimized. The samples prepared under the optimal conditions showed the adsorption capacity of 30.46 mg/g and the separation degree of 2.90. Thereafter, the structures of modified TiO2 and surface molecularly imprinted polymer of solanesol (SA-SMIP) obtained at optimal conditions were characterized by Fourier transform infrared spectroscopy (FTIR) adopting original TiO2 and non-surface imprinted molecular polymer (NSMIP) as references. Finally, based on the samples prepared under the optimal conditions, the adsorption kinetics and isotherm were investigated in detail, and some useful results were obtained and discussed systemically.
Results and discussion
Characterization of the structure of modified TiO2
Figure 1 shows the FTIR spectra of original TiO2 and modified TiO2. For original TiO2, the strong absorbency around 3430 cm−1 related to the stretch vibration of O–H on the surface of TiO2, the peak at 1636 cm−1 corresponded to the bending vibration of O–H, the peak at 1415 cm−1 was associated to antisymmetric stretching vibration of CO32− in TiCO3, and the peaks at 675 and 534 cm−1 could be ascribed to the symmetric stretching vibration and antisymmetric stretching vibration of Ti–O. The above signals indicated that the original material was TiO2 with some TiCO3 impurity. After TiO2 modified with KH570, the spectrum of modified TiO2 showed new absorption bands at both 2913 and 2852 cm−1, which could be assigned as the vibration of –CH2 in KH570. Besides, modified TiO2 also showed another new peak at 1083 cm−1, which was the asymmetric stretching vibration of Si–O–Si [31]. In all, the above FTIR results indicated that KH570 was grafted on the surface of TiO2 successfully.

The FTIR spectra of TiO2 and modified TiO2.
Characterization of the structure of SA-SMIP
Figure 2 presents FTIR spectra of SA-SMIP and NSMIP, the strong absorbency occurred at about 3440 cm−1 was the stretch vibration of O–H in HEMA and H2O. The peak displayed at about 2950 cm−1 could be assigned as the stretching vibration of C–H in –CH3 and –CH2 of HEMA and KH570. The peak at about 1727 cm−1 was the stretching vibration of C=O in the HEMA and KH570. The peak at 1639 cm−1 corresponded to the stretch vibration of C=O (which are produced by residual acrylic acid) and bending vibration of O–H, and the peak at 1396 cm−1 could be attributed to the in-plane bending vibration of C–H. Compared with the modified TiO2, all peaks related to HEMA and KH570 simultaneously increased at a large degree, while Si–O–Ti-specific stretch vibration peak at about 1086 cm−1 and Ti–O-specific stretch vibration peak at 656 and 535 cm−1 decreased and found to be too small, which indicated that HEMA had been successfully combined in the prepared SA-SMIP. Moreover, the peak at around 1165 cm−1 (νC–N) in SA-SMIP shifted to 1155 cm−1 in NSMIP, the peak at 1639 cm−1 in SA-SMIP shifted to 1633 cm−1 in NSMIP, and the peak at 1731 cm−1 in SA-SMIP shifted to 1727 cm−1 in NSMIP and increased obviously; all the above results implied that there were some difference in the arrangement of functional groups between NSMIP and SA-SMIP.

The FTIR spectra of SA-SMIP and NSMIP.
The adsorption kinetics of SA-SMIP
Figure 3 displays the adsorption kinetics of SA-SMIP in SA solution. It could be found that the adsorption capacity (Qt) increased first and then remained basically unchanged. This phenomenon could be attributed to the combined action of slower diffusion speed and the distribution equilibrium. Due to the special design and preparation process, there were many active sites, including complete imprinted cavities (CIC) and defective imprinted cavities (DIC) [30], in SA-SMIP matrix, all of which were at their high energy state because of the suffered non-equilibrium valence bond, active sites needed to absorb some compounds to decrease their energy level, SA-SMIP would absorb SA naturally. However, due to the high cross-linking degree of SA-SMIP, the steric resistance of SA suffered as it diffused into SA-SMIP matrix was high, which would slow down the diffusion rate of SA. The deeper the diffusion range meant that the slower the diffusion rate; the increased degree of Q decreased with prolonging t naturally. Besides, the distribution equilibrium of SA in SA-SMIP matrix and solution was another reason. Just as mentioned above, the active sites in SA-SMIP contained CIC and DIC; because the affinity of CIC to SA was higher, it would adsorb SA first. At this condition, the equilibrium constant was larger, added to the quantities of SA adsorbed on SA-SMIP was small, the desorption rate could be neglected. As a result, Q increased quickly at the beginning of adsorption. With prolonging t, the adsorption shifted from CIC to DIC gradually, because of the weaker affinity to SA, the equilibrium constant decreased gradually, added to the increased quantities of adsorbed SA, the desorption rate increased gradually, and the increase degree of Qt decreased correspondingly. As the adsorption rate decreased to equal with the desorption rate, Qt kept as a constant. In this work, Qt arrived at a constant after 6 h, but it arrived at 95% of Qe after 2.5 h, both of which were far shorter than that of SA-SMIP samples prepared by solution polymerization and suspension polymerization, which implied that most of the active sites distributed on the outer surface and shallow strata of SA-SMIP. Because nearer the active sites to solution was, the smaller the steric resistance and the shorter distance of SA diffusion were.

The relationship between Qt and t.
To study the adsorption kinetics mechanism in detail, the above experimental data were fitted with pseudo-first order [32] and pseudo-second order equations [33], and the results of their regression curve and the regression equations are shown in Fig. 4 and Table I (rows 1 and 2). It could be observed that both the goodness of fit and Qe to experimental data of pseudo-second order model were better than those of pseudo-first order model, which meant the adsorption of SA-SMIP was a pseudo-second order process.

Results of experiment data analyzed by pseudo-first-order model and pseudo-second order model.
Based on the above kinetics analysis result, the adsorption control step was explored further. According to the reports, pseudo-second order model related control step model could be classified as diffusion and adsorption reaction aspects; the diffusion aspect could be further divided into homogeneous particle diffusion model [34] and intraparticle diffusion model [35]. Based on the above three models, this work explored the control step in detail as follows.
The homogeneous particle diffusion model equation is given as follows:
where X(t) is the fraction of solute adsorbed at time t, X(t) = Qt/Qe. Qe (mg/g) is the amount of SA adsorbed at equilibrium, and Qt (mg/g) is the amount of SA adsorbed at any time t. De represents the effective diffusion coefficient of SA in the sorbent phase (m2 s), r is the radius of the adsorbent particle, which were assumed to be spherical (m), and Z is an integer.
The Vermeulen’s approximation [34] of Eq. (1) fits the whole range 0 < X(t) < 1 was more convenient for practical use on spherical particles.
The Eq. (2) could be further simplified to cover most of the data points for calculating effective particle diffusivity by using the following expression [34]:
Intraparticle diffusion model [35] is given as follows:
where Qt (mg/g) is the quantity of adsorption at time t (h), Ki is the intraparticle diffusion rate constant, and C is a constant.
As the adsorption reaction was control step, which we named as the reaction control adsorption, the relationship between Qt and t for second order kinetics could be expressed as follows.
where A is SA molecular, B represents active sites in SA-SMIP, and P stands for SA adsorbed by SA-SMIP. According to the above expression, the differential equation between Qt and t is shown in Eq. (6):
where m denotes the mass decrease of SA in solution at time t, Qt (mg/g) is the adsorption capacity of 1.0 g of SA-SMIP at time t, C0V0 (mg) is the mass of SA in original solution, b is the active sites in 1.0 g of SA-SMIP, which could be expressed approximately by the statured adsorption capacity and could be obtained from adsorption thermodynamics experiment, m′ represents the mass used for adsorption experiment, and k is the rate constant.
Assigning m′ = 1.0 g, Eq. (6) would change to Eq. (7):
Separated variables and the form could be expressed as Eq. (8):
The left part of Eq. (8) could not be integrated directly and should be calculated using following method:
Assign
then
Comparing the coefficient of two side of equation, the following relationship could be obtained:
Plugging the value of X and Y into Eq. (8) and integrating two sides of it, Eq. (9) was arrived at as follows.
Equation (9) displays the relationship between Qt and t as adsorption reaction was the rate determining step. The results of the models that satisfied with pseudo-second order are shown in Figs. 3 and 4 and Table I (rows 3–5). It could be found the goodness of fit of the reaction control adsorption model was the largest, while that of intraparticle diffusion model was the smallest; besides, Qe calculated from the equation also displayed the same result, all of which indicated that the adsorption was mainly controlled by adsorption reaction (Fig. 5).

Results of experiment data analyzed by the homogenous particle diffusion and the intraparticle diffusion model.
In fact, the adsorption of SA-SMIP to SA was a reaction control adsorption, which could be understood easily. Because SA-SMIP was prepared by a special design, the active sites were mainly distributed on the surface, which meant the diffusion resistance was low, and the diffusion distance was short relatively, which SA could arrive at active sites in with a speeder rate. Because of the high cross-linking, the steric resistance of SA-SMIP matrix was high, the orientation adjustment of SA in SA-SMIP matrix would be relative difficult, which meant that the reaction adsorption would be a slow and time-consuming process and became the control step of adsorption.
Actually Fig. 6 has not still fit well with the experimental data, especially at the beginning stage, which meant that the adsorption was not controlled by the reaction adsorption alone, and some other factors were also influenced the adsorption. Because the adsorption essence was a mass transfer process between two phases of solid–liquid, and the adsorption could not happen as until solution arrived at, which meant that the diffusion also played an important role in the adsorption process, especially at the beginning of adsorption. Based on the above thoughts, the experimental results were further analyzed through subsection mode, i.e., the adsorption beginning stage was explored by the diffusion model, while the later stage was explored by the reaction control adsorption model, and results are shown in Figs. 7 and 8 and Table II. It could be found the homogeneous particle diffusion model and the reaction control adsorption model could express the experimental results well for the adsorption beginning stage and the adsorption later stage, respectively, because the goodness of fit for two stages was very near to 1, which implied that the whole adsorption was controlled by two aspects, the beginning stage was the homogeneous particle diffusion and the later stage was the reaction control adsorption.

Result of experiment data analyzed by the adsorption reaction control model.

Results of the beginning adsorption stage experiment data analyzed by the homogenous particle diffusion model and the intraparticle diffusion model.

Results of the later adsorption stage experiment data analyzed by the reaction.
The beginning stage was controlled by the homogeneous particle diffusion model, which could also be understood easily. Because SA-SMIP is a high cross-linked polymer material, the high cross-linking degree made pores in SA-SMIP matrix constrict to a degree that solution could not enter directly; added to the short diffusion distance, the swelling of SA-SMIP would help gradual film diffusion process to occur naturally.
The adsorption isotherm of SA-SMIP
The relationship of the equilibrium adsorption capacity (Qe) of SA-SMIP and the feed concentration of SA (C0) is shown in Fig. 9. It displays the equilibrium adsorption capacity (Qe) increased with the feed concentration (C0) of SA until the saturated equilibrium adsorption reached as C0 increased to 4.4 g/L. As C0 was low, the quantities of SA were small. At this condition, almost all absorbed SAs were entered into CIC of SA-SMIP because of the high affinity of CIC to SA, and the increased degree of Qe was also large because the equilibrium constant of CIC was high. With the increase in C0, majority of CICs were occupied by SA gradually, and the adsorption would be varied to DIC gradually. Due to the low affinity of DIC to SA and the increased adsorption amounts, the desorption rate increased, which would lead to the decrease the increasing degree of Qe, and Qe increased slower and slower gradually. As adsorption rate decreased to equal with the desorption rate, Qe would be kept as a constant.

The relationship between Qe and C0.
To verify the above analysis, the Scatchard model was introduced to analyze the binding and recognition abilities of SA-SMIP to SA. The Scatchard equation [36] is shown as follows:
where Qmax (mg/g) denotes the saturated adsorption capacity, Qe (mg/g) is the equipment adsorption capacity, Kd (mg/L) is the equilibrium dissociation constant of the binding sites, and Ce (mg/L) stands for the equilibrium concentration of SA.
The results of regression curves and equation are shown in Fig. 10 and Table III (rows 1 and 2), respectively, from which it could be found that experimental data could be regressed well by two linear equations, which documented that there were two sorts of binding sites obviously, i.e., CIC and DIC, in SA-SMIP. According to the slope and intercept of the related linear equation, Kd and Qmax of different binding sites were obtained, and related Ce and Qe calculated are also presented in Table III, from which it could be found that Ce and Qe of the first binding site were 0.2612 g/L and 26.65 mg/g, respectively, which were smaller obviously than those of the second binding sites (the related Ce and Qe were 1.7060 g/L and 42.80 mg/g, respectively), and implied that the first binding site was CIC and the second was DIC. This conclusion was in accordance with the expression that the adsorption was first carried out at CIC and then transferred to DIC gradually [30]. Besides, comparing R2 of two binding sorts, it could be found that the value of the first was 0.9982, which was much higher than that of the second, and strengthened the conclusion that the first equation is related to the binding site of CIC. Because CICs in SA-SMIP were all same and could adsorb SA specially, the interaction force was same naturally, and R2 was closed to 1 accordingly. Whereas some defects always existed in DIC, and the defect degree was also different, which would lead to the higher difference of interaction force [37], and R2 displayed a value far from 1 correspondingly.

Results of experiment data analyzed by the Scatchard model.
To further explore the adsorption feature of different binding sites in SA-SMIP, the Langmuir and Freundlich models [38] were used to analyze the experimental data related to each adsorption sort, and the regression equation is shown as follows:
Freundlich model:
where Qe (mg/g) is the equilibrium adsorption capacity, 1/n is the Freundlich constant, Ce (mg/mL) represents the equilibrium concentration, and KF corresponds to the adsorption equilibrium constant.
Langmuir model:
where Ce (mg/mL) is the equilibrium concentration, Qe (mg/g) is the equilibrium adsorption capacity, Qm (mg/g) represents the maximum saturated adsorption capacity, and KL corresponds to the Langmuir constant.
The results are presented in Figs. 11(a) and 11(b) and Table IV. It could be found the adsorption feature of both of CIC and DIC was more accordance with the Langmuir model, which could be understood easily. For the adsorption of CIC, because the special imprinted cavities structure, a CIC could only combine with an SA molecule, which was satisfied with the basic theoretical hypothesis of monolayer absorption. Besides, the interaction force of each CIC was almost the same because of the same imprinted cavities structure. Thus, the adsorption of SA on CIC was satisfied with all the basic theoretical hypothesis of Langmuir model completely, i.e., the uniformity of surface and the monolayer absorption, and the adsorption of CIC would be consistent with Langmuir model inevitably. For the adsorption of DIC, on the one hand, there was an obvious difference in the structure of DIC, which would lead to the different adsorption force inevitably, and was more suitable to the first basic theoretical hypothesis of Freundlich model, i.e., the nonuniformity of surface. On the other hand, because DIC was resulted from the dissociation of the self-assembly, the structure of DIC would be still confined to adsorption one SA molecule, added to the high cross-linking of SA-SMIP matrix, the confine of space was strengthened. As a result, the adsorption of DIC was satisfied with the second basic theoretical hypothesis of Langmuir model, i.e., the monolayer absorption. However, the experimental results showed the adsorption of SA-SMIP fits Langmuir model well, which means that the influence authority of adsorption layer was higher than that of uniformity of surface.

Results of adsorption feature for two binding sites analyzed by Langmuir and Freundlich models. (a) CIC and (b) DIC.
Adsorption thermodynamics
The thermodynamic experiment in this work was carried out under an isothermal condition, and the enthalpy change (ΔH0) and entropy change (ΔS0) could not be calculated directly. But the Gibbs free energy change (ΔG0) could be estimated [39]. The equation was as follows:
where R is the gas constant [8.314 J/(mol K)], T (K) is the adsorption temperature, T in this experiment is 313 K, and Qe/Ce are obtained from the Scatchard equation.
From Eq. (13), ΔG0 at each equilibrium concentration could be calculated, the variation trend of estimated Gibbs free energy is shown in Fig. 12, and its values are calculated and summarized in Table V. The negative values of ΔG0 indicated that the adsorption process was spontaneous in a certain concentration range and more favorable at higher concentration.

The relationship between Gibbs free energy and Qe/Ce (313 K).
As is known to all, the adsorption enthalpy is usually a result of changing internal energy, large or small depending on the interaction between sorbent and substrate. With the increase in the concentration of SA, the adsorption capacity increased gradually, which meant that the interaction between sorbent and substrate at the process of adsorption became strong, which confirmed the endothermic nature of the processes, and ΔH0 is greater than zero [40]. Because of the presence of CIC and DIC in the adsorption system, with the increase in the concentration of SA, the adsorption sites were occupied gradually, and during this process, some side reactions would occur unexpectedly, such as the collapse of imprinted cavities. The increased in SA molecular and unexpected side reactions induced the complexity of the adsorption system. It can be understood as an entropy increase process, so that the change in adsorption entropy is greater than zero. On analyzing Eq. (14), the same conclusion can be confirmed.
Influence of preparation conditions on adsorption performance of SA-SMIP
Influence of initiator on adsorption performance of SA-SMIP
Figure 13 shows the influence of initiator on QSA, QTA, and D. It could be concluded that with the increase in azodiisobutyronitrile (AIBN), QSA, QTA, and D increased first and then decreased, which all reached a maximum at nAIBN/nHEMA = 2%. As the amount of initiator was low, the quantity of free radical and the polymerization rate were lower, the polymerization of monomers was insufficient, which would produce a large amount of soluble polymer, oligomer, and monomer [41]. Because the above components would run off in the process of extraction, the self-assembly system would be destroyed and large amount of the imprinted cavities would be changed to DIC and more free active loci (FAL). Due to the strong interaction among FAL, the deformation and collapse of SA-SMIP matrix would occur with an obvious degree. As a result, QSA, QTA, and D presented a low value. With the increase in AIBN, the polymerization tended to be sufficient, and the quantities of soluble polymer, oligomer, and monomer in the system would decrease because the rapid reaction rate would provide enough energy for the polymerization of monomers. At this condition, the cross-linking degree of SA-SMIP would be improved at a large state, and the perfection and stability of imprinted cavities increased. Because DIC has more affinity to SA [30], QSA displayed a higher value than QTA correspondingly. However, as AIBN exceeded the optimal quantity, the explosive polymerization would happen. Because explosive polymerization would produce large amount of soluble oligomer, which could run off during extraction process, DIC and FAL increased inevitably. As discussed above, the increase in FAL related to the increase in pores collapse and reduction of selectivity, and the decrease in QSA, QTA, and D could be understood naturally.

Influence of AIBN on the adsorption performance of SA-SMIP.
Influence of polymerization temperature on adsorption performance of SA-SMIP
Figure 14 displays the influence of temperature (T) on QSA, QTA, and D. It could be found that QSA, QTA, and D first increased and then decreased, and QSA and D reached their maximum values when T = 64 °C, while QTA reached its maximum values when T = 60 °C. At the low temperature, the initiator could not trigger the polymerization sufficiently because small amount of active initiator would result in the slow reaction rate for both polymerization and cross-linking [42], which would lead a majority of monomer and soluble polymer existed in the network of SA-SMIP. Because monomer and soluble polymer would be eluted from SA-SMIP, the self-assembly system would be destroyed, and large amount of DIC and FAL would be produced. Due to the increased difficulty of phase separation, added to the small cross-linking degree, the collapsed degree of pores would kept at a high level, and QSA and QTA showed a low value correspondingly. At the same time, because of the low selectivity of CIC, D would also be at a low level accordingly. With the increase in temperature, the number of activated molecules in the initiator increased, both the polymerization and cross-linking reactions were speeded up, more and more monomers would polymerize; meanwhile, the cross-linking degree would increase, which would facilitate the formation of CIC. As a result, DIC and FAL decreased gradually. Because decreased FAL meant the decreased pores collapse degree, the efficient active sites increased naturally and QSA and QTA increased gradually. Besides, because the increased CIC related to the increased selectivity of SA-SMIP to SA, the increase degree of QSA was higher than that of QTA and D increased naturally. With the further increase in temperature, the majority of initiators would be at the active state, which would quickly decompose and produce numerous radical and further trigger large amount of monomer to react, and as a result, explosive polymerization and stepwise cross-linking would happen because large reaction heat could not be released. Owing to the explosive polymerization and unwanted cross-linking, CIC would be destroyed, while large quantities of DIC would be produced. Because the produced DIC would lead to certain decrease in QSA accordingly, D showed a decrease trend with the further increase in T.

Influence of polymerization temperature (T) on the adsorption selectivity of SA-SMIP.
Influence of nEGDMA/nHEMA on adsorption performance of SA-SMIP
Figure 15 shows the relationships among nEGDMA/nHEMA and QSA, QTA, and D. It could be found that with the increase in nEGDMA/nHEMA, QSA and D increased and reached a maximum at the ratio of nEGDMA/nHEMA = 5 and 6, respectively, while QTA displayed a reversed variation trend with that of QSA. The cross-linking degree of SA-SMIP was the main reason to the above phenomena [43]. At low nEGDMA/nHEMA, the quantities of nEGDMA were small, which means that the cross-linking degree of synthesized SA-SMIP was low. At this condition, the rigidity of imprinted loci in SA-SMIP matrix was poor, and they would deform the imprinting cavity in the process of drying and extraction, which would lead to low QSA and D relatively. Besides, low cross-linking degree would also result in large amount of uncross-linked polymer, which would run off from the original region in extraction process and caused a more serious deformation of the imprinting cavity. Thus, QSA and D presented a lower value naturally. With the increase in EGDMA, the cross-linking degree of SA-SMIP increased, the rigidity of imprinted loci strengthened, while the uncross-linked polymer reduced, the amount of CIC increased, which would increase the affinity of SA-SMIP to SA at a large degree because the predetermined shape of cavities and distribution of functional groups in cavities in the preparation process were well conserved. As a result, QSA and D increased, while QTA decreased. However, as EGDMA exceeded the optimal quantity, severe cross-linking degree of polymers causes a large extent of internal stresses, which would make CIC deform its shape [44], and further resulted in the decreased affinity of imprinted cavity to SA and QSA decreased. While for TA, due to the smaller molecule size, it could still enter into the above deformed cavities and be adsorbed and QTA increased naturally. As for D related EGDMA was larger than that of QSA, which could be ascribed to the decreased DIC and increased CIC.

Influence of nEGDMA/nHEMA on the adsorption performance of SA-SMIP (nEGDMA/nHEMA: the molar ratio of EGDMA to HEMA).
Influence of polymerization time on adsorption performance of SA-SMIP
Figure 16 presents the influence of polymerization time on QSA, QTA, and D. It could be found that with the increase in reaction time, QSA, QTA, and D increased first and then decreased, but the maximum value related time was different, QSA and D related time was 28 h, while that for QTA was 24 h. The above phenomenon could be attributed to that there existed two types of polymerization reaction during the SA-SMIP preparation, namely chain polymerization and stepwise polymerization [45]; in contrast to stepwise polymerization, chain polymerization was more conducive to generate CIC, but during the process of SA-SMIP synthesis, stepwise polymerization with slower reaction rate and higher reaction energy occurred inevitably, which was not conducive to generate stable CIC. At the start of polymerization, chain reaction was not sufficient to form a stable cross-linking network, and there were many soluble components in the system, and the extraction of SA process would change the position of the soluble components and even remove them. Because both of the above cases would destroy the imprinted cavities and increase the level of FAL, the phase separation between SA-SMIP and solvent would be difficult and the pores collapse degree increased [14], and QSA, QTA, and D showed a low value correspondingly. With prolonging t, the polymerization and cross-linking reactions tended to be completed and the quantity of soluble components decreased, which means the majority of CICs would be reserved. Because of the decreased quantity of soluble components, FAL would decrease, the phase separation would become easier, and the pores collapse degree would reduce, and QSA, QTA, and D increased relatively. As the polymerization time prolongs to a given time, all self-assembly groups would be fixed at their original position gradually because of the increased polymer molecular weight and cross-linking degree; thus, CIC would increase and DIC would decrease quickly. Meanwhile, because the reaction rate of chain polymerization was much faster than that of stepwise polymerization, the influence of stepwise polymerization on CIC formation was negligible under these circumstances, and the deformation of CIC could be ignored accordingly. Due to the special selectivity of SA-SMIP to SA, QSA and D still increased, while QTA decreased. However, with the further increase in t, the type of cross-linking reaction changed from chain cross-linking to stepwise cross-linking among the remained acrylic acid and HEMA, because the stepwise cross-linking would destroy the structure of CIC and produce DIC, and FAL would increase correspondingly. Due to the weakened phase separation ability and the increased pores collapse, QSA, QTA, and D decreased.

Influence of polymerization time (t) on the adsorption performance of SA-SMIP.
Influence of suspended dispersant agent on adsorption performance of SA-SMIP
Figure 17 presents the influence of the amount of span80 on QSA, QTA, and D; it could be found that with the increase in Span80, QSA and D reached a maximum at mSpan80/mHEMA = 384.2%, while the maximum of QTA related mSpan80/mHEMA = 115.3%. The reason could be attributed to the interaction among SA, HEMA, and water. Because the solubility of SA and HEMA was much smaller in water than in chloroform, they mainly existed in chloroform. However, due to the presence of hydroxyl group in SA and the hydroxyl group and ester group in HEMA, both of which could interact strongly with water through hydrogen bond, the self-assembly between SA and HEMA becomes more difficult. As a result, the prepared SA-SMIP could form CIC hardly. Span80, an esterification derivative of sorbitol, was always used as a stabilizer of surface polymerization due to the suitable hydrophilicity [46]. During the preparation of SA-SMIP, the function of Span80 was to isolate SA and HEMA from water and help the formation of stable monomer droplet that contained modified TiO2 because Span80 was more hydrophilic than SA and HEMA. Thus, the variation of QSA, QTA, and D with Span80 could be understood easily. As the amount of Span80 was low, monomer droplets could not be isolated from water completely, and SA and HEMA, which are exposed outside, would interact with water, which would damage the self-assembly structure and produce more FAL. Because higher FAL content means difficult phase separation of reaction system and higher degree of pores collapse, the efficient active sites were lower and QSA and QTA presented a lower value naturally. As for D, because the dissociation of self-assembly structure would decrease the amount of CIC while increase the amount of DIC, which means that the special selectivity of sample decreased, and D also presented a low value naturally. With the increase in Span80, monomer droplets were gradually isolated sufficiently, and the stability of monomer droplets was also improved gradually. Because the increased stable monomer droplets means the decreased damage degree of self-assembly system, the quantities of CIC increased, while DIC and FAL decreased, which would lead to the decreased pores collapse degree. As a result, QSA, QTA, and D increased correspondingly. As the amount of Span80 increased to a certain degree, the self-assembly system could almost be maintained sufficiently, the majority of active sites would be CIC. Because of the special selectivity of CIC to SA, QSA and D would still increase, while QTA start to decrease. However, as Span80 exceeded the optimal quantity, the droplet of monomer and modified TiO2 would be split into smaller size because more quantities of Span80 means more micelle produced in the reaction system at the same condition, and more micelle further means smaller size of reactive droplets. Due to the decrease in droplets size, more self-assembly group would be seen on the outer side of prepared SA-SMIP, which would be damaged in the process of extraction and drying and resulted in the decrease in efficient active sites, and QSA, QTA, and D decreased correspondingly.

Influence of the amount of Span80 on the adsorption performance of SA-SMIP (mSpan80/mHEMA: the mass ratio of Span80 to HEMA).
Influence of nSA/nHEMA on adsorption performance of SA-SMIP
Figure 18 shows the influence of nSA/nHEMA on QSA, QTA, and D. It could be found that QSA, QTA, and D presented a maximum with the increase in nSA/nHEMA, but the maximum related nSA/nHEMA was different. QSA, QTA, and D related nSA/nHEMA was 0.25, 0.26, and 0.2, respectively. At low value of nSA/nHEMA, HEMA was surplus after self-assembly. Due to the high solubility of HEMA in chloroform, excessive HEMA would distribute in the obtained SA-SMIP matrix random and elevate the level of FAL. Sourced from the interaction among groups of free HEMA, the phase separation between SA-SMIP and chloroform was difficult. Thus, the collapse level of pores would be large during the process of extraction and drying, which would lead to amount of active sites, i.e., CIC and DIC, lose efficacy, and QSA and QTA presented a low value naturally. With the increase in nSA/nHEMA, surplus HEMA would decrease after self-assembly. Due to the decreased FAL, the phase separation ability between SA-SMIP and chloroform increased, which would reduce the degree of pores collapse, as a result, and the number of efficient CIC and DIC in samples increased, and QSA and QTA increased naturally. However, as nSA/nHEMA increased to an unsuitable degree, HEMA would be insufficient to form the complete self-assembly system with SA. At this condition, the self-assembly system would produce imperfection and even defect gradually because of the decrease in the coordination number. As a result, the quantity of CIC decreased and D decreased correspondingly. But for QSA and QTA, because of the self-assembly equilibrium, the increase in SA in a given range would increase DIC, while decrease FAL reversely, which would decrease the collapse level of pores to certain extent and lead to the increase in efficient active sites, and QSA and QTA still displayed an increase trend. With the further increase of SA, the self-assembly system would be destroyed and became more and more unstable gradually, which would lead to the phase separation between chloroform and SA-SMIP became difficult again, the collapse degree of pores increased again relatively, and QSA and QTA decreased naturally.

Influence of nSA/nHEMA on the adsorption performance of SA-SMIP (nSA/nHEMA: the molar ratio of SA to HEMA).
Comparison on the effects of different preparation conditions on adsorption performance of SA-SMIP
Table VI shows the effect of different conditions, such as initiator, polymerization temperature, nEGDMA/nHEMA, polymerization time, suspended dispersant agent, and nSA/nHEMA, on the adsorption performance of SA-SMIP. It could be found that the maximums of QSA under different conditions were almost the same, the difference was only 1.40 mg/g, however, the maximums of QTA under different conditions were different, the difference was 12.22 mg/g. Among the above conditions, the conditions corresponding to the maximum and minimum values of QTA were SA concentration and polymerization time, respectively. The reasons for the above phenomenon could be referred to the influence of time and nSA/nHEMA on adsorption performance of SA-SMIP.
Conclusion
In this work, SA-SMIP was prepared by reversed phase suspension polymerization using modified titanium dioxide (TiO2) as carrier, SA as template, azodiisobutyronitrile (AIBN) as initiator, HEMA and EGDMA as functional monomer and cross-linker, span-80 and polyvinyl alcohol (PVA) as surfactant, and chloroform as solvent. The operating conditions were investigated and optimized, and optimal conditions were obtained as follows: the ratio of nSA/nHEMAA/nEGDMA was 0.25:1:5, the ratio of nAIBN/nHEMA was 2%, the ratio of mspan-80/mHEMA was 384.2%, and the reaction temperature and time were 64 °C and 28 h, respectively. Under optimal conditions, the prepared sample displayed an excellent separation performance, i.e., the adsorption capacity of SA-SMIP was 30.64 mg/g, the separation degree of SA-SMIP in the solution of SA and TA with molar ratio of nSA/nTA = 1/3 was arrived at 2.90. Thereafter, structures of modified TiO2 and SA-SMIP obtained at optimal conditions were characterized by FTIR adopting original TiO2 and NSMIP as reference, and the results implied that both the modified TiO2 and the SMIP were prepared successfully. Finally, based on the samples prepared under the optimal conditions, the adsorption kinetics and isotherm were investigated and analyzed. Adsorption kinetics results indicated the adsorption of SA-SMIP to SA was a pseudo-second order process, and the adsorption of beginning stage was controlled by homogeneous particle diffusion, but the later stage was controlled by adsorption reaction process. Adsorption isotherm results documented that there were two sorts of bonding sites, CIC would adsorb SA preferentially, DIC could not adsorb SA until the adsorption of CIC was finished. The adsorption for two bonding sites could be well expressed by Langmuir model, but CIC could be expressed better.
Experimental
Materials and equipment
Titanium dioxide (TiO2, rutile type) was obtained from Lanzhou Guangyuan Chemical Co., Ltd. (China, Lanzhou). Silane coupling agent KH570 (AR, >98%) was supplied by Wuhan Chenxi Chemical Co., Ltd. (China, Wuhan). Methanol (CH3OH, AR, >99.5%) was purchased from Shanghai Chunzhang Fine Chemical Research Institute (China, Shanghai). Hydrochloric acid (HCl, 37.5%) was supplied by Dongguan Shengneng Chemical Research Institute (China, Dongguan). Ammonium molybdate [(NH4)2MoO4, 56.5%] was purchased from Tianjin Chemical Reagent Factory Kaida Chemical Plant (China, Tianjin). Sulfuric acid (H2SO4, AR, 98%) was obtained from Dongguan Shengneng Chemical Industry Co., Ltd. (China, Dongguan). L-ascorbic acid (vitamin C, AR, >99.0%) was purchased from Tianjin Deen Chemical Reagent Co., Ltd. (China, Tianjin). PVA was received from Tianjin Guangfu Fine Chemical Research Institute (China, Tianjin). Span80 (C24H44O6) was received from Tianjin Baishi Chemical Industry (China, Tianjin). EGDMA (AR, 98%) was purchased from Liaoning Fushun Chemical Industry (China, Liaoning). SA (95%) was supplied by Shanghai Jiachen Chemical Industry (China, Shanghai). Chloroform (CHCl3, AR, 98%) was obtained from Shanghai Baolong Chemical Industry (China, Shanghai). TA (C30H62O, 90%) was purchased from Huzhou Sifeng Natural Wax Refinery (China, Huzhou). HEMA (99%) was supplied by Tianjin Chemical Reagent Research Institute (China, Tianjin). AIBN (98%) was obtained from Jinan Zhengyu Chemical Industry (China, Ji’nan), and other presented regents in this paper were used as received. The structures of SA, TA, and KH570 are shown in Fig. 19.

Structural formulas of SA, TA, and KH570.
The instruments used in this study were as follows: 752N UV spectrophotometer (Shanghai Precision and Scientific Instrument Co., China) and FTIR (Nicolet NEXUS 670; Thermo Nicolet Corporation, America).
Preparation of stock solution
Preparation of KH570 stock solution
0.5000 g of KH570 was added to a 100 mL beaker and dissolved with methyl alcohol, then transferred it into a 100 mL volumetric flask and diluted with methyl alcohol to volume, and a standard stock solution of 5 g/L KH570 was obtained.
Preparation of SA stock solution
At first, 5 g of SA was added to a 1000 mL beaker and dissolved with chloroform, then transferred it into a 1000 mL volumetric flask completely and diluted with chloroform to volume, and SA stock solution of 5 g/L was obtained.
Preparation of TA stock solution
Ten grams of TA was dissolved with chloroform in a 1000 mL beaker, then the solution carefully transferred into a 1000 mL volumetric flask completely and diluted with chloroform to the volume, and TA stock solution of 10 g/L was obtained.
Determination of maximum absorbance wavelengths
The maximum absorbency of SA and TA was detected with a 752N spectrophotometer (Shanghai Precision and Scientific Instrument Co., China, Shanghai) by full wavelength scanning method using chloroform as reference. The wavelength scope was fixed on 200–380 nm and the scan interval was 2 nm, and the obtained results are shown in Fig. 20. The figure displays the maximum absorbency related wavelengths of SA and TA were 328 and 244 nm, respectively. At the maximum absorbency wavelength of one compound, the absorbency of the other was almost zero, which meant the concentration determination of each compound in solution could be carried out at its maximum absorbance wavelength directly.

The maximum wavelength of SA and TA.
Preparation of standard curves
Preparation of KH570 standard curve
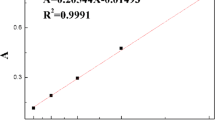
A series of KH570 with concentration from 0.6000 g/L to 1.0000 g/L were prepared, and the absorbency of each standard solution was measured with silicon–molybdenum blue photometric method at 660 nm [47]; the relationship between concentration (C) and absorbency (A) was curved and regressed, and results showed a well linear relationship between C and A (A = 1.0438C − 0.5499, R2 = 0.9994) in the concentration range of 0.6–1, which documented the variation of concentration could be determined directly as the concentration was fell in the range.
Preparation of SA standard curve
Based on the prepared stock solution, a series of SA with concentration from 0.2000 to 3.0000 g/L were prepared, and the absorbency of each solution was determined at 328 nm. After that, the relationship between concentration (C) and absorbency (A) was curved and regressed, and the result showed a well linear relationship between C and A (A = 0.2889C + 0.0149, R2 = 0.9993) in the concentration range of 0.2–3, which documented the variation of concentration could be determined directly as the concentration was fell in the range.
Preparation of TA standard curve
Based on the prepared stock standard solution, a series of TA with concentration from 2.0000 g/L to 12.0000 g/L was prepared, and the absorbency of each solution was determined at 244 nm. Thereafter, the relationship between concentration (C) and absorbency (A) was curved and regressed, and the result showed a well linear relationship between C and A (A = 0.03749C − 0.00932, R2 = 0.9990) in the concentration range of 2–12, which documented the variation of concentration could be determined directly as the concentration was fell in the range.
Modification of TiO2
Thirty milliliters of 5.00 g/L of KH570 methanol solution was added into a dried 100 mL conical flask, which had been loaded with 1 g dried TiO2 particles. After nitrogen purge for 30 min, the conical flask was fixed in a 40 °C oil bath equipped with a magnetic stirrer and reacted for 24 h under nitrogen atmosphere. Thereafter, the sample was filtrated and then washed with isopropanol till the absorbance of elution was smaller than 0.001, as it was detected with silicon–molybdenum blue photometric method (during detection, the reference was prepared with the same way as silicon–molybdenum blue photometric method, except the elution solution was altered by analytical isopropanol). Finally, the obtained particles were dried at 60 °C under a vacuum condition (vacuum degree, 0.8–0.9) for another 24 h and then stored in desiccators for further use.
Characterization of graft ratio
The graft ratios (Gr) were characterized by silicon–molybdenum blue photometric method [47]. After determining the absorbance of original solution and raffinate, the concentration of each of them was calculated, then Gr was calculated using Eq. (15).
where Gr stands for the graft ratio of KH570 on TiO2; C0 and C1 (g/L) are the concentration of KH570 in original and raffinate solution, respectively; 30 (mL) is the volume of KH570; and W (g) is the mass of TiO2. Under the above given conditions, Gr of prepared sample was 34.13%.
Characterization of the structure of modified TiO2
The structure of modified TiO2 was characterized by FTIR (Nicolet NEXUS 670; American Nicolet Corp.). During this process, FTIR of unmodified TiO2 was also presented to help the analysis of modified TiO2. The samples used for FTIR characterization were dried at 120 °C for 48 h to ensure most of water be evaporated, then it was grounded into a proper powder, prepared to tablet with potassium bromide, and finally scanned at the wavelength range of 4000–400 cm−1.
Synthesis of SA-SMIP
First, 3 g of PVA was dissolved in 120 mL distilled water under 95 °C and cooled the solution (1#) to room temperature. Then, 0.5 g of Span80 was injected into a beaker with 30 mL chloroform (CHCl3), which was solution 2#. Thereafter, well-mixed solution 1# and 2# and added it into a 250 mL flask with three necks which with 1 g modified TiO2. Finally, 5 mL (100 mmol/L) SA chloroform solution and 10 mL (200 mmol/L) HEMA were added in turn. After the mixture was agitated for 10 min, 20 mL 500 mmol/L chloroform solution of EGDMA was added and purging nitrogen to the flask for 30 min, then 20 mL (2 mmol/L) AIBN chloroform solution was added through the constant pressure funnel. After that, the flask was immersed in a 64 °C water bath for 28 h. Finally, the obtained polymer microsphere was filtered using a 200-mesh nylon gauze and extracted with a mixture of methanol/AA (9:1, v/v) in a Soxhlet extractor until the absorbency of the mixture at 328 nm kept as a constant. Drying the extracted particles at 60 °C under a vacuum condition (vacuum degree, 0.8–0.9) for 48 h, the obtained SA-SMIP sample was stored in desiccators for further use.
NSMIPs were prepared and treated simultaneously under the same conditions except SA was not added.
Characterization of SA-SMIP and NSMIP
FTIR (Nicolet Corporation) was conducted to explore the structural difference between SA-SMIP and NSMIP, and the method was similar as that introduced in section “Synthesis of SA-SMIP”.
Determination of adsorption selectivity
0.20 g of SA-SMIP was added into a 150 mL conical flask, which contained with 60 mL of above prepared SA and TA stock solution mixtures with the volume ratio vSA/vTA = 1/1 and stayed for 6 h; after that, SA-SMIP was removed and the absorbance of raffinate was detected at 328 nm and 244 nm. Thereafter, the adsorption capacity and separation degree of SA-SMIP to SA and TA were calculated with Eqs. (16), (17), and (18).
where QSA and QTA (mg/g) stand for the adsorption capacity of SA-SMIP to SA and TA, respectively, C and C0 (g/L) are the initial concentration and concentration of SA and TA after adsorption for 6 h, 60 (mL) is the volume of the mixture solution, and W (g) stands for the mass of SA-SMIP.
where D stands for the separation degree of SA-SMIP to SA and TA.
Determination of adsorption kinetics
A series of 0.20 g SA-SMIP were sealed into 200-mesh nylon bags (10 × 60 mm) and marked first, and then, these nylon bags were loaded into a series of 500 mL flasks marked with the same code. Thereafter, 240 mL of 5 g/L SA solution was added into each flask and ensured the tea bag be submersed. Fixed these flasks in a 40 °C water bath, and after 0.5 h, the tea bags were removed according to the number of mark with 0.5 h interval in turn. The absorbency of each raffinate was tested immediately, and the adsorption capacity (Qt) was calculated according to Eq. (19):
where C0 and C (g/L) are the initial concentration and concentration of SA in solution at time t and W (g) stands for the mass of SA-SMIP.
Study of adsorption isotherm
A series of 500 mL flasks were marked, and then, each flask was loaded with 0.2 g SA-SMIP and 240 mL SA solution with the concentration range of 0.8–5.6 g/L. After that, these flasks were put into a 40 °C water bath for 6 h. Removing SA-SMIP in flasks, the concentration of each raffinate was tested immediately. Thereafter, the equilibrium adsorption capacity (Qe) was calculated as given in Eq. (20):
where Ce is the equilibrium concentration of SA and W2 (g) stands for the mass of SA-SMIP.
References
R. Alleti, J. Vagner, D.C. Dehigaspitiya, V.E. Moberg, N.G.R.D. Elshan, N.K. Tafreshi, N. Brabez, C.S. Weber, R.M. Lynch, and V.J. Hruby: Synthesis and characterization of time-resolved fluorescence probes for evaluation of competitive binding to melanocortin receptors. Bioorg. Med. Chem. 21, 5029–5038 (2013).
W. Zhong, W. Wang, Z. Kong, B. Wu, L. Zhong, X. Li, J. Yu, and F. Zhang: Coenzyme Q10 production directly from precursors by free and gel-entrapped Sphingomonas sp. ZUTE03 in a water-organic solvent, two-phase conversion system. Appl. Microbiol. Biotechnol. 89, 293–302 (2011).
N. Yan, Y. Liu, D. Gong, Y. Du, H. Zhang, and Z. Zhang: Solanesol: A review of its resources, derivatives, bioactivities, medicinal applications, and biosynthesis. Phytochem. Rev. 14, 403–417 (2015).
M.A. Taylor and P.D. Fraser: Solanesol: Added value from solanaceous waste. Phytochemistry 72, 1323–1327 (2011).
R.S. Hu, J. Wang, H. Li, H. Ni, Y.F. Chen, Y.W. Zhang, S.P. Xiang, and H.H. Li: Simultaneous extraction of nicotine and solanesol from waste tobacco materials by the column chromatographic extraction method and their separation and purification. Sep. Purif. Technol. 146, 1–7 (2015).
H. Chen, S. Liu, L. Ji, T. Wu, F. Ma, Y. Ji, Y. Zhou, M. Zheng, and G. Huang: Associations between Alzheimer’s disease and blood homocysteine, vitamin B12, and folate: a case-control study. Curr. Alzheimer Res. 12, 88–94 (2015).
C. Zhao, C. Li, and Y. Zu: Rapid and quantitative determination of solanesol in Nicotiana tabacum by liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 44, 35–40 (2007).
C.J. Zhao, L.I. Chun-Ying, F.U. Yu-Jie, and Z.U. Yuan-Gang: Extraction and determination of solanesol in waste tobacco leaves by ultrasonic and HPLC. Chin. J. Appl. Chem. 22, 1265 (2005).
D.S. Tang, L. Zhang, H.L. Chen, Y.R. Liang, J.L. Lu, H.L. Liang, and X.Q. Zheng: Extraction and purification of solanesol from tobacco(I). Extraction and silica gel column chromatography separation of solanesol. Sep. Purif. Technol. 56, 291–295 (2007).
Y. Zhao and Q. Du: Separation of solanesol in tobacco leaves extract by slow rotary counter-current chromatography using a novel non-aqueous two-phase solvent system. J. Chromatogr. 1151, 193–196 (2007).
X. Ma, Z. Meng, L. Qiu, J. Chen, Y. Guo, D. Yi, T. Ji, H. Jia, and M. Xue: Solanesol extraction from tobacco leaves by flash chromatography based on molecularly imprinted polymers. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 1020, 1–5 (2016).
T. Inanan, N. Tüzmen, S. Akgöl, and A. Denizli: Selective cholesterol adsorption by molecular imprinted polymeric nanospheres and application to GIMS. Int. J. Biol. Macromol. 92, 451–460 (2016).
X. Yang, J. Liu, H. He, L. Zhou, C. Gong, X. Wang, L. Yang, J. Yuan, H. Huang, L. He, B. Zhang, and Z. Zhuang: SiO2 nanoparticles induce cytotoxicity and protein expression alteration in HaCaT cells. Part. Fibre Toxicol. 7, 1 (2010).
S. Wu, L. Yao, G. Yu, J. Guan, C. Pan, D. Yong, X. Xiang, and Z. Wang: Facile preparation of dibenzoheterocycle-functional nanoporous polymeric networks with high gas uptake capacities. Macromolecules 47, 2875–2882 (2014).
M. Esfandyari-Manesh, M. Javanbakht, E. Shahmoradi, R. Dinarvand, and F. Atyabi: The control of morphological and size properties of carbamazepine-imprinted microspheres and nanospheres under different synthesis conditions. J. Mater. Res. 28, 2677–2686 (2013).
Y. Wenming, C. Yang, X. Xiaoling, Z. Zhiping, L. Lukuan, and X. Wanzhen: Preparation of indole surface molecularly imprinted polymer by atom transfer radical emulsion polymerization and its adsorption performance. J. Mater. Res. 28, 2666–2676 (2013).
Y. Fu and Y. Yue: Preparation and adsorption selectivity of rutin molecularly imprinted polymers. J. Appl. Polym. Sci. 123, 903–912 (2011).
H. Kim, K. Kaczmarski, and G. Guiochon: Mass transfer kinetics on the heterogeneous binding sites of molecularly imprinted polymers. Chem. Eng. Sci. 60, 5425–5444 (2005).
A. Mehdinia, S. Dadkhah, K.T. Baradaran, and A. Jabbari: Design of a surface-immobilized 4-nitrophenol molecularly imprinted polymer via pre-grafting amino functional materials on magnetic nanoparticles. J. Chromatogr. 1364, 12–19 (2014).
M. Yang, Y. Zhang, S. Lin, X. Yang, Z. Fan, L. Yang, and X. Dong: Preparation of a bifunctional pyrazosulfuron-ethyl imprinted polymer with hydrophilic external layers by reversible addition-fragmentation chain transfer polymerization and its application in the sulfonylurea residue analysis. Talanta 114, 143–151 (2013).
Y. Li, X. Li, C. Dong, Y. Li, P. Jin, and J. Qi: Selective recognition and removal of chlorophenols from aqueous solution using molecularly imprinted polymer prepared by reversible addition-fragmentation chain transfer polymerization. Biosens. Bioelectron. 25, 306–312 (2009).
L. Wang, M. Zhou, Z. Jing, and A. Zhong: Selective separation of lead from aqueous solution with a novel Pb(II) surface ion-imprinted sol-gel sorbent. Microchim. Acta 165, 367–372 (2009).
H. Bagheri, K. Molaei, A.A. Asgharinezhad, H. Ebrahimzadeh, and M. Shamsipur: Magnetic molecularly imprinted composite for the selective solid-phase extraction of p-aminosalicylic acid followed by high performance liquid chromatography with ultraviolet detection. J. Sep. Sci. 39, 4166–4174 (2016).
C. Lai, M.M. Wang, G.M. Zeng, Y.G. Liu, D.L. Huang, C. Zhang, R.Z. Wang, P. Xu, M. Cheng, and C. Huang: Synthesis of surface molecular imprinted TiO2/graphene photocatalyst and its highly efficient photocatalytic degradation of target pollutant under visible light irradiation. Appl. Surf. Sci. 390, 368–376 (2016).
R. Gao, X. Mu, J. Zhang, and Y. Tang: Specific recognition of bovine serum albumin using superparamagnetic molecularly imprinted nanomaterials prepared by two-stage core–shell sol–gel polymerization. J. Mater. Chem. B 2, 783–792 (2014).
A. Martín-Esteban: Molecularly-imprinted polymers as a versatile, highly selective tool in sample preparation. TrAC, Trends Anal. Chem. 45, 169–181 (2013).
G. Ertürk and B. Mattiasson: Molecular imprinting techniques used for the preparation of biosensors. Sensors 17, 288 (2017).
S. Sadeghi, M. Jahani, and F. Belador: The development of a new optical sensor based on the Mn doped ZnS quantum dots modified with the molecularly imprinted polymers for sensitive recognition of florfenicol. Spectrochim. Acta, Part A 159, 83–89 (2016).
Y.M. Ren, J. Yang, W.Q. Ma, J. Ma, J. Feng, and X.L. Liu: The selective binding character of a molecular imprinted particle for Bisphenol A from water. Water Res. 50, 90–100 (2014).
O.S. Muddineti, B. Ghosh, and S. Biswas: Current trends in using polymer coated gold nanoparticles for cancer therapy. Int. J. Pharm. 484, 252–267 (2015).
J.C. Liu, M.J. Xu, L. Tao, and B. Li: Effect of surface-modified ammonium polyphosphate with KH550 and silicon resin on the flame retardancy, water resistance, mechanical and thermal properties of intumescent flame retardant polypropylene. Ind. Eng. Chem. Res. 54, 9733–9741 (2015).
J.P. Simonin: On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics. Chem. Eng. J. 300, 254–263 (2016).
R. Coşkun and S. Akdeniz: Functionalization of poly(ethylene terephthalate) fibers by grafting of maleic acid/methacrylamide monomer mixture. Fibers Polym. 11, 1111–1118 (2010).
C. Valderrama, J.L. Cortina, A. Farran, X. Gamisans, and F.X. de las Heras: Kinetic study of acid red dye removal by activated carbon and hyper-cross-linked polymeric sorbents Macronet Hypersol MN200 and MN300. React. Funct. Polym. 68, 718–731 (2008).
N. Balkaya and H. Cesur: A kinetic study on cadmium adsorption from aqueous solutions by pre-conditioned phosphogypsum. Desalin. Water Treat. 57, 2515–2521 (2016).
H. Yu, Z. Chen, Y. Fu, L. Kang, M. Wang, and X. Du: Synthesis and optimization of molecularly imprinted polymers for quercetin. Polym. Int. 61, 1002–1009 (2012).
T. Huo, Z. Chen, W. Meng, J. Long, X. Liu, and X. Du: Preparation of glutathione molecularly imprinted polymer microspheres by reversed phase suspension polymerization. Polym.-Plast. Technol. Eng. 54, 889–898 (2015).
S. Zhong, C. Zhou, X. Zhang, H. Zhou, H. Li, X. Zhu, and Y. Wang: A novel molecularly imprinted material based on magnetic halloysite nanotubes for rapid enrichment of 2,4-dichlorophenoxyacetic acid in water. J. Hazard. Mater. 276, 58–65 (2014).
J. Pan, Y. Hang, G. Wei, H. Ou, P. Huo, W. Xue, X. Zou, and C. Li: Selective adsorption of 2,6-dichlorophenol by surface imprinted polymers using polyaniline/silica gel composites as functional support: Equilibrium, kinetics, thermodynamics modeling. Chem. Eng. J. 172, 847–855 (2011).
S. Li, X. Huang, M. Zheng, W. Li, and K. Tong: Molecularly imprinted polymers: Thermodynamic and kinetic considerations on the specific sorption and molecular recognition. Sensors 8, 2854–2864 (2008).
C.J. Percival, S. Stanley, A. Braithwaite, M.I. Newton, and G. Mchale: Molecular imprinted polymer coated QCM for the detection of nandrolone. Analyst 127, 1024–1026 (2002).
T. Chen, X.G. Sun, W. Xiao, X.J. Liu, W. Zhang, K. Ma, and Y.R. Zhu: Optimization of microwave-assisted extraction of solanesol from potato leaves and stems. Med. Chem. Res. 19, 732–742 (2010).
R.A.A. Muzzarelli: Chitosan composites with inorganics, morphogenetic proteins and stem cells, for bone regeneration. Carbohydr. Polym. 83, 1433–1445 (2011).
K. Kabiri, H. Omidian, S.A. Hashemi, and M.J. Zohuriaan-Mehr: Synthesis of fast-swelling superabsorbent hydrogels: Effect of crosslinker type and concentration on porosity and absorption rate. Eur. Polym. J. 39, 1341–1348 (2003).
R. Suedee, V. Seechamnanturakit, B. Canyuk, C. Ovatlarnporn, and G.P. Martin: Temperature sensitive dopamine-imprinted (N,N-methylene-bis-acrylamide cross-linked) polymer and its potential application to the selective extraction of adrenergic drugs from urine. J. Chromatogr. 1114, 239–249 (2006).
X.L. Wang, X.Z. Yuan, H.J. Huang, L.J. Leng, L. Hui, P. Xin, W. Hou, L. Yan, and G.M. Zeng: Study on the solubilization capacity of bio-oil in diesel by microemulsion technology with span80 as surfactant. Fuel Process. Technol. 118, 141–147 (2014).
Y.H. Wang, J.P. Lin, Y.H. He, X. Lu, Y.L. Wang, and G.L. Chen: Microstructure and mechanical properties of high Nb containing TiAl alloys by reactive hot pressing. J. Alloys Compd. 461, 367–372 (2008).
Acknowledgments
This work was supported by the Natural Science Foundation of China (Grant No. 51563015).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Duan, C., Chen, Z., Liu, X. et al. Noble surface molecularly imprinted polymer modified titanium dioxide toward solanesol adsorption selectivity study. Journal of Materials Research 34, 3271–3287 (2019). https://doi.org/10.1557/jmr.2019.148
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2019.148