
Abstract
A new molecularly imprinted polymer was prepared on the surface of ethylenediamine-poly(styrene–divinylbenzene) particles by thermal polymerization method using 1-methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide ([EOM][Tf2N]) as template, acrylamide as functional monomer, and chloroform as porogen. The influence of porogen, monomer, and the ratio of [EOM][Tf2N] to monomer and cross-linker was systematically investigated, and the microstructure was characterized by Fouier transform infrared spectrum and scanning electron microscopy. The selective recognition performance of the imprinted polymer for target was investigated by the static adsorption and HPLC. It was shown that the surface imprinted material had excellent selectivity towards [EOM][Tf2N] and the short chain imidazolium ionic liquids, but its adsorption towards the molecules without imidazole ring was quite low. The maximum binding capacities of [EOM][Tf2N] on the imprinted and non-imprinted polymers were 137.2 and 23.4 µmol g−1, respectively. The adsorption equilibrium was achieved in 35 min, and the kinetic adsorption process could be described by pseudo-second order model. From high pressure liquid chromatography measurements, it was found that using the imprinted polymer, 84–91 % [EOM][Tf2N], 10 % 2,4-dichlorophenol, and 3 % N-butylpyridinium chloride could be separated from the mixtures of [EOM][Tf2N] + 2,4-dichlorophenol and [EOM][Tf2N] + N-butylpyridinium chloride, respectively. By contrast, the non-imprinted polymer could adsorb only less than 10 % [EOM][Tf2N], 10 % 2,4-dichlorophenol, and 3 % N-butylpyridinium chloride, respectively, from the mixtures. The imprinted polymer coupled with HPLC was applied for the selective extraction and determination of [EOM][Tf2N] in complex mixture samples.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ionic liquids are new magic green solvents. Due to the unique properties such as good solubility, negligible vapor pressure, high thermal stability and high ionic conductivity, they have attracted great interest for application in chemical synthesis, extraction, dissolution and among others [1–4]. Although there is no air pollution, some drawbacks of ionic liquids have attracted much attention in recent years. On the one hand, many environmentally unfriendly toxic organic solvents were always used in the synthesis of ionic liquids [5]. On the other hand, toxicity of some ionic liquids has been measured referred to different aquatic organisms [6] and human cell lines [7]. Because most ionic liquids have significant solubility in aquatic medium, their discharge into soil and water environment is not negligible with the wide application of ionic liquids. This can cause water environmental pollution and related risks [8–10]. Hence, it is of certain importance to effectively determine trace ionic liquids in environmental samples.
Some traditional analytical methods, such as spectroscopy [11] and HPLC [12], have been used for the determination of organic pollutants. However, due to the interference of the coexisting matrix and the low concentration of ionic liquids in environmental samples, it is necessary to use complicated pre-concentration steps for eliminating any matrix components and improving the detection limit. To this end, many researchers have introduced the molecular imprinted technique to circumvent these limitations [13–16].
Molecular imprinting technology is one of the most promising strategies among the various approaches studied for producing biomimetic receptors for molecular recognition [17]. Molecularly imprinted polymer (MIP) is a template-directed polymerization process. The polymer is based on the assembly of a cross-linked polymer matrix around an imprint molecule that is held in place, either covalently or non-covalently, by judiciously chosen functional monomers [8]. As the template was removed, a binding site which is complementary in size, geometry, and functional groups orientation to the template was remained in the polymer matrix [18, 19]. Therefore, the key feature of MIP is their engineered selectivity for binding to an analyte of interest [20]. In addition, compared to other recognition systems, MIP possesses many promising characteristics, such as low cost and easy synthesis, high stability to harsh-chemical and physical conditions, and excellent reusability [21]. MIP has been widely applied in chromatographic separation [22], solid phase extraction [23, 24], chemical sensors [25], and catalysis etc. [26].
The MIP was traditionally prepared by bulk polymerization. Thus, the removal of template becomes an extremely exhausting task, and the crushing of bulk polymers is a time-consuming and wasteful process that results in irregular sizes and shapes, where this irregularity leads to poor recognition performance and low capacity [27]. Instead, surface imprinted polymer can be applied to avoid the disadvantages arising from bulk polymerized MIP. A fundamental strategy of surface imprinting is to locate the molecularly imprinted sites on the surface of supported material, thereby improving sensitivity for the recognition of target molecules [20]. In comparison with the bulk imprinting, the transfer of template molecules is much easier, and the adsorption and desorption processes are less restricted [28]. Thus, the attention of researchers has been attracted to surface imprinting technique to obtain regularly shaped imprinted polymers, and to improve the adsorption properties [29].
1-Methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide ([EOM][Tf2N]) is a kind of ionic liquid containing ether group. The introduced ethyl group can increase the ionic conductivity and decrease the glass transition temperature of the ionic liquid. This highlights the importance of ionic liquids and their derivatives for the development of new electrolyte materials [30, 31]. Therefore, [EOM][Tf2N] is used as an important electrolyte in the field of electrochemistry. However, the determination of trace [EOM][Tf2N] in environmental system is still a difficult task. To selectively recognize [EOM][Tf2N] ionic liquid from environmental samples, we designed and synthesized a new imprinted polymer, in this work, using [EOM][Tf2N] as the template molecule on the surface of ethylenediamine-poly(styrene–divinylbenzene) particles. The as-prepared imprinted material was found to have a high specific adsorption performance for [EOM][Tf2N] and its structural analogs with imidazole ring, and could easily reach an adsorption equilibrium. Moreover, by coupling with high pressure liquid chromatography, the imprinted polymer is suitable for separation and determination of template molecule from the interference containing six-membered ring compounds, such as phenolic and pyridine substances.
Results and discussion
Optimization of the preparation conditions
Effect of the solvent
In the polymerization process, solvent could influence the strength of non-covalent interaction and the performance of MIP. To this end, three porogens (methanol, chloroform, and acetonitrile) were tested for the synthesis of imprinted polymers in this work, and the imprinting factor (β) [32] was chosen to investigate the selective recognition ability of the MIP and NIP for the template molecule. The higher β value stands for the better imprinting effect. The result was shown in Table 1.
Here, Q MIP and Q NIP (µmol g−1) stand for the binding capacity of [EOM][Tf2N] on the MIP and NIP materials.
It was found from Table 1 that higher binding capacity and imprinting factor (3.99) were only obtained in chloroform. Due to the strong polarity of methanol and acetonitrile, these two solvents could interfere with the H-bond interaction between template and functional monomer, and the binding ability of the polymers prepared in these two solvents was weak. Therefore, chloroform was selected as the optimum porogen in this work.
Effect of the functional monomer
A good functional monomer can create the molecular recognition sites by interaction with template, and maintain suitable shape and size when the template is removed. The widely used monomers, such as methacrylic acid, acrylamide, and 4-vinylpyridine, are capable of forming hydrogen bonds with the electron-acceptor group. To choose the optimal monomer, UV absorption spectra for 0.1 mmol dm−3 of 1-methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide in methanol, 0.6 mmol dm−3 of functional monomer (methacrylic acid, acrylamide, or 4-vinylpyridine) in methanol, and the mixture of template with each functional monomer were determined, respectively. As examples, the UV spectra of [EOM][Tf2N] and acrylamide were shown in Fig. 1.

UV absorption spectra of [EOM][Tf2N] and acrylamide in methanol. Adsorption conditions: 1, C [EOM][Tf2N] = 0.1 mmol dm−3; 2, C acrylamide = 0.6 mmol dm−3; 3, the mixture of 1 and 2; 4, the sum of adsorption value of 1 and 2. T = 25 °C, time = 40 min
It was shown that the maximum absorption wavelength of [EOM][Tf2N] underwent red-shift when the functional monomer was added to the solution of [EOM][Tf2N]. At the wavelength of 222 nm, the absorbance value of the mixture of [EOM][Tf2N] and acrylamide was only 74 % of the summation of the two components. However, the absorbance values (not listed) of the mixture of [EOM][Tf2N] with methacrylic acid and 4-vinylpyridine were 77 and 80 % of their summation, respectively. Moreover, the results in Table 2 showed that the binding capacities and β value (4.3) of the imprinting polymer synthesized using acrylamide as functional monomer was maximum among the three kinds of imprinting materials. This difference was resulted from the pre-arrangement between [EOM][Tf2N] and acrylamide through hydrogen bond between –O– of [EOM][Tf2N] and –NH2 of acrylamide, 3-H of [EOM][Tf2N] and =O of acrylamide, as well as the ion–dipole interaction between imidazolium cation of [EOM][Tf2N] and =O of acrylamide. These interactions of [EOM][Tf2N] with acrylamide were stronger than those of [EOM][Tf2N] with the other two monomers, which led to the highest affinity of MIP towards the target analyte. Therefore, acrylamide was selected as functional monomer in the next experiments.
Ratio of template to functional monomer could affect imprinting result and adsorption properties of the polymers. If more [EOM][Tf2N] was retained onto MIP, more binding sites could be formed for binding target molecules [33]. The binding properties of imprinted polymer prepared at different molar ratios (1:2, 1:4, and 1:6) but the same amount of crosslinking agent were evaluated, and the results were given in Table 3. It can be seen that the binding capacities of MIP towards [EOM][Tf2N] increased with the increase of the monomer amount. However, excess of monomer resulted in difficult polymerization, which might be detrimental to template-functional monomer interaction and led to poor polymer yield and long post-treatment period [16]. Therefore, molar ratio of 1:6 ([EOM][Tf2N] to acrylamide) was selected for the further test.
Selection of cross-linker
The selectivity is greatly influenced by the nature and amount of cross-linking agent used in the synthesis of the imprinted polymer. Ethylene dimethacrylate is a common cross-linker used in non-covalent molecular imprinting, which can serve stabilized binding sites and impart mechanical stability to the polymer matrix [34]. Thus, ethylene dimethacrylate was chosen as the cross-linker to prepare the imprinted polymer. To improve the selectivity of MIP, three different molar ratios of template to cross-linker were tested where the amount of functional monomer was fixed. As can be seen from Table 3, more binding sites were formed with the increase of the amount of ethylene dimethacrylate, and the adsorption performance was enhanced also. However, when the molar ratio was higher than 1:20, the performance was decreased. This might be explained from the fact that with the increase of the cross-linker concentration, the degree of cross-linking increased and restricted the transfer of template molecules. To enable the cavities to maintain three-dimensional structure complementary in both shape and chemical functionality to that of the template molecule after the removal of template, 1:6:20 of template to monomer and ethylene dimethacrylate was selected to enhance recognition properties of MIP towards template.
Characterization of the molecularly imprinted polymer
Infrared spectra analysis
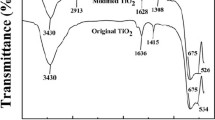
The Infrared spectra of MIP, NIP, and DVB particles were recorded in the range of 400–4,000 cm−1 by using KBr pellet method (Fig. 2). It was shown that the peak at 2,947 cm−1 was the stretching vibrations of –CH3 and –CH2 of ethylene dimethacrylate, the band at 1,723 cm−1 was resulted from the stretching vibration of –COOH, and the relatively strong absorption band at 1,138 cm−1 was due to the stretching vibrations of C–O–C of ethylene dimethacrylate. In addition, no characteristic absorption peaks were observed for C=C and C=N of the imidazole ring in MIP. This suggests that the template molecules have been eluted completely, and no template molecules were retained on the MIP.

IR spectrum of the polymers: a DVB; b NIP; and c MIP
SEM analysis
The SEM images of MIP, NIP, and DVB particles were shown in Fig. 3. It is clear that particles of MIP exhibited a more porous and rough structure, but those of NIP and DVB were smooth and there were no surface cavities, which reveled that the addition and elution of template molecules resulted in the imprinting cavities on the surface of MIP particles during the polymerization process.

SEM images of the polymers: a imprinted polymer; b non-imprinted polymer; c DVB particles
By comparing the size of MIP, NIP, and DVB particles, it could be observed that the diameters of MIP, NIP, and DVB particles were similar to each other. This suggested that the imprinting layer was very thin due to polymerization reaction occurring on the surface of DVB particles, and the surface imprinted polymers were prepared successfully.
Adsorption characteristics of MIP and NIP
Adsorption isotherm
The adsorption capacity is an important factor in evaluation of the recognition performance of the imprinted polymer. To measure the adsorption behavior of polymer materials, adsorption isotherms were determined at the concentration range of [EOM][Tf2N] from 0.3 to 10.0 mmol dm−3. As shown in Fig. 4, the adsorption capacity of [EOM][Tf2N] on the MIP and NIP increased with the increase of initial concentration, but MIP exhibited relatively higher adsorption capacity than that of NIP. When the concentration was beyond 7.0 mmol dm−3,the binding capacity of MIP was close to saturation, and the gap of the adsorption capacity between MIP and NIP appeared to be larger at the high concentration of [EOM][Tf2N]. The results demonstrated that the imprinted cavities were formed in MIP after the removal of the template molecules, which were spatially oriented to [EOM][Tf2N] and complementary to [EOM][Tf2N] in term of size and shape. And there were no specific imprinted cavities in NIP so that the binding capacity was quite low.

Equilibrium adsorption isotherms of [EOM][Tf2N] on MIP and NIP. Adsorption conditions: V = 2 cm3, m MIP = m NIP = 15 mg, C [EOM][Tf2N] = 0.3–10.0 mmol dm−3, T = 25 °C, time = 40 min
The most commonly used isotherm is described by Langmuir equation (Eq. 2) [14]. For this adsorption model, the maximum amount of substrate bounded on the adsorbent in the equilibrium solution can be calculated by
where Q max (µmol g−1) is the maximum binding capacity towards [EOM][Tf2N], C eq (mmol dm−3) is equilibrium concentration of [EOM][Tf2N], and B is a constant.
From Eq. (2), the Q max values of [EOM][Tf2N] were calculated to be 137.2 and 23.4 µmol g−1 for MIP and NIP, respectively. The observed significant difference suggested that hydrogen-bonding and ionic-dipole interactions between template and monomer were established, which resulted in the tight combination between them by self-assembly and led to specific imprinted cavities in MIP. However, there were no imprinted cavities in NIP, so that their binding capacity was lower. Consequently, it is definite that specific imprinting cavities in MIP play an important role in recognizing the template molecule.
Adsorption kinetics
Adsorption kinetic experiments were carried out in a solution containing 0.5 mmol dm−3 ionic liquid for the bounding of [EOM][Tf2N] on MIP and NIP at different time intervals. The adsorption curve in Fig. 5 showed the relationship between the adsorption capacities and the equilibrium time in the range of 0–250 min. It can be seen that the adsorption capacities of [EOM][Tf2N] increased rapidly in the first 30 min, because the presence of a large number of high-affinity binding sites on the surface of MIP enabled the template to bind and recognize easily during the initial adsorption process [35]. After 35 min, the adsorption capacities increased slowly and eventually reached adsorption equilibrium. The adsorption trend on NIP and MIP was similar. However, the adsorption capacities on NIP were relatively low due to the lack of the imprinting cavities, and the adsorption process was mainly non-specific adsorption.

Adsorption kinetic of [EOM][Tf2N] onto the MIP and NIP. Adsorption conditions: V = 2 cm3, m MIP = m NIP = 15 mg, C [EOM][Tf2N] = 0.2 mmol dm−3, T = 15 °C
To examine the controlling mechanism of sorption process, the experimental data were fitted by pseudo-first order (Eq. 3) and pseudo-second order kinetic models (Eq. 4), respectively [33].
where K a and K b are the rate constants, Q t (µmol g−1) is the binding capacity at given time t (min), and Q e (µmol g−1) is the binding capacity at equilibrium. It is clear that only if ln(Q e − Q t ) vs. t or t/Q t vs. t gives a linear plot, the model is applicable. Here, we found that the pseudo-second order model fitted the experimental data well (t/Q t = 0.18389 + 0.06835t, r 2 = 0.9927), but the pseudo-first order kinetic model could not describe the time dependence of the binding capacity since no such a linear relationship was observed. This indicated that two types of sorption, i.e. physisorption and chemisorption, coexited for the sorption on the MIP, and the adsorption process was controlled by chemisorption.
Binding specificity of the MIP and NIP polymers
The specific recognition of the polymer materials was identified by static adsorption experiments. To this end, 15.0 mg of the polymer was added into 1.5 cm3 of methanol containing 0.5 mmol dm−3 of [EOM][Tf2N] or its structural analogs such as [C4mim][CH3SO3], [C4mim][Tf2N], [EOM][PF6], [TOM][PF6], N-butylpyridinium chloride, phenol, and 2,4-dichlorophenol (Fig. 6).

Structures of [EOM][Tf2N] and its analogues
Results in Table 4 illustrated that the MIP had a similar recognition performance towards [EOM][Tf2N], [C4mim][CH3SO3], [C4mim][Tf2N], [EOM][PF6], and [TOM][PF6]. From Fig. 6, it could be seen that the template molecule, [C4mim][CH3SO3], [C4mim][Tf2N], [EOM][PF6], and [TOM][PF6] contained similar structure of imidazole ring, and hydrogen bonding and ionic-dipole interactions could be established between imidazolium cation and functional monomer. Therefore, the bounding amount of MIP towards [EOM][Tf2N], [C4mim][CH3SO3], [C4mim][Tf2N], [EOM][PF6], and [TOM][PF6] did not show much difference. However, the adsorption capacity of [EOM][Tf2N] on MIP was much higher than that of the other four imidazolium ionic liquids, and ΔQ value (ΔQ is the difference of binding capacities on MIP and NIP, ΔQ = Q MIP − Q NIP) [14] towards [EOM][Tf2N] was the maximum among the five compounds. This indicated that the imprinting cavities on MIP were complementary to the structure of [EOM][Tf2N], and the highest adsorption capacity was resulted from the specific adsorption. Though [C4mim][CH3SO3], [C4mim][Tf2N], [EOM][PF6], and [TOM][PF6] owned the same imidazole ring, and the imprinted polymer could adsorb them to some extent, their structure and size were not match with [EOM][Tf2N] completely so that ΔQ value was lower. In addition, as shown in Table 4, the adsorption performance of the MIP towards the compounds without imidazole ring, such as N-butylpyridinium chloride, phenol, and 2,4-dichlorophenol were very poor. Because the structure of six-membered ring compounds is very different from that of the imidazolium ionic liquids, the MIP could not recognize them effectively. These results implied that the “imprinting cavities” were key factor in the binding process, and the MIP prepared in this work had a good imprinting effect for short chain imidazolium ionic liquids.
Selectivity evaluation of MIP and NIP
To further demonstrate the application of the prepared MIP, common pollutants 2,4-dichlorophenol and N-butylpyridinium chloride (Pyn) were used as the interfering substances. The extraction experiments were conducted by addition of 50.0 mg of MIP to the mixed solution containing 0.5 mmol dm−3 [EOM][Tf2N] and 0.1 mmol dm−3 2,4-dichlorophenol, and 50.0 mg of MIP to the mixture containing 0.5 mmol dm−3 [EOM][Tf2N] and 0.5 mmol dm−3 Pyn, under the condition of incubation for 1 h at room temperature. As a contrast, 50.0 mg of NIP was treated by the same procedure as that of MIP. The supernatants were analyzed by HPLC at a flow rate of 0.5 cm3 min−1. The mixture of methanol and water (35/15, v/v) was used as mobile phase for the determination of the mixture of [EOM][Tf2N] and 2,4-dichlorophenol, while the mixture of methanol, water, and acetic acid (5/45/0.0045, v/v/v) was used as mobile phase for the determination of the mixture of [EOM][Tf2N] and Pyn.
The chromatograms for the mixed solutions of [EOM][Tf2N] + 2,4-dichlorophenol and [EOM][Tf2N] + Pyn in methanol with and without extraction treatment by MIP and NIP were shown in Fig. 7. It was found that 84–91 % [EOM][Tf2N], 10 % 2,4-dichlorophenol, and 3 % Pyn could be separated from the two mixed solutions by MIP. By contrast, when NIP was used as adsorbent, only less than 10 % [EOM][Tf2N] and 2,4-dichlorophenol and 3 % Pyn were adsorbed, respectively. These results showed that [EOM][Tf2N] could be selectively recognized by MIP, but the recognition performance of MIP towards 2,4-dichlorophenol and Pyn was very poor. The possible reason was that the structure of 2,4-dichlorophenol and Pyn could not match with the template molecule, which led to the low adsorption capacity of MIP for these compounds. Therefore, a good selective separation performance of [EOM][Tf2N] from six-membered ring compounds was achieved using MIP.

Chromatograms for the mixtures: a [EOM][Tf2N] + 2,4-dichlorophenol; and b [EOM][Tf2N] + Pyn. (1) By direct injection of the mixture; (2) supernatant of mixture extracted by NIP; (3) supernatant of mixture extracted by MIP. Chromatographic conditions of the C18 reversed-phase column: detection wavelength, 216 nm; mobile phase, methanol–water (35/15, v/v) for chromatogram (a) and methanol–water-acetic acid (5/45/0.0045, v/v/v) for chromatogram (b); flow rate, 0.5 cm3 min−1; column temperature 30 °C
Conclusions
In this study, [EOM][Tf2N] molecularly imprinting polymer was synthesized on the surface of ethylenediamine-poly(styrene–divinylbenzene) particles. The selective adsorption performance and its application in the selective extraction of [EOM][Tf2N] from coexisting substance were investigated. From the above experimental results, the following conclusions have been drawn.
-
1.
The imprinted polymer was prepared in chloroform using acrylamide as functional monomer, and the imprinting factor was the highest for the MIP synthesized at the molar ratio of 1:6:20 (the template:functional monomer:cross-linker).
-
2.
The adsorption equilibrium of the imprinted material could be achieved within 35 min, and the pseudo-second order model fitted the kinetic adsorption curves.
-
3.
The surface imprinted polymer showed high selectivity and adsorption capacity towards short chain imidazolium ionic liquids, and the imprinted polymer could be coupled with HPLC for the selective separation of [EOM][Tf2N] from the six-membered ring compounds, and its analytical determination as well.
Experimental
Ethylenediamine-poly(styrene–divinylbenzene) (DVB) particles (40–60 μm) were purchased from Shanghai Boshi Biotech Co., Ltd (Shanghai, China). The ionic liquids of 1-methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide ([EOM][Tf2N]), 1-butyl-3-methylimidazolium methanesulfonate ([C4mim][CH3SO3]), 1-butyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide ([C4mim][Tf2N]), 1-methoxyethyl-3-methylimidazolium hexafluorophosphate ([EOM][PF6]), 1-methoxymethyl-3-methylimidazolium hexafluorophosphate ([TOM][PF6]), and N-butylpyridinium chloride (Pyn) were obtained from Shanghai Cheng Jie Chem. Co. LTD (Shanghai, China). Methacrylic acid, acrylamide, 4-vinylpyridine, and ethylene dimethacrylate were purchased from Acros Co. (New Jersey, USA) and distilled before use. The analytical reagent grade of methanol, acetonitrile, acetic acid, and chloroform were obtained from Tianjin Kermel Chem. Co. (Tianjin, China). Azobisisobutyronitrile and 2,4-dichlorophenol were from Beijing Chem. Co. (Beijing, China) and the former was recrystallized before use. The chromatographic purity methanol was obtained from Zhengzhou Guoda Chem. Co. (Henan, China). Ultra pure water was prepared from Milli-Q purification system (MilliPore, USA). Chromatographic purity reagents and ultra pure water were used in the HPLC analysis, and deionized water was used in other experiments.
A T1810 UV–vis spectrophotometer (Beijing, China) and a high pressure liquid chromatography (HPLC, Waters, USA) equipped with a PDA 2998 UV–Vis detector and a reversed-phase C18 column (4.6 × 150 mm) were used in the present work. The scanning electron microscope (SEM) micrographs of the sorbents were obtained at 20.0 kV on a JSM-5610LV scanning electron microscopy (JEOL, Japan). Infrared spectra (IR) in KBr were recorded at 4,000–400 cm−1 using a Perkin-Elmer 983 infrared spectrophotometer (Norwalk, USA). A THZ-82 thermostatic oscillator (Jiangsu, China) was used for the preparation of the polymer.
Preparation of the ionic liquid imprinted polymer materials
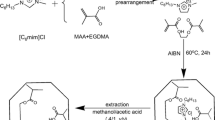
In this work, the polymer materials were prepared by thermal polymerization. First, 0.2097 g of [EOM][Tf2N], 0.2132 g of acrylamide, and 0.5 g of DVB (the carrier) particles were added into 50 cm3 of chloroform. After the mixture was shaken for 6 h at room temperature for pre-polymerization, ethylene dimethacrylate and azobisisobutyronitrile were added. Next, the mixed solution was purged with nitrogen for 10 min to remove oxygen, and then was sealed for polymerization at 60 °C for 24 h in a water bath thermostatic oscillator. Then, the resultants were extracted with a mixed solvent of methanol/acetic acid (8:2, v/v) for 48 h at 80 °C until the template molecule, 1-methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide could not be detected in the extraction solvent. The obtained particles were dried at 60 °C for 24 h. As a reference, the non-imprinted polymer (NIP) was synthesized using the same procedure but without the template. The route for preparation of the imprinted polymer was shown in Fig. 8.

Schematic illustration of the preparation of [EOM][Tf2N] molecularly imprinted polymer
Static adsorption experiments
The performance of selective adsorption, the optimum preparation conditions and the adsorption dynamics of the polymer materials were studied by static adsorption experiments. To evaluate the above parameters, 15.0 mg of the MIP and NIP particles were added into 1.5 cm3 of [EOM][Tf2N] methanol solutions, respectively. The mixtures were allowed to stand for 1 h at room temperature. Then, [EOM][Tf2N] in the supernatant was determined by UV–Vis spectrophotometer. The binding capacity (Q/µmol g−1) of [EOM][Tf2N] on the polymer was calculated by the following equation
where C 0 and C e (mmol dm−3) are the initial and the final concentrations of 1-methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide or its analogs, V (cm3) is the volume of solution, and m (mg) is the mass of MIP or NIP particles.
HPLC evaluation experiments
The HPLC experiments were carried out to evaluate the ability of selective separation of the prepared polymer for the template molecule and the interfering substance. In the static adsorption experiment, 20.0 mg of the imprinted and non-imprinted polymer materials were added into 1.5 cm3 of the mixed solution (the mixture of [EOM][Tf2N] + 2,4-dichlorophenol and [EOM][Tf2N] + N-butylpyridinium chloride), respectively. The target analyte in the mixed solution before and after adsorption by polymer materials were analyzed by the HPLC with a flow rate of 0.5 cm3 min−1 at 30 °C, and the UV–Vis detector was operated at 216 nm.
References
Dupont J, Souza RF, Suarez PA (2002) Chem Rev 102:3667
Myasoedova GV, Molochnikova NP, Mokhodoeva OB, Myasoedov BF (2008) Anal Sci 24:1351
Pei Y, Wang J, Xuan X, Fan J, Fan M (2007) Environ Sci Technol 41:5090
Li Z, Liu X, Pei Y, Wang J, He M (2012) Green Chem 14:2941
Gouveia W, Jorge TF, Martins S, Meireles M, Carolino M, Cruz C, Almeida TV, Araujo ME (2014) Chemosphere 104:51
Luis AGP, Irabien A (2010) J Mol Liq 152:28
Das RN, Roy K (2014) Chemosphere 104:170
Carlo P, Cinzia C, Daniela P, Michela G, Francesca A, Gianfranca M, Luigi I (2006) Green Chem 8:238
Randall JB, Michael AB, Michelle AE, Gary AL (2005) Environ Toxicol Chem 24:87
Coleman D, Spulak M, Garcia MT, Gathergood N (2012) Green Chem 14:1350
Fan Y, Zhang S, Wang Q, Li J, Fan H, Shan D (2013) Spectrochim Acta A 105:297
Qi P, Lin Z, Li J, Wang C, Meng W, Hong H, Zhang X (2014) Food Chem 164:98
Gao X, Fan J, Wang X, Zhang Y (2013) Acta Chim Sinica 71:1411
Zhu G, Fan J, Gao Y, Gao X, Wang J (2011) Talanta 84:1124
Gao X, Fan J, Zhu G, Wang X, Wang J (2013) J Sep Sci 36:3277
Zhu G, Fan J, Gao X, Wang J (2013) Adsorpt Sci Technol 31:791
Tan L, Kang C, Xu S, Tang Y (2013) Biosens Bioelectron 48:216
Muhammad T, Cui L, Jide W, Piletska EV, Guerreiro AR, Piletsky SA (2012) Anal Chim Acta 709:98
Krupadam RJ, Patel GP, Balasubramanian R (2012) Environ Sci Pollut Res 19:1841
Tan L, Li W, Li H, Tang Y (2014) J Chromatogr A 1336:59
Chen L, Li B (2012) Anal Methods 4:2613
Luo X, Zhan Y, Huang Y, Yang L, Tu X, Luo S (2011) J Hazard Mater 187:274
Faghihian H, Ghanbari Adivi F (2012) Adsorpt Sci Technol 30:205
Shen X, Zhu L, Wang N, Ye L, Tang H (2012) Chem Commun 48:788
Bali Prasad B, Jauhari D, Tiwari MP (2013) Biosens Bioelectron 50:19
Yuan Y, Wang Y, Huang M, Xu R, Zeng H, Nie C, Kong J (2011) Anal Chim Acta 695:63
Hu J, Feng T, Li W, Zhai H, Liu Y, Wang L, Hu C, Xie M (2014) J Chromatogr A 1330:6
Freitas LA, Vieira AC, Mendonca JA, Figueiredo EC (2014) Analyst 139:626
Poma A, Turner AP, Piletsky SA (2010) Trends Biotechnol 28:629
Dobbelin M, Azcune I, Bedu M, Luzuriaga AR, Genua A, Jovanovski V, Cabanero G, Odriozola I (2012) Chem Mat 24:1583
Lewandowski AA, Mocek S (2009) J Power Sources 194:601
Jia X, Xu M, Wang Y, Ran D, Yang S, Zhang M (2013) Analyst 138:651
Lopez Mdel M, Perez MC, Garcia MS, Vilarino JM, Rodriguez MV, Losada LF (2012) Anal Chim Acta 721:68
Yan HY, Row KH (2006) Int J Mol Sci 7:155
Chen D, Deng J, Liang J, Xie J, Huang K, Hu C (2013) Anal Methods 5:722
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (Grant No. 20977025 and 21377036) and Science and Technology Department of Henan Province (Grant No. 13A610538 and 132102210436).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhu, G., Wang, X., Gao, X. et al. Preparation of surface molecularly imprinted polymer and selective extraction of 1-methoxyethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide. Monatsh Chem 146, 431–440 (2015). https://doi.org/10.1007/s00706-014-1342-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-014-1342-2