
Abstract
Hot-melt extrusion has found extensive application as a feasible pharmaceutical technological option over recent years. HME applications include solubility enhancement, taste masking, and sustained drug release. As bioavailability enhancement is a hot topic of today’s science, one of the main applications of HME is centered on amorphous solid dispersions. This review describes the most significant aspects of HME technology and its use to prepare solid dispersions as a drug formulation strategy to enhance the solubility of poorly soluble drugs. It also addresses molecular and thermodynamic features critical for the physicochemical properties of these systems, mainly in what concerns miscibility and physical stability. Moreover, the importance of applying the Quality by Design philosophy in drug development is also discussed, as well as process analytical technologies in pharmaceutical HME monitoring, under the current standards of product development and regulatory guidance.

Graphical Abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The high-throughput screening methodology created many new drug candidates with low aqueous solubility in the last decades, classified as II or IV by the Biopharmaceutical Classification Systems (BCS) [1]. The low aqueous solubility of these molecules is typically the bottleneck for absorption, which leads to low bioavailability (BA) and justifying their failure as therapeutic agents. Diverse approaches have been employed to overcome solubility barriers, such as reducing particle size, amorphous solid dispersions, lipid-based strategies, surfactants, and cyclodextrins, among others [2, 3].
Amorphous solid dispersions (ASD) have been recognized to optimize the solubility of poorly soluble materials [2, 3]. Therefore, significant effort has been devoted to understanding solid dispersions lately, in various aspects, such as manufacturing processes, polymeric carriers’ applications, and the physical properties of prepared systems. Considered complex formulations, a complete understanding of the physical structure and chemical properties is essential to predict solubility, BA, and even stability of the solid dispersion.
ASDs are the outcome of the kinetic entrapment of the amorphous active compound, where it is molecularly solubilized in the carrier. These systems have an improved dissolution rate, but they also tend to revert to the more stable thermodynamic form, the crystalline [4]. Indeed, this is the primary concern of ASD, which leads to phase separation and recrystallization and can eventually affect product performance on dissolution [5,6,7,8,9].
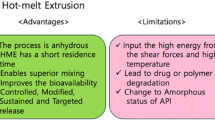
Besides hot-melt extrusion (HME), preparation techniques of ASDs include spray drying, freeze-drying, and supercritical fluid drying. HME found its place in the pharmaceutical area, and many researchers embraced this technique due to its promising performances [10, 11]. The extruder applies energy through shear and temperature to the drug and the thermoplastic excipients. The energy produced by the combination of temperature and friction can overpass the crystal lattice energy and turn the polymer molten. During extrusion, the material is simultaneously mixed and dispersed.
Comparing to other manufacturing processes for ASD, HME presents unique characteristics that justify the high interest of formulation scientists and pharmaceutical companies over the world. It allows continuous manufacturing, is solvent-free, is relatively fast, and requires a narrow footprint. Nonetheless, the high process temperatures, the requirement for downstream processing, and the large energy consumption are significant drawbacks. Besides, there are not many excipients with thermoplastic properties approved for pharmaceutical applications, and the metastable nature of the final drug product is always challenging. In the daily routine of laboratories and factories, some technical difficulties still exist, and the full potential of HME is yet to be met, like any breakthrough innovation [12].
In this review, the use of HME in the pharmaceutical industry is discussed. The focus is on bridging of HME technology with pharmaceutical development to better understand the benefits and fragilities for poorly soluble compounds. Steps and tools for the development of a successful HME product are discussed. Furthermore, this work intends to shed light on Quality by Design (QbD) principles for developing HME formulations, promoting the adoption of these concepts in both academic and industry settings.
PRINCIPLES of HME
At the end of the eighteenth century, HME was invented and applied in the manufacturing of lead pipes. It has been used in other industries since then, like plastic, rubber, and food. The technology was also found useful in the pharmaceutical industry for the robust manufacturing of very different Drug Delivery Systems [10,11,12]. Current interest is rising exponentially, with over 500 papers published during the last 10 years.
Despite the enormous potential of HME for solubility enhancement [13], few are on the market so far, but this tendency is clearly changing. Companies are now specialized in HME, including Abbvie through the Meltrex® technology, and Grünenthal through the use of Intac® formulation. SOLIQS, now a brand of Abbvie, has developed Meltrex® formulation and redeveloped Kaletra®. Kaletra® (lopinavir/ritonavir) is a well-known example of a formulation that represents the impact of HME in product performance. In addition to the BA enhancement, the redeveloped HME product brought significant benefits for patients with a reduced dosage, frequency of administrations, and improved stability at room temperature [4], and this was recognized by the Food and Drug Administration USA (FDA) through a fast-track approval [12]. The HME technology has been used by Abbvie since then for the development of many other products already in the market, as Mavyret®, Norvir®, Viekira Pak®, and Venclexta®. Similarly, Grünenthal GmbH developed an abuse-deterrent formulation to prevent drug abuse focusing on opioids, the Intac® technology, using high-molecular weight polyethylene oxide (PEO) mixtures that lead to an end product with high resistance to crushing through HME. This technology has been licensed to other companies, namely Endo Pharmaceuticals for Opana® ER (Oxymorphone HCl) and Janssen for Nucynta® (tapentadol). HME is generally a sought solution for abuse deterrence as the solid forms are not crushable or chewable [12].
In HME, the components are transformed by heat and mechanical stress into a new material of constant shape and density [3, 9]. This process involves compacting, blending, and dispersing a mixture of excipients and drug substance by two rotating screws through the heated barrel [14]. At the end of the barrel, there is a die, dictating the shape of the extruded system [12]. The theory behind HME technology (Fig. 1) can be summarized step-by-step as follows [4, 9, 12]: feeding through a hopper, mixing and kneading, flowing, venting, extrusion from the die, and downstream processing.

HME as an efficient processing method for solid dispersions and possible obtainable pharmaceutical forms: flakes, powder, pellets, tablets, films, and two-layered forms through co-extrusion
HME works under high temperatures to soften the blend, and the different barrel sections are demarcated with specific temperatures [15]. After feeding, the material is conveyed by the rotating screws while it is melted, mixed, suffers kneading and dispersion. Mixing is a crucial step during HME and may be classified as distributive or dispersive. Distributive mixing is related to drug homogeneity within the blend, whereas dispersive mixing means particle size reduction and molecular distribution [9]. Overall, HME aims to produce an intimately blended end product, the extrudate, where all the materials are mixed to the molecular level. Twin-screw extrusion offers several benefits over single screw and is preferred in pharmaceutical processes. It provides an intense mixing of the components (high kneading and dispersing capability), easier feeding, a lower potential to overheat, and shorter residence time [11, 14].
Over the past 20 years, extruders’ manufacturers worked in meeting the particular requirements of the pharmaceutical industry. The core unit and principles are similar to extruders used for plastics, but the main requirement is to follow the current Good Manufacturing Practices (GMP). Individual parts of the extruder must be built from a special type of stainless steel to avoid reactions or adsorption with the formulation. There are also FDA-approved lubricating oils that should be used, as well as water-cooled tubing [9, 14]. This technology is still under implementation in the pharmaceutical industry, specifically in adjusting documentation on cleaning, specifications, and validations [9].
Process analytical technologies (PATs) have been the focus of both regulators and the pharmaceutical industry. Along with the well-known International Conference on Harmonization (ICH) guidelines Q8 (R2), Q9, Q10, and Q11 [16,17,18,19] emphasizing QbD and continuous manufacturing, FDA also issued the “PAT–A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance” guideline [20], regarded as the core of this concept [21]. PAT has been applied in HME to improve control and real-time analysis [4, 22]. Many techniques are currently available to measure and control process parameters such as product temperature, feed rate, screw speed, pressure, as well as product characteristics like residual crystallinity, drug dispersion/homogeneity, and drug particle size or concentration [23]. Rheology and several spectroscopic techniques, such as optical, ultrasonic, electrical, UV-VIS, Raman, and infrared, have been applied for HME production [9, 15, 24, 25]. Besides, alternative techniques have demonstrated adequate capacity to control low amounts of crystallinity, such as terahertz, dielectric, NMR, and ultrasonic spectroscopies. These techniques were not used in HME so far, but they are likely to be applied in the future [23, 26].
The selection of the analytical method depends on the intended application but also practicability during measurement (measuring equipment or probe), the extent of physical and chemical information provided, the complexity of the required data analysis (not rarely demanding multivariate analysis), and its cost. The development of these real-time measurement methods is similar to any other analytical method for product release or stability testing and requires a full validation as described in the ICH guidance Q2(R1), to ensure that it fits for its intended use. Reports of real PAT applications within HME in the pharmaceutical industry are still limited [4]. PAT tools and their application in HME processes have been thoroughly reviewed [23, 26,27,28].
POLYMERS IN HME
The carrier is usually made from meltable substances, either polymeric (more common) or non-polymeric (like lipids). After the HME process, they function as drug depots or release retardants [9, 11]. Essential prerequisites are their thermal stability and thermoplastic behavior. Nonetheless, due to the usually short residence time, most thermolabile drugs are not excluded from HME processing [29]. Polymeric carriers must be thermoplastic and thermally stable. Other relevant characteristics include suitable Tg or Tm (usually in the range of 50 to 180°C), low hygroscopicity, and low toxicity since large amounts are required. The preferred carriers are the ones with high miscibility with the compound because a higher drug load may be achieved. Characteristics like lipophilicity and hydrogen bonding groups are also requisites for high solubilization [30]. Polymeric materials can be biodegradable or non-biodegradable, from natural or synthetic sources.
Natural polymers are valuable sources for pharmaceutical and biomedical applications. However, their degradation is usually based on enzymes at a hardly predictable rate [15]. Synthetic polymers were developed to modulate and improve physicochemical properties, which will ultimately control the products’ performance. The necessity for using biodegradable excipients was identified by advancements in tissue engineering, gene therapy, and controlled release of drugs [15]. The goal of these materials is to perform a predetermined task, as drug release, through their slow degradation. Therefore, biodegradable polymers should be biocompatible (free of endotoxins, non-toxic, carcinogenic, immunogenic, or inflammatory) and have adequate mechanical, physicochemical, and thermal behavior. Moreover, they should present suitable degradation kinetics and resistance to sterilization methods, if required by the dosage form [15]. Non-biodegradable polymers have also been widely applied in very different systems, from oral formulations to transdermal films, implants, and scaffolds for tissue engineering.
Physicochemical properties (as aqueous solubility, viscosity, or Tm/Tg) command the choice of a specific polymer. Table I summarizes the characteristics of the most common natural polymers and derivatives tested in HME processes. Table II summarizes the characteristics and uses of the most common synthetic biodegradable polymers (processing temperatures are not mentioned as they depend heavily on the structure of the specific polymer). Table III presents an overview of the most common synthetic non-biodegradable polymers with applications reported in HME processes.
PHARMACEUTICAL DEVELOPMENT UNDER QbD PRINCIPLES
The QbD philosophy promoted an in-depth knowledge of products and manufacturing processes. It initiates right at the beginning of the pharmaceutical development but continues during commercial production, as defended by the FDA and the European Medicine Agency (EMA) [31, 32]. According to the ICH Q8(R2), “quality cannot be tested into a product but must be incorporated by design” [16]. QbD replaced quality by testing and is a better approach.
Essential elements of the pharmaceutical development under QbD principles, as defined by the ICH Q8(R2) guideline, are the Quality Target Product Profile (QTPP), the Critical Quality Attributes (CQAs), the Risk Assessment, and the Design Space. Both formulation and processing conditions may be considered critical (critical material or process parameters) and govern drug product CQAs [16]. Several studies have described the relationship between formulation and process parameters using QbD and rational approaches [33,34,35,36]. Although the primary aim of preliminary studies is to develop a formulation and preparation process subjected to further optimization, critical process parameters (CPPs) should be identified from the early beginnings [1, 4, 37].
By ICH Q8(R2), a design space is the “multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality”, i.e., meeting the CQAs. A design space is beneficial since no regulatory change is required to work within the defined range of input variables, which can be both material attributes and process parameters [38]. The constant assessment of potential hazards through risk assessments is a crucial concept of QbD. Fishbone diagrams are useful as a starting point as they provide an overview of the system. Some examples applied to HME have been published [11, 38,39,40,41,42,43]. An example applied to the initial risk assessment of an HME product is depicted in Fig. 2. This approach should be complemented with a more detailed tool, as the Failure Mode and Effects Analysis (FMEA) or the Risk Estimation Matrix (REM), which have also been applied to HME [37, 42].

Fishbone diagram for an HME process based on ICH Q8(R2) recommendations [16]
HME is now a well-understood industrial process, and CPPs for each product may be readily determined [44]. The most common CQAs for ASDs manufactured by HME are acceptable levels of chemical degradation, adequate levels of residual crystallinity, suitable solubility, and dissolution rate [45]. CQAs are deeply affected by an intricate interaction of processing parameters and material attributes. CPPs for extrusion are usually temperature, residence time, screw speed, screw design, feeding rate, shear stress, or specific energy [44, 46]. Although easy testing may occur during steady-state production, the definition of CPPs is not straightforward because some factors are distributional in nature, as shear stress, barrel temperature, and residence time. Therefore, they are usually controlled through process temperatures, screw speed, screw design, and feed rate [44].
One of the most relevant tools to develop a design space is the Design of Experiments (DoE), which is essential to decouple all the complex interactions of input variables and allow a complete process understanding [21, 38, 47]. Several DoE studies have been published for ASDs, and some factors described to have a primary role for the physical stability of the formulation, such as drug load, polymer type, and physicochemical characteristics such as molecular weight [39, 48]. For instance, Pawar and his group developed an efavirenz HME formulation based on a QbD approach, where the combination of HPMCAS and Soluplus with 30% of the drug was optimized based on mathematical modeling [49]. Other studies have also highlighted the influence of process parameters, including screw speed, temperature, feeding rate, and screw design on product quality [47, 50,51,52,53,54]. In the vast majority of literature reports on HME and DoE, the most common studied response is dissolution [38, 39]. Some studies also consider physical stability in the statistical analysis, using formulation and process variables with long-term and accelerated stability [48]. Principles of HME formulation and process development are reviewed in the following sections, including scale-up and QbD issues.
FORMULATION DEVELOPMENT
The majority of excipients used in HME products are also applied for common solid forms. They may be matrix carriers, bulking agents, release modifying agents, thermal lubricants, antioxidants, or others. The selection of the excipients conveys specific characteristics to the HME-based formulation [11]. One of the specificities of this type of formulations is the relatively high amount of polymers, sometimes higher than the approved quantities in the Inactive Ingredient Database from the FDA. In some cases, toxicological studies may be required.
Drug properties may be either positive or harmful to the HME formulation and process. At the beginning of the development, a thorough drug characterization must be performed, including thermal, chemical, and physical properties [14]. Some drug characteristics are relevant for a quick assessment of the feasibility of amorphous formulations and the suitability of HME as the processing technology. For instance, drugs with very high Tm (> 250°C), thermal instability, or high melt viscosity are usually not recommended for the HME process. Other characteristics are usually considered, namely the number of hydrogen acceptors or donors to establish intermolecular interactions with the polymer, the solubility in different solvents (aqueous and organic), solubility in biorelevant media, the Tm and Tg (its ratio is preferred below 1.3 [4]), logP, particle size distribution, among others.
For the development of any solid dispersion, the pre-formulation is a critical stage. The selection of processing conditions is highly influenced by the degradation of the materials and rheological properties of the blend. Drug and carrier properties should be deeply evaluated, as drug solubility in different solvents, drug solubility in polymeric solutions, Tm of the drug, Tg of polymer, drug–polymer miscibility, melt viscosity, and thermal stability of the blend [4]. The selection of potential carriers relies on the drug miscibility in the polymeric matrix, polymer physical properties, the stability of the composition, and other prerequisites of final dosage forms. Additionally, functional excipients as stabilizers, surfactants, antioxidants, plasticizers (usually added to reduce Tg and melt viscosity, smoothing the extrusion process), diluents, release modifiers, and processing aids can also be included in the HME formulation [9, 11]. All these properties will impact the process parameters that should be thoroughly studied and defined in the design space.
The drug and the polymeric carriers may suffer chemical transformations during HME processes [14]. Solvolysis and oxidation are two common mechanisms for the degradation of drugs. Nonetheless, solvolysis is rarely an issue, as HME is a solvent-free process [14]. Oxidation has been described due to peroxides remaining after the polymer synthesis or on polymer oxidation. For instance, excessive temperatures needed for under-plasticized cellulose-based polymers (as HPC) may lead to polymer oxidation [9]. Antioxidants should be considered if oxidative degradation of drugs or carriers is likely to occur. According to Lang et al., mechanisms of chemical degradation may be classified into main or side-chain reactions [55]. The main-chain reactions include cross-linking and scissions of the polymer backbone [56, 57], whereas side-chain comprehend cyclization and elimination [55]. Examples of polymer degradation during thermal treatment in HME have been reviewed [55] and are summarized in Table IV. Another common risk for HME-based formulations is drug–polymer interactions, often triggered by thermal and mechanical energy that accelerate these reactions. Some of these incompatibility cases are well described in the literature [69, 70].
Physical Stability Considerations
The stability of HME products has been demonstrated to be related to the characteristics of carriers, the physical state of the compound, packaging materials, and storage conditions. Although HME formulations usually have good long-term stability [9], amorphous compositions are metastable and tend in nature to the most thermodynamically favored state through recrystallization [55]. This is one of the most common problems observed with ASDs, in which the drug reverts into the crystalline form on storage.
Both thermodynamic and kinetic factors determine the physical stability of this type of composition [71, 72]. Thermodynamic factors are related to the thermodynamic stability of ASDs and control the occurrence of recrystallization. As the dispersion of solvates in solvents, ASDs should be thermodynamically stable if the drug load is below the saturation concentration. Therefore, thermodynamic factors are related to miscibility and polymer solubility concepts [73], discussed in the next section (drug–polymer miscibility/solubility), and are responsible for nucleation and crystal growth [74]. The kinetic factors are, on the other hand, related to kinetic stability and therefore to molecular mobility and the rate of crystal growth [72, 74]. ASDs with high drug loading may be thermodynamically unstable but may be physically stable over enough time for clinical use within a specific timeframe (translated into shelf life). These kinetic factors include the Tg of drugs, polymers and their blends, the molecular mobility of drugs and their intrinsic properties of physical stability (like the glass-forming ability), and the intermolecular interactions between the drug and polymers, discussed hereafter. These factors are highly affected by the environmental conditions and the preparation process [73]. Thorough revisions of these thermodynamic and kinetic concepts controlling the physicochemical properties of ASDs are available in the literature [72,73,74].
The storage of ASDs 50°C below Tg is commonly accepted to decrease the risk of recrystallization, owing to reduced molecular mobility [75]. Nonetheless, molecular mobility still occurs below this point due to β-relaxations, and 50°C may not be enough taking into account typical storage time for pharmaceuticals [76]. Therefore, the characterization of the β-relaxation is crucial since amorphous products are usually stored at temperatures where relaxation is driven mainly by the β-process (below Tg). This characterization is typically performed with calorimetry (differential calorimetric screening (DSC) or isothermal microcalorimetry) or dielectric spectroscopy [76].
Two main approaches are generally considered to increase the physical stability of amorphous formulations, as reviewed by Baghel and colleagues [77] and first by Janssens and Mooter [78]. In one, polymers kinetically stabilize the amorphous systems through the reduction of the molecular mobility, “freezing” the drug and blocking any molecular movement. The addition of polycarbophil, poly(vinylpyrrolidone) (PVP) K25, or HPMC may be used as crystallization inhibitors [9]. On the other, molecular mobility is reduced by intermolecular interactions, which provide stability through the decrease of the thermodynamic energy of the system. These interactions are typically van der Waals, H-bonding, hydrophobic, electrostatic, and rarely ionic. Although weak, its sum is often enough to stabilize solid dispersions.
A number of equations were developed to predict molecular mobility. The three most commonly used are the Arrhenius equation, the Kohlrausch–Williams–Watts (KWW) equation, and the Adam–Gibbs (AG) equation. The Arrhenius equation may be applied to crystallization data to estimate the long-term physical stability of ASDs [79]. Zhu et al. managed to measure the impact of moisture and polymers on the recrystallization of ritonavir, which was well described by the parameters of the Arrhenius model. The model seemed feasible for estimating the long-term physical stability based on short-term data generated under accelerated conditions [79]. Bhardwaj et al. also correlated physical strength to molecular mobility in itraconazole in the amorphous form. The group identified β-relaxations responsible for its physical instability, which exhibited Arrhenius behavior, temperature dependent over the entire experimental temperature [80]. Miyanishi and his group evaluated the recrystallization of a nifedipine ASD, showing that the solid dispersion would need to be stored at −20°C to maintain its performance for at least 3 years [81]. Despite the wide use of this equation, the Arrhenius model is not always accurate. Amorphous polymers act as strong glasses exhibiting (near) Arrhenius behavior, and most of the small drugs act as fragile glasses which deviate significantly from the Arrhenius behavior [82]. In these cases, fragility parameters are preferred through the KWW or the AG equations.
The KWW equation links the “relaxation recovery enthalpy” to the average relaxation time constant (τ) and a stretch parameter (β) [78, 83]. The KWW equation has been mainly applied in single-component compositions [84, 85] and some complex systems [86]. However, the predictive capability for physical stability was demonstrated to be somewhat limited, as in studies performed with celecoxib [87, 88]. Its performance is usually acceptable for single-component systems but often fails when multicomponent systems are evaluated due to the increased complexity. Although still widely used, the predictive ability of the KWW equation is considered nowadays limited [82]. To overcome some of the handicaps of the KWW equation, the non-linear AG equation was proposed [78]. Mao et al. outlined a straightforward method based on DSC to assess the relaxation time [89]. The AG equation has been successfully used for the calculation of relaxation times and correlation with the physical stability of ASDs. Literature reports studies on indomethacin [90], salicin, felodipine and nifedipine [91], indomethacin, felodipine, griseofulvin, citric acid, ketoconazole and nifedipine [89], and even mixtures of phenobarbital and nifedipine in a PVP matrix [92]. What is still to be clarified is how these concepts may be related to a multicomponent system, in complex formulations. Although there was a clear improvement over the KWW equation, the AG theory did not always predict the physical stability of ASD accurately, as in the simvastatin case [93]. Clear limitations of the AG equation are that the relaxation process is not exponential in nature, and other entropic contributions besides configurational are not taken into account. Nonetheless, it seems that either calculated from KWW or AG approaches, the stability may still be predicted appropriately, at least qualitatively [82].
Drug–Polymer Miscibility/Solubility
It is known that solutes and solvents are miscible only within specific percentage ranges, which also applies to the case of drugs and polymers [55]. A single-phase ASD system is usually preferred due to improved physical stability compared to a multi-phase system [55, 94]. Moreover, a low percentage of hydrophilic polymers in drug-rich phases decrease the release rate of poorly soluble compounds [55]. High drug–polymer miscibility is needed to lower the risk of recrystallization, and there are several approaches to evaluate properly this issue.
The Gordon–Taylor equation is used to predict the Tg of amorphous dispersions [95]. Deviations from the theoretical Tg are usually an indication of intermolecular interactions within the components. A positive deviation generally suggests that the number and strength may be greater than in the physical mixture due to, for instance, H-bonding, and a negative deviation is generally a sign of loss of interactions after mixing [96]. Several studies report the use of the Gordon–Taylor (or Fox) equation to predict the miscibility of drug–polymer compositions. For instance, Nair et al. determined the influence of interactions on the Tg of various drug–PVP blends, namely propranolol hydrochloride, acetaminophen, griseofulvin, naproxen, carbamazepine, or salicylamide [96]. Moreover, molecular interactions based on the deviation between experimental and theoretical Tg within four drug–amino acid systems were recently studied [97]. Another interesting study by Rask et al. reported increasing positive deviations with increasing copovidone ratios, suggesting strong interactions [98]. In another study, various grades of HPMC were used to produce ASDs of itraconazole by HME, and the theoretical Tg was compared with the experimental results [99].
Miscibility may also be predicted based on the calculation of the three-dimensional solubility Hansen parameters (δ). Compounds with comparable δ values are probably miscible [100]. Precisely, three Hansen parameters are calculated for each molecule, measured in MPa0.5: the energy from dispersion forces between molecules (δd), the energy from the dipolar intermolecular force between molecules (δp), and the energy from hydrogen bonds between molecules (δh). Then, the total δ is calculated through the combination of solubility parameters. Group contribution methods may be applied, like the one by Hoftyzer and Van Krevelen [3, 101], or the more recently developed by Just and Sievert [102]. The literature considers a cut-off value for the difference in δ of less than 7 MPa0.5 for good miscibility [4, 55, 100, 103]. This method is widely applied for ASDs. Forster et al. evaluated two model drugs and some excipients to predict the formation of glassy solutions. Miscibility was determined experimentally by DSC and thermomicroscopy, and the experimental results met the Hanssen predictions [103]. Another study by Baghel et al., with dipyridamole and cinnarizine, predicted successfully the miscibility of binary mixtures tested [104]. Carbamazepine and Soluplus miscibility was correctly estimated based on Hansen parameters by Djuris et al. [105]. Zhang and colleagues selected the proper carriers for HME for the drug baicalein also using the described method [106]. Although widely applied, this approach presents limitations, and for systems involving long-range interactions (such as ionic) or highly directional (as H-bonds), this approach may not work. Moreover, it is based on a pure chemical approach and does not consider crystal lattice energy [3, 82, 104]. Yoo et al. studied a multicomponent amorphous system that showed ASD formation, regardless of the Hansen results [107]. In another study, the predicted Hansen parameters demonstrated a poor correlation with the experimental results [96]. Hence, in compositions with strong interactions, miscibility will probably be rated too low if assessed by the Hansen approach.
The Melting Point Depression (MPD) theory is also applied to predict miscibility. The basic principle is that the melting point of a drug decreases if it is miscible with a carrier, as it becomes a thermodynamically favorable phenomenon. The polymer that reduces the melting point the most is the more probable to be miscible with the drug [108]. Therefore, the theory of Flory–Huggins was adopted to evaluate drug–polymer solubility through the calculation of the interaction parameter (χ) [109]. Several successful examples are available from the literature. Marsac et al. estimated the χ from MPD data for two compounds, nifedipine and felodipine, when blended with PVP K-12, and the theoretical results were in accordance with the experimental data [110]. Also, Tian et al. determined the χ for felodipine with Soluplus and HPMCAS using the MPD method, demonstrating limited miscibility [111]. The miscibility of carbamazepine and Soluplus was successfully estimated based on the Flory–Huggins theory by Djuris et al. [105], and Yang et al. used a theoretical model based on the MPD to successfully predict the solubility of paracetamol in poly(ethylene oxide) (PEO) [108]. To apply the MPD method, both the compound and the polymer need to be chemically stable over the studied range of temperature [3], and enough molecular interactions are required for the depression in the Tm be perceived in the DSC. Besides, this method is more suitable for systems where the drug has a Tm significantly higher than the Tg of the polymer [82]. However, the most significant handicap is that the calculation of χ is linear only at low percentages of polymer and, therefore, best applied to high drug loading systems [82].
Phase diagrams are another valuable tool for the development of ASDs. They are built with the Flory–Huggins theory and the link between χ and temperature [55]. It generates a curve between unstable and metastable regions, called spinodal [112]. Several examples may be found in the literature [35, 104, 111, 113, 114]. For instance, Thakral and Thakral investigated the miscibility of PEG 6000 with 83 drugs [113]. Baghel et al. presented a phase diagram of four systems, considered to provide a reasonable estimation of physical stability [104]. Li and colleagues constructed a phase diagram for the blend of felodipine and Eudragit® EPO and concluded that these diagrams are useful also to select processing temperature for HME manufacturing to ensure complete miscibility [35]. Phase diagrams of albendazole–polymer compositions were also used to assess the feasibility of HME and spray drying [114]. Phase diagrams are temperature dependent, and the miscibility of the drug–polymer system may change with slight variations in the product temperature [82].
HME PROCESS DEVELOPMENT
The HME process has been demonstrated as crucial to guarantee the CQAs of the drug product. For the first extrusion tests, the definition of general processing conditions is needed. These conditions rely on the physicochemical characteristics of drug and excipients and the phases to be considered. One of the two regimes may be chosen: miscibility or solubilization regime [115]. In the miscibility concept, the extrusion temperature is higher than the Tm, which requires a screw design able to provide higher distributive mixing to spread the two liquids. Here, residence time and shear stress are less significant for the efficacy of mixing. When considering the later, where the extrusion temperature is lower than the Tm, higher specific energy input and aggressive screw designs are needed to guarantee enough shear and residence time. However, as shown by Maddineni et al., excessively harsh designs may result in unnecessary impurities [116]. The choice of screw design may become easier when the polymer melt viscosity is known. The modular design of the screws permits different configurations [117] through the use of forwarding or reverse conveying elements, kneading blocks, and other structures (Fig. 3) [14]. Moreover, when dealing with thermo-sensitive materials, reducing the residence time, or lowering the processing temperatures should be considered [118]. In summary, through the careful analysis of the collected data, it is possible to select the initial process parameters probably very close to the final or optimal.

General characteristics of screws and details of screw elements. Di is the inner diameter of the screw, and Do is the outer diameter
The next stage is focused on assessing the manufacturability, solubility, and stability of prepared ASDs. It should focus on processing temperatures, screw speed, melt pressure, and motor load. Most HME systems provide measurements in real time for these parameters, which are used to rank order performance. The first evaluation is purely visual, considering the presence of crystallinity when the extrudate is not seen as a transparent glass [47]. This evaluation should be supported by polarized light microscopy, complemented by other analytical technologies, such as DSC. Pressure and motor load are always evaluated in every extrusion test, but its study and optimization through processing parameters are usually performed later, along with selecting the prototype. The extrudate is then milled, and its performance is deeply characterized. Typical attributes evaluated in this stage are dissolution rate, chemical degradation, solid-state, and physical stability, where ideal systems will have no change during storage. In case the compound is a BCS class IV, an in-depth characterization may be necessary, where testing in animal models is especially recommended [1, 112]. Results from each issue (manufacturability, solubility, and physicochemical stability) allow the selection of the preferred system. In general, solubility is considered primacy. The next topic for evaluation is physical stability, as options still exist for enhancing physical resilience, for instance, through restrictive storage conditions. Lastly, manufacturability is assessed. Excessive motor loads or high levels of impurities may be further improved with slight changes in formulation or process parameters.
As soon as the prototype formulation and process are identified, the product development enters the optimization stage. At this step, process parameters and formulation characteristics require careful evaluation, namely the process length, the screw speed, the barrel temperature profile, the screw design (which controls the residence time), the specific energy, and the feed material properties (as particle size, water content, morphology, crystal habit, bulk density). Others include the feed configuration (fed together or separately, at the same position or different parts of the barrel), the downstream processing method, and finally, the scale-up [1, 47] (Fig. 4). Besides the contribution of each individual variable, interactions between them should be studied during process development, as they are common in HME and may influence the product CQAs heavily. DoE is one of the main tools in this stage. Considered state-of-the-art in the pharmaceutical industry, DoE should support the laboratory runs, analyze the impact of input variables on CQAs, decouple multivariate interactions, and optimize the lead formulation and the final processing parameters [119].

Process development in three main stages: preliminary extrusion tests, process development, and process optimization
When the aim is to improve solubility, dissolution behavior is the main CQA and is thoroughly evaluated during pharmaceutical development. The goal is to predict dissolution, permeation [71], and eventually recrystallization in the GI tract due to supersaturation [120,121,122], to optimize formulation development. This is usually evaluated through biorelevant dissolution experiments, where drug permeability may be assessed as well by artificial or cell membranes coupled in the dissolution system [123]. Simulated gastric and intestinal media under non-sink conditions are usually used to mimic the GI tract, where the effect of excipients in the drug solubilization, supersaturation, and its maintenance (including the potential to inhibit precipitation in vivo) may be properly assessed [122]. The results are then used, together with pharmacokinetic data, to build an in vivo–in vitro correlation (IVIVC), or a physiologically based pharmacokinetic (PBPK) model [71, 124]. The drug release from ASD is complex and requires a proper characterization and a careful evaluation of all the in vitro data to predict its in vivo performance. Moreover, innovative predictive tools are still required in the field of enabling formulations [125, 126], and the combination of in vitro and in silico data will be decisive to support rational formulation development.
FORMULATION OF FINISHED DOSAGE FORMS
Molten materials are conveyed to the downstream equipment for final dosage form preparation. This may involve milling, pelletization, calendaring, or tableting/encapsulation (Fig. 5) [11, 15, 127]. Cooling the extrudate may be performed with air, nitrogen, on conveyors, rolls, or even with water. Optimizing the cooling rate is of foremost importance to obtain the required amorphicity. Rapid cooling would form a relatively low crystallinity level (being an amorphous or molecularly dispersed product), whereas slow cooling would result in crystal growth [15]. The shape of the extrudate is molded by the die. Circular dies are the most common and are used for pellets and granules. Films and patches require flat dies, and annular dies are dedicated to tubing and co-extrusion. The molten blend can also be used in injection molding [9], which can result in a tablet or a capsule shape, or into customized designs, as adhesives, vaginal tablets or rings, eye inserts, or others. All of this can be performed in a single continuous process, which can potentially decrease overall costs during production.

Downstream processing equipment: a cooling calender, b mill, c pelletizer, d shaping calender, e injection molding, f film extrusion, and g co-extrusion
The most common downstream processing for oral administration is milling, to be finally converted into dosage forms like granulates, tablets, or capsules. The particle size of granules impacts the process capability. However, especially when poorly soluble compounds are concerned, it has an important impact on bioperformance, as generally speeds up the drug release rate. Therefore, it is an important parameter to understand and control. For certain extrudate compositions (for instance, cellulose-based), milling can be challenging. The selection of the type of milling technique depends on the material characteristics and the target mean size and size distribution of the resultant powder. For extrusion materials, hammer or pin mills are usually preferred (impact mills). The final size distribution is generally smaller with pin mills (15–30 μm) than when using hammer mills (20–60 μm) [128]. For solid pharmaceutics, like tablets and capsules, the material flow is crucial, and fine particles are usually avoided. Therefore, a granulation step is, in some cases, added to the process as this range of particle sizes is relatively low to ensure a predictable powder flow.
However, the disintegration time may become too long if the milled extrudate is filled directly into capsules or compressed into tablets. This is because polymers have high binding and gelling properties, and they are present at high levels in the formulation, which leads to the formation of non-dispersible lumps when in contact with water [129]. In such cases, water-insoluble excipients, spacers among polymeric ASD particles, should be used. Best results were found with microcrystalline cellulose and crospovidone, but inorganic excipients as dicalcium phosphate may be used [130]. Another strategy is to use highly soluble ingredients to promote the formation of a porous system when in contact with water, triggering a faster drug release from the extruded matrix, using, for instance, mannitol or lactose. Typically, final dosage forms containing ASDs require disintegrant amounts of 5 to 20%, higher than usual. Crospovidone or another disintegrant with limited swelling performance is recommended to avoid the formation of gelified lumps when in contact with water.
Changes in the mechanical properties of the components during extrusion make ASDs less compressible than physical mixtures [129]. Molecular mobility is deferred due to the low free volume during extrusion, which leads to a compact product and prevents a further decrease in density during compression [131]. Therefore, extragranular excipients with suitable compactibility are essential to achieve a tablet with adequate pharmacotechnical properties, namely hardness, friability, and disintegration time. Sufficient lubrication is also crucial to avoid mechanical problems during compression processes, as picking and sticking.
SCALE-UP
Process scale-up is part of product development and enables large-scale and commercial production, assuring drug product CQAs simultaneously [55]. There is not much published information about HME scale-up, but some reports have proposed a number of models. The upscale of continuous processes is considered more straightforward than batch processes. Using the same equipment and process parameters, scale-up is assured by longer running times [11, 21]. However, moving to a larger extruder demands a complete characterization of the process and end product to guarantee that it has not changed.
For this purpose, several scale-up models have been tried over the years. Carley and McKelvey presented in 1953 the first scale-up method, based on an adiabatic concept [132], later recovered by Nakatani [133]. The influence of heat transfer in an HME scale-up was reported by Schenkel, Maddock, and Chung [134]. Others extended previous laws to a whole non-isothermal and non-Newtonian situation [135] or tried to relate the effects of processing conditions (throughput rate and screw speed) on different scales [136]. New scale-up rules were later developed and published by Bigio and Wang [137].
Methodologies had evolved until today. The geometric similarity between extruders is considered crucial to ensure the scale-to-scale production of HME materials with similar properties [21]. This is referred not only to the likeness of screw design (similar conveying, distributive, and dispersive sections) but also to the screw geometry itself, as it can have implications on shear stress input and residence times [138]. When the extruders at both scales are geometrically similar, scale-up should be relatively straightforward [21]. Simple relationships between processing parameters may be used to provide a target throughput at a larger scale. The main HME process scale-up theories are known as the Volumetric Scale-up, the Heat Transfer Scale-up, and the Power Scale-up, widely discussed in the literature [26, 47] and briefly compared hereafter.
The three theories culminate with the calculation of the targeted process throughput (and consequently the feed rate) [26]. The Volumetric Scale-up focuses on maintaining the same degree of fill of the extruder. It is considered for geometrically similar extruders with different diameters. In a simplified approach, the screw speed is kept constant, and the targeted process throughput is then calculated. The Heat Transfer Scale-up should be applied when the process is limited by the heat transfer of the barrel. The process throughput is then calculated while maintaining the heat transfer rate and, preferably, the screw speed. For the Power Scale-up, the process specific energy input is critical. This is the case of the production of high-energy compositions and should be kept constant when scaling up. In these cases, the specific energy of large-scale equipment can be calculated and used to determine the target throughput (which maintains the SE input constant and, preferably, also the screw speed) [47, 55, 139]. For instance, a volumetric approach was used to upscale an HME process from laboratory to clinical scale [140], with minimum consumption of drug during the whole study.
Nonetheless, heat and mass transfer restrictions may happen, mainly when the difference between scales is too wide. When confronted with these limitations, adjustments are needed to maintain the CQAs. The specific energy, residence time, and product temperature are the most important factors to keep steady [138]. This may be accomplished by regulating HME process parameters, at this stage mainly by the feed rate (based on calculated throughput). The screw speed, the product temperature, and screw design should only be adjusted if calculations led to excessive motor torque [47], or if differences in CQAs arise due to decreased heat and mass transfer at the larger scale [138]. However, since kneading elements are usually required for the production of ASDs, screw configuration changes should not be the first approach and must be considered carefully.
The process setup and scale-up through advanced modeling is the latest development in this challenging task, used as a first approach to predict scale-up parameters. The calculations by software require raw material data, which can be difficult to provide for mixtures of components that evolve during the HME process from a solid to a non-Newtonian fluid [21]. However, advanced software tools are being developed. Ludovic® and XimeX-TSE® (both now marketed by SC-Consultants, France) are simulation commercial software packages dedicated to HME processes. They modulate and optimize the HME process through an in-depth analysis of the evolution of materials. Some applications are already available in the literature [141,142,143], although additional work is still required. Alternative modeling strategies were also recently described, as the validated computational framework developed by Matić et al. [71]. Despite the innovative power of software and modeling in the scale-up of HME processes, skilled operators remain essential [21] to successful scale-up in real manufacturing plants.
The next step is the manufacturing of clinical batches in a GMP environment. The QbD approach enables a simplified process of transferring technology due to a clear identification of critical process parameters and material attributes, as well as its mechanistic knowledge of the impact on the CQAs. The selection of the GMP manufacturing site should be taken into account from the early development, as the capabilities must be considered and thoroughly evaluated, like the type of equipment, screw configuration, and batch size [71]. A detailed control strategy determining acceptable limits for critical parameters and attributes supported by a rigorous risk assessment is key for a successful transfer for the pilot batch scale.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
The emerging trends in the high-throughput screening for drug discovery have led to new but very lipophilic drugs, with high molecular weight and poor BA. The numbers are astonishing, with about 40% of already approved drugs and almost 90% of compounds under development demonstrating poor or very poor water solubility [144]. Pharmaceutical scientists and pharmaceutical industries, along with the process, physical, mechanical, and chemical engineers, have worked in the last years in solutions, and novel drug delivery technologies have emerged to allow the formulation and oral administration of these high potency poorly soluble compounds.
In this context, HME came out as a novelty for product development and represents a promising technology to enhance solubility and absorption. As a matter of fact, an increasing number of companies are implementing HME to answer the low solubility of compounds. This technology has been used successfully for already approved products and many others under development, including medical devices. The interest of the pharmaceutical industry in HME is easily justified as a solvent-free, continuous, and cost-effective technology, creating robust processes for a variety of pharmaceutical forms, as oral solids, oral films, topical, ophthalmic inserts, and implants. The consistency and reproducibility of the continuous process is also a significant benefit of HME. Moreover, extrusion is suitable for high potency compounds, very common nowadays. The current advances in solid dispersions and HME technology and the knowledge built around material and polymer sciences applied to pharmaceutics enabled formulation scientists to solve complex problems of BA.
Soon, it is highly expected that HME technology becomes a more prominent approach for pharmaceutical companies to solve solubility and BA issues of their drug pipeline. FDA acknowledged the exceptional flexibility of HME to QbD concepts and PAT tools, both enabling real-time control to ensure the consistency of the end products. This feature is becoming more important and should place HME as a central technology in pharmaceutical manufacturing. However, the trend seems to be the specialization of companies and human resources, instead of generalized implementation, due to the several specificities of this technology discussed in this review, applied to both developers and manufacturers. In conclusion, HME is undoubtedly leading the change of the traditional manufacturing but also of the formulation with new excipients and redesigned properties, and equipment, including optimization of extruders, downstream processing, and further developments in PAT tools.
Abbreviations
- AG:
-
Adam–Gibbs
- ASD:
-
Amorphous solid dispersions
- BA:
-
Bioavailability
- BCS:
-
Biopharmaceutical Classification Systems
- CPP:
-
Critical process parameter
- CQA:
-
Critical Quality Attribute
- DoE:
-
Design of Experiments
- DSC:
-
Differential Calorimetric Screening
- FDA:
-
Food and Drug Administration USA
- Tg :
-
Glass transition temperature
- HME:
-
Hot-melt extrusion
- HPC:
-
Hydroxypropyl cellulose
- HPMC:
-
Hydroxypropyl methylcellulose (hypromellose)
- HPMCAS:
-
Hypromellose acetate succinate
- ICH:
-
International Conference on Harmonization
- KWW:
-
Kohlrausch–Williams–Watts
- MPD:
-
Melting point depression
- Tm :
-
Melting temperature (Tm)
- PATs:
-
Process Analytical Technologies
- PEG:
-
Polyethylene glycol
- PVP:
-
Poly(vinylpyrrolidone)
- QbD:
-
Quality by Design
References
Lakshman JP. Formulation, Bioavailability, and manufacturing process enhancement: novel applications of melt extrusion in enabling product development. In: Repka MA, Langley N, Di Nunzio J, editors. Melt extrusion: materials, technology and drug product design: Springer; 2013.
Verma S, Rudraraju VS. A systematic approach to design and prepare solid dispersions of poorly water-soluble drug. AAPS PharmSciTech. 2014;15(3):641–57. https://doi.org/10.1208/s12249-014-0093-z.
Pina MF, Zhao M, Pinto JF, Sousa JJ, Craig DQ. The influence of drug physical state on the dissolution enhancement of solid dispersions prepared via hot-melt extrusion: a case study using olanzapine. J Pharm Sci. 2014;103(4):1214–23. https://doi.org/10.1002/jps.23894.
Shah S, Maddineni S, Lu J, Repka MA. Melt extrusion with poorly soluble drugs. Int J Pharm. 2013;453(1):233–52. https://doi.org/10.1016/j.ijpharm.2012.11.001.
Becker K, Salar-Behzadi S, Zimmer A. Solvent-free melting techniques for the preparation of lipid-based solid oral formulations. Pharm Res. 2015;32(5):1519–45. https://doi.org/10.1007/s11095-015-1661-y.
Gao P, Shi Y. Characterization of supersaturatable formulations for improved absorption of poorly soluble drugs. AAPS J. 2012;14(4):703–13. https://doi.org/10.1208/s12248-012-9389-7.
Sarode AL, Sandhu H, Shah N, Malick W, Zia H. Hot melt extrusion for amorphous solid dispersions: temperature and moisture activated drug-polymer interactions for enhanced stability. Mol Pharm. 2013;10(10):3665–75. https://doi.org/10.1021/mp400165b.
Lu M, Guo Z, Li Y, Pang H, Lin L, Liu X, et al. Application of hot melt extrusion for poorly water-soluble drugs: limitations, advances and future prospects. Curr Pharm Des. 2014;20(3):369–87. https://doi.org/10.2174/13816128113199990402.
Repka MA, Shah S, Lu J, Maddineni S, Morott J, Patwardhan K, et al. Melt extrusion: process to product. Expert Opin Drug Deliv. 2012;9(1):105–25. https://doi.org/10.1517/17425247.2012.642365.
Wilson M, Williams MA, Jones DS, Andrews GP. Hot-melt extrusion technology and pharmaceutical application. Ther Deliv. 2012;3(6):787–97. https://doi.org/10.4155/tde.12.26.
Patil H, Tiwari RV, Repka MA. Hot-melt extrusion: from theory to application in pharmaceutical formulation. AAPS PharmSciTech. 2016;17(1):20–42. https://doi.org/10.1208/s12249-015-0360-7.
Maniruzzaman M, Boateng JS, Snowden MJ, Douroumis D. A review of hot-melt extrusion: process technology to pharmaceutical products. ISRN Pharm. 2012;2012:436763–9. https://doi.org/10.5402/2012/436763.
Chivate A, Garkal AD, Dhas NL, Mehta DTA. Hot melt extrusion: an emerging technique for solubility enhancement of poorly water soluble drugs. PDA J Pharm Sci Technol. 2021:pdajpst.2019.011403. https://doi.org/10.5731/pdajpst.2019.011403.
Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, et al. Pharmaceutical applications of hot-melt extrusion: part I. Drug Dev Ind Pharm. 2007;33(9):909–26. https://doi.org/10.1080/03639040701498759.
Stankovic M, Frijlink HW, Hinrichs WL. Polymeric formulations for drug release prepared by hot melt extrusion: application and characterization. Drug Discov Today. 2015;20(7):812–23. https://doi.org/10.1016/j.drudis.2015.01.012.
ICH. Q8 (R2) Pharmaceutical Development. August 2009.
ICH. Q9 Quality Risk Management. November 2005.
ICH. Q10 Pharmaceutical Quality System. June 2008.
ICH. Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). May 2012.
FDA. Guidance for industry PAT—a framework for innovative pharmaceutical manufacturing and quality assurance. September 2004.
Maniruzzaman M, Nokhodchi A. Continuous manufacturing via hot-melt extrusion and scale up: regulatory matters. Drug Discov Today. 2017;22(2):340–51. https://doi.org/10.1016/j.drudis.2016.11.007.
Kallakunta VR, Sarabu S, Bandari S, Tiwari R, Patil H, Repka MA. An update on the contribution of hot-melt extrusion technology to novel drug delivery in the twenty-first century: part I. Expert Opin Drug Deliv. 2019;16(5):539–50. https://doi.org/10.1080/17425247.2019.1609448.
Hitzer P, Bauerle T, Drieschner T, Ostertag E, Paulsen K, van Lishaut H, et al. Process analytical techniques for hot-melt extrusion and their application to amorphous solid dispersions. Anal Bioanal Chem. 2017;409(18):4321–33. https://doi.org/10.1007/s00216-017-0292-z.
Wesholowski J, Prill S, Berghaus A, Thommes M. Inline UV/Vis spectroscopy as PAT tool for hot-melt extrusion. Drug Deliv Transl Res. 2018;8(6):1595–603. https://doi.org/10.1007/s13346-017-0465-5.
Kelly AL, Gough T, Isreb M, Dhumal R, Jones JW, Nicholson S, et al. In-process rheometry as a PAT tool for hot melt extrusion. Drug Dev Ind Pharm. 2017;44:1–7. https://doi.org/10.1080/03639045.2017.1408641.
Repka MA, Bandari S, Kallakunta VR, Vo AQ, McFall H, Pimparade MB, et al. Melt extrusion with poorly soluble drugs—an integrated review. Int J Pharm. 2018;535(1-2):68–85. https://doi.org/10.1016/j.ijpharm.2017.10.056.
Gryczke A. Hot-melt extrusion process design using process analytical technology. In: Repka AM, Langley N, DiNunzio J, editors. Melt extrusion: materials, technology and drug product design. New York: Springer New York; 2013. p. 397–431.
Netchacovitch L, Thiry J, De Bleye C, Chavez PF, Krier F, Sacré PY, et al. Vibrational spectroscopy and microspectroscopy analyzing qualitatively and quantitatively pharmaceutical hot melt extrudates. J Pharm Biomed Anal. 2015;113:21–33. https://doi.org/10.1016/j.jpba.2015.01.051.
Huang S, O'Donnell KP, Delpon de Vaux SM, O'Brien J, Stutzman J, Williams RO 3rd. Processing thermally labile drugs by hot-melt extrusion: the lesson with gliclazide. Eur J Pharm Biopharm. 2017;119:56–67. https://doi.org/10.1016/j.ejpb.2017.05.014.
Chavan RB, Rathi S, Jyothi V, Shastri NR. Cellulose based polymers in development of amorphous solid dispersions. Asian J Pharm Sci. 2019;14(3):248–64. https://doi.org/10.1016/j.ajps.2018.09.003.
Sangshetti JN, Deshpande M, Zaheer Z, Shinde DB, Arote R. Quality by design approach: regulatory need. Arab J Chem. 2017;10:S3412–S25. https://doi.org/10.1016/j.arabjc.2014.01.025.
Mishra V, Thakur S, Patil A, Shukla A. Quality by design (QbD) approaches in current pharmaceutical set-up. Expert Opin Drug Deliv. 2018;15(8):737–58. https://doi.org/10.1080/17425247.2018.1504768.
Penumetcha SS, Gutta LN, Dhanala H, Yamili S, Challa S, Rudraraju S, et al. Hot melt extruded Aprepitant-Soluplus solid dispersion: preformulation considerations, stability and in vitro study. Drug Dev Ind Pharm. 2016;42(10):1609–20. https://doi.org/10.3109/03639045.2016.1160105.
Aho J, Edinger M, Botker J, Baldursdottir S, Rantanen J. Oscillatory shear rheology in examining the drug-polymer interactions relevant in hot melt extrusion. J Pharm Sci. 2016;105(1):160–7. https://doi.org/10.1016/j.xphs.2015.11.029.
Li S, Tian Y, Jones DS, Andrews GP. Optimising drug solubilisation in amorphous polymer dispersions: rational selection of hot-melt extrusion processing parameters. AAPS PharmSciTech. 2016;17(1):200–13. https://doi.org/10.1208/s12249-015-0450-6.
Chan SY, Qi S, Craig DQ. An investigation into the influence of drug-polymer interactions on the miscibility, processability and structure of polyvinylpyrrolidone-based hot melt extrusion formulations. Int J Pharm. 2015;496(1):95–106. https://doi.org/10.1016/j.ijpharm.2015.09.063.
Simoes MF, Pinto RMA, Simoes S. Hot-melt extrusion in the pharmaceutical industry: toward filing a new drug application. Drug Discov Today. 2019;24(9):1749–68. https://doi.org/10.1016/j.drudis.2019.05.013.
Gupta A, Khan M. Hot-melt extrusion: an FDA perspective on product and process understanding. In: Douroumis D, editor. Hot-melt extrusion: pharmaceutical applications: John Wiley & Sons, Ltd; 2012. p. 323-31.
Chaves LL, Vieira AC, Reis S, Sarmento B, Ferreira DC. Quality by design: discussing and assessing the solid dispersions risk. Curr Drug Deliv. 2014;11(2):253–69. https://doi.org/10.2174/1567201811666140211110943.
Pawar J, Suryawanshi D, Moravkar K, Aware R, Shetty V, Maniruzzaman M, et al. Study the influence of formulation process parameters on solubility and dissolution enhancement of efavirenz solid solutions prepared by hot-melt extrusion: a QbD methodology. Drug Deliv Transl Res. 2018;8(6):1644–57. https://doi.org/10.1007/s13346-018-0481-0.
Desai PM, Hogan RC, Brancazio D, Puri V, Jensen KD, Chun JH, et al. Integrated hot-melt extrusion–injection molding continuous tablet manufacturing platform: effects of critical process parameters and formulation attributes on product robustness and dimensional stability. Int J Pharm. 2017;531(1):332–42. https://doi.org/10.1016/j.ijpharm.2017.08.097.
Patwardhan K, Asgarzadeh F, Dassinger T, Albers J, Repka MA. A quality by design approach to understand formulation and process variability in pharmaceutical melt extrusion processes. J Pharm Pharmacol. 2015;67(5):673–84. https://doi.org/10.1111/jphp.12370.
Zhang L, Mao S. Application of quality by design in the current drug development. Asian J Pharm Sci. 2017;12(1):1–8. https://doi.org/10.1016/j.ajps.2016.07.006.
Markarian J. Defining design space in hot-melt extrusion. Pharm Technol; 2012 [cited 2019 2 Ago]; Available from: http://www.pharmtech.com/defining-design-space-hot-melt-extrusion.
Butreddy A, Bandari S, Repka MA. Quality-by-design in hot melt extrusion based amorphous solid dispersions: an industrial perspective on product development. Eur J Pharm Sci. 2021;158:105655. https://doi.org/10.1016/j.ejps.2020.105655.
Islam MT, Maniruzzaman M, Halsey SA, Chowdhry BZ, Douroumis D. Development of sustained-release formulations processed by hot-melt extrusion by using a quality-by-design approach. Drug Deliv Transl Res. 2014;4(4):377–87. https://doi.org/10.1007/s13346-014-0197-8.
Thiry J, Krier F, Evrard B. A review of pharmaceutical extrusion: critical process parameters and scaling-up. Int J Pharm. 2015;479(1):227–40. https://doi.org/10.1016/j.ijpharm.2014.12.036.
Wu JX, van den Berg F, Sogaard SV, Rantanen J. Fast-track to a solid dispersion formulation using multi-way analysis of complex interactions. J Pharm Sci. 2013;102(3):904–14. https://doi.org/10.1002/jps.23409.
Pawar J, Tayade A, Gangurde A, Moravkar K, Amin P. Solubility and dissolution enhancement of efavirenz hot melt extruded amorphous solid dispersions using combination of polymeric blends: a QbD approach. Eur J Pharm Sci. 2016;88:37–49. https://doi.org/10.1016/j.ejps.2016.04.001.
Lang B, McGinity JW, Williams RO 3rd. Dissolution enhancement of itraconazole by hot-melt extrusion alone and the combination of hot-melt extrusion and rapid freezing—effect of formulation and processing variables. Mol Pharm. 2014;11(1):186–96. https://doi.org/10.1021/mp4003706.
Saerens L, Ghanam D, Raemdonck C, Francois K, Manz J, Kruger R, et al. In-line solid state prediction during pharmaceutical hot-melt extrusion in a 12 mm twin screw extruder using Raman spectroscopy. Eur J Pharm Biopharm. 2014;87(3):606–15. https://doi.org/10.1016/j.ejpb.2014.03.002.
Reitz E, Vervaet C, Neubert RH, Thommes M. Solid crystal suspensions containing griseofulvin—preparation and bioavailability testing. Eur J Pharm Biopharm. 2013;83(2):193–202. https://doi.org/10.1016/j.ejpb.2012.09.012.
Baronsky-Probst J, Moltgen CV, Kessler W, Kessler RW. Process design and control of a twin screw hot melt extrusion for continuous pharmaceutical tamper-resistant tablet production. Eur J Pharm Sci. 2016;87:14–21. https://doi.org/10.1016/j.ejps.2015.09.010.
Chen M, Lu J, Deng W, Singh A, Mohammed NN, Repka MA, et al. Influence of processing parameters and formulation factors on the bioadhesive, temperature stability and drug release properties of hot-melt extruded films containing miconazole. AAPS PharmSciTech. 2014;15(3):522–9. https://doi.org/10.1208/s12249-013-0029-z.
Lang B, McGinity JW, Williams RO 3rd. Hot-melt extrusion—basic principles and pharmaceutical applications. Drug Dev Ind Pharm. 2014;40(9):1133–55. https://doi.org/10.3109/03639045.2013.838577.
Crowley MM, Zhang F, Koleng JJ, McGinity JW. Stability of polyethylene oxide in matrix tablets prepared by hot-melt extrusion. Biomaterials. 2002;23(21):4241–8. https://doi.org/10.1016/s0142-9612(02)00187-4.
Gallet G, Carroccio S, Rizzarelli P, Karlsson S. Thermal degradation of poly(ethylene oxide–propylene oxide–ethylene oxide) triblock copolymer: comparative study by SEC/NMR, SEC/MALDI-TOF-MS and SPME/GC-MS. Polymer. 2002;43(4):1081–94. https://doi.org/10.1016/s0032-3861(01)00677-2.
Watanabe T, Okabayashi M, Kurokawa D, Nishimoto Y, Ozawa T, Kawasaki H, et al. Determination of primary bond scissions by mass spectrometric analysis of ultrasonic degradation products of poly(ethylene oxide-block-propylene oxide) copolymers. J Mass Spectrom. 2010;45(7):799–805. https://doi.org/10.1002/jms.1771.
Shojaee S, Cumming I, Kaialy W, Nokhodchi A. The influence of vitamin E succinate on the stability of polyethylene oxide PEO controlled release matrix tablets. Colloids Surf B: Biointerfaces. 2013;111:486–92. https://doi.org/10.1016/j.colsurfb.2013.06.038.
Carrasco F, Pagès P, Gámez-Pérez J, Santana OO, Maspoch ML. Processing of poly(lactic acid): characterization of chemical structure, thermal stability and mechanical properties. Polym Degrad Stab. 2010;95(2):116–25. https://doi.org/10.1016/j.polymdegradstab.2009.11.045.
Zhang T, Zhou S, Gao X, Yang Z, Sun L, Zhang D. A multi-scale method for modeling degradation of bioresorbable polyesters. Acta Biomater. 2017;50:462–75. https://doi.org/10.1016/j.actbio.2016.12.046.
Liu W-C, Halley PJ, Gilbert RG. Mechanism of degradation of starch, a highly branched polymer, during extrusion. Macromolecules. 2010;43(6):2855–64. https://doi.org/10.1021/ma100067x.
Hughey JR, Keen JM, Miller DA, Brough C, McGinity JW. Preparation and characterization of fusion processed solid dispersions containing a viscous thermally labile polymeric carrier. Int J Pharm. 2012;438(1-2):11–9. https://doi.org/10.1016/j.ijpharm.2012.08.032.
Lu G, Kalyon DM, Yilgör I, Yilgör E. Rheology and extrusion of medical-grade thermoplastic polyurethane. Polym Eng Sci. 2003;43(12):1863–77. https://doi.org/10.1002/pen.10158.
Dong Z, Choi DS. Hydroxypropyl methylcellulose acetate succinate: potential drug-excipient incompatibility. AAPS PharmSciTech. 2008;9(3):991–7. https://doi.org/10.1208/s12249-008-9138-5.
Alexy P, Lacı́k I, Šimková B, Bakoš D, Na P, Liptaj T, et al. Effect of melt processing on thermo-mechanical degradation of poly(vinyl alcohol)s. Polym Degrad Stab. 2004;85(2):823–30. https://doi.org/10.1016/j.polymdegradstab.2004.02.011.
Lin S-Y, Yu H-L, Li M-J. Formation of six-membered cyclic anhydrides by thermally induced intramolecular ester condensation in Eudragit E film. Polymer. 1999;40(12):3589–93. https://doi.org/10.1016/s0032-3861(98)00488-1.
Lin S-Y, Yu H-L. Thermal stability of methacrylic acid copolymers of Eudragits L, S, and L30D and the acrylic acid polymer of carbopol. J Polym Sci A Polym Chem. 1999;37(13):2061–7. https://doi.org/10.1002/(sici)1099-0518(19990701).
Hughey JR, DiNunzio JC, Bennett RC, Brough C, Miller DA, Ma H, et al. Dissolution enhancement of a drug exhibiting thermal and acidic decomposition characteristics by fusion processing: a comparative study of hot melt extrusion and KinetiSol dispersing. AAPS PharmSciTech. 2010;11(2):760–74. https://doi.org/10.1208/s12249-010-9431-y.
Stroyer A, McGinity JW, Leopold CS. Solid state interactions between the proton pump inhibitor omeprazole and various enteric coating polymers. J Pharm Sci. 2006;95(6):1342–53. https://doi.org/10.1002/jps.20450.
Matić J, Paudel A, Bauer H, Garcia RAL, Biedrzycka K, Khinast JG. Developing HME-based drug products using emerging science: a fast-track roadmap from concept to clinical batch. AAPS PharmSciTech. 2020;21(5):176. https://doi.org/10.1208/s12249-020-01713-0.
Jelić D. Thermal stability of amorphous solid dispersions. Molecules. 2021;26(1):238. https://doi.org/10.3390/molecules26010238.
Lin X, Hu Y, Liu L, Su L, Li N, Yu J, et al. Physical stability of amorphous solid dispersions: a physicochemical perspective with thermodynamic, kinetic and environmental aspects. Pharm Res. 2018;35(6):125. https://doi.org/10.1007/s11095-018-2408-3.
Pandi P, Bulusu R, Kommineni N, Khan W, Singh M. Amorphous solid dispersions: an update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int J Pharm. 2020;586:119560. https://doi.org/10.1016/j.ijpharm.2020.119560.
Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm Res. 1995;12(6):799–806. https://doi.org/10.1023/a:1016292416526.
Laitinen R, Lobmann K, Strachan CJ, Grohganz H, Rades T. Emerging trends in the stabilization of amorphous drugs. Int J Pharm. 2013;453(1):65–79. https://doi.org/10.1016/j.ijpharm.2012.04.066.
Baghel S, Cathcart H, O'Reilly NJ. Polymeric amorphous solid dispersions: a review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J Pharm Sci. 2016;105(9):2527–44. https://doi.org/10.1016/j.xphs.2015.10.008.
Janssens S, Van den Mooter G. Review: physical chemistry of solid dispersions. J Pharm Pharmacol. 2009;61(12):1571–86. https://doi.org/10.1211/jpp/61.12.0001.
Zhu DA, Zografi G, Gao P, Gong Y, Zhang GGZ. Modeling physical stability of amorphous solids based on temperature and moisture stresses. J Pharm Sci. 2016;105(9):2932–9. https://doi.org/10.1016/j.xphs.2016.03.029.
Bhardwaj SP, Arora KK, Kwong E, Templeton A, Clas SD, Suryanarayanan R. Correlation between molecular mobility and physical stability of amorphous itraconazole. Mol Pharm. 2013;10(2):694–700. https://doi.org/10.1021/mp300487u.
Miyanishi H, Nemoto T, Mizuno M, Mimura H, Kitamura S, Iwao Y, et al. Evaluation of crystallization behavior on the surface of nifedipine solid dispersion powder using inverse gas chromatography. Pharm Res. 2013;30(2):502–11. https://doi.org/10.1007/s11095-012-0896-0.
Shah N, Sandhu H, Choi DS, Chokshi H, Malick AW. Amorphous solid dispersions: theory and practice. New York: Springer; 2014.
Gupta SS, Solanki N, Serajuddin AT. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion, IV: Affinisol HPMC HME polymers. AAPS PharmSciTech. 2016;17(1):148–57. https://doi.org/10.1208/s12249-015-0426-6.
Mao C, Chamarthy SP, Pinal R. Time-dependence of molecular mobility during structural relaxation and its impact on organic amorphous solids: an investigation based on a calorimetric approach. Pharm Res. 2006;23(8):1906–17. https://doi.org/10.1007/s11095-006-9008-3.
Surana R, Pyne A, Rani M, Suryanarayanan R. Measurement of enthalpic relaxation by differential scanning calorimetry—effect of experimental conditions. Thermochim Acta. 2005;433(1-2):173–82. https://doi.org/10.1016/j.tca.2005.02.014.
Hasegawa S, Ke P, Buckton G. Determination of the structural relaxation at the surface of amorphous solid dispersion using inverse gas chromatography. J Pharm Sci. 2009;98(6):2133–9. https://doi.org/10.1002/jps.21573.
Bansal SS, Kaushal AM, Bansal AK. Enthalpy relaxation studies of two structurally related amorphous drugs and their binary dispersions. Drug Dev Ind Pharm. 2010;36(11):1271–80. https://doi.org/10.3109/03639041003753847.
Kakumanu VK, Bansal AK. Enthalpy relaxation studies of celecoxib amorphous mixtures. Pharm Res. 2002;19(12):1873–8. https://doi.org/10.1023/a:1021453810624.
Mao C, Prasanth Chamarthy S, Byrn SR, Pinal R. A calorimetric method to estimate molecular mobility of amorphous solids at relatively low temperatures. Pharm Res. 2006;23(10):2269–76. https://doi.org/10.1007/s11095-006-9071-9.
Karmwar P, Graeser K, Gordon KC, Strachan CJ, Rades T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int J Pharm. 2011;417(1-2):94–100. https://doi.org/10.1016/j.ijpharm.2010.12.019.
Mao C, Chamarthy SP, Pinal R. Calorimetric study and modeling of molecular mobility in amorphous organic pharmaceutical compounds using a modified Adam-Gibbs approach. J Phys Chem B. 2007;111(46):13243–52. https://doi.org/10.1021/jp072577.
Aso Y, Yoshioka S, Kojima S. Molecular mobility-based estimation of the crystallization rates of amorphous nifedipine and phenobarbital in poly(vinylpyrrolidone) solid dispersions. J Pharm Sci. 2004;93(2):384–91. https://doi.org/10.1002/jps.10526.
Berthier L, Coslovich D. Novel approach to numerical measurements of the configurational entropy in supercooled liquids. Proc Natl Acad Sci U S A. 2014;111(32):11668–72. https://doi.org/10.1073/pnas.1407934111.
Qian F, Huang J, Hussain MA. Drug-polymer solubility and miscibility: stability consideration and practical challenges in amorphous solid dispersion development. J Pharm Sci. 2010;99(7):2941–7. https://doi.org/10.1002/jps.22074.
Forster A, Hempenstall J, Tucker, Rades T. The potential of small-scale fusion experiments and the Gordon-Taylor equation to predict the suitability of drug/polymer blends for melt extrusion. Drug Dev Ind Pharm. 2001;27(6):549–60. https://doi.org/10.1081/ddc-100105180.
Nair R, Nyamweya N, Gönen S, Martı́nez-Miranda LJ, Hoag SW. Influence of various drugs on the glass transition temperature of poly(vinylpyrrolidone): a thermodynamic and spectroscopic investigation. Int J Pharm. 2001;225(1-2):83–96. https://doi.org/10.1016/s0378-5173(01)00767-0.
Jensen KT, Larsen FH, Lobmann K, Rades T, Grohganz H. Influence of variation in molar ratio on co-amorphous drug-amino acid systems. Eur J Pharm Biopharm. 2016;107:32–9. https://doi.org/10.1016/j.ejpb.2016.06.020.
Rask MB, Knopp MM, Olesen NE, Holm R, Rades T. Influence of PVP/VA copolymer composition on drug-polymer solubility. Eur J Pharm Sci. 2016;85:10–7. https://doi.org/10.1016/j.ejps.2016.01.026.
O'Donnell KP, Woodward WH. Dielectric spectroscopy for the determination of the glass transition temperature of pharmaceutical solid dispersions. Drug Dev Ind Pharm. 2015;41(6):959–68. https://doi.org/10.3109/03639045.2014.919314.
Greenhalgh DJ, Williams AC, Timmins P, York P. Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci. 1999;88(11):1182–90. https://doi.org/10.1021/js9900856.
Van Krevelen DW, Te Nijenhuis K. Chapter 7—Cohesive properties and solubility. In:Properties of polymers. 4th ed. Amsterdam: Elsevier; 2009. p. 189–227.
Just S, Sievert F, Thommes M, Breitkreutz J. Improved group contribution parameter set for the application of solubility parameters to melt extrusion. Eur J Pharm Biopharm. 2013;85(3 Pt B):1191–9. https://doi.org/10.1016/j.ejpb.2013.04.006.
Forster A, Hempenstall J, Tucker I, Rades T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int J Pharm. 2001;226(1-2):147–61. https://doi.org/10.1016/s0378-5173(01)00801-8.
Baghel S, Cathcart H, O'Reilly NJ. Theoretical and experimental investigation of drug-polymer interaction and miscibility and its impact on drug supersaturation in aqueous medium. Eur J Pharm Biopharm. 2016;107:16–31. https://doi.org/10.1016/j.ejpb.2016.06.024.
Djuris J, Nikolakakis I, Ibric S, Djuric Z, Kachrimanis K. Preparation of carbamazepine-Soluplus solid dispersions by hot-melt extrusion, and prediction of drug-polymer miscibility by thermodynamic model fitting. Eur J Pharm Biopharm. 2013;84(1):228–37. https://doi.org/10.1016/j.ejpb.2012.12.018.
Zhang Y, Luo R, Chen Y, Ke X, Hu D, Han M. Application of carrier and plasticizer to improve the dissolution and bioavailability of poorly water-soluble baicalein by hot melt extrusion. AAPS PharmSciTech. 2014;15(3):560–8. https://doi.org/10.1208/s12249-013-0071-x.
Yoo SU, Krill SL, Wang Z, Telang C. Miscibility/stability considerations in binary solid dispersion systems composed of functional excipients towards the design of multi-component amorphous systems. J Pharm Sci. 2009;98(12):4711–23. https://doi.org/10.1002/jps.21779.
Yang M, Wang P, Gogos C. Prediction of acetaminophen's solubility in poly(ethylene oxide) at room temperature using the Flory-Huggins theory. Drug Dev Ind Pharm. 2013;39(1):102–8. https://doi.org/10.3109/03639045.2012.659188.
Marsac PJ, Shamblin SL, Taylor LS. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm Res. 2006;23(10):2417–26. https://doi.org/10.1007/s11095-006-9063-9.
Marsac PJ, Konno H, Taylor LS. A comparison of the physical stability of amorphous felodipine and nifedipine systems. Pharm Res. 2006;23(10):2306–16. https://doi.org/10.1007/s11095-006-9047-9.
Tian Y, Booth J, Meehan E, Jones DS, Li S, Andrews GP. Construction of drug-polymer thermodynamic phase diagrams using Flory-Huggins interaction theory: identifying the relevance of temperature and drug weight fraction to phase separation within solid dispersions. Mol Pharm. 2013;10(1):236–48. https://doi.org/10.1021/mp300386v.
Shah SM, Jain AS, Kaushik R, Nagarsenker MS, Nerurkar MJ. Preclinical formulations: insight, strategies, and practical considerations. AAPS PharmSciTech. 2014;15(5):1307–23. https://doi.org/10.1208/s12249-014-0156-1.
Thakral S, Thakral NK. Prediction of drug-polymer miscibility through the use of solubility parameter based Flory-Huggins interaction parameter and the experimental validation: PEG as model polymer. J Pharm Sci. 2013;102(7):2254–63. https://doi.org/10.1002/jps.23583.
Hengsawas Surasarang S, Keen JM, Huang S, Zhang F, McGinity JW, Williams RO 3rd. Hot melt extrusion versus spray drying: hot melt extrusion degrades albendazole. Drug Dev Ind Pharm. 2017;43(5):797–811. https://doi.org/10.1080/03639045.2016.1220577.
DiNunzio JC, Martin ZFC, McGinity JW. Melt Extrusion. In: Williams III RO, Watts AB, Miller DA, editors. Formulating poorly water soluble drugs. New York: Springer; 2012. p. 331–62.
Maddineni S, Battu SK, Morott J, Majumdar S, Murthy SN, Repka MA. Influence of process and formulation parameters on dissolution and stability characteristics of Kollidon(R) VA 64 hot-melt extrudates. AAPS PharmSciTech. 2015;16(2):444–54. https://doi.org/10.1208/s12249-014-0226-4.
Mendonsa N, Almutairy B, Kallakunta VR, Sarabu S, Thipsay P, Bandari S, et al. Manufacturing strategies to develop amorphous solid dispersions: an overview. J Drug Deliv Sci Technol. 2020;55:101459. https://doi.org/10.1016/j.jddst.2019.101459.
Dinunzio JC, Brough C, Hughey JR, Miller DA, Williams RO 3rd, McGinity JW. Fusion production of solid dispersions containing a heat-sensitive active ingredient by hot melt extrusion and Kinetisol dispersing. Eur J Pharm Biopharm. 2010;74(2):340–51. https://doi.org/10.1016/j.ejpb.2009.09.007.
Yu LX. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. 2008;25(4):781–91. https://doi.org/10.1007/s11095-007-9511-1.
Elkhabaz A, Sarkar S, Simpson GJ, Taylor LS. Characterization of phase transformations for amorphous solid dispersions of a weakly basic drug upon dissolution in biorelevant media. Pharm Res. 2019;36(12):174. https://doi.org/10.1007/s11095-019-2718-0.
Elkhabaz A, Sarkar S, Dinh JK, Simpson GJ, Taylor LS. Variation in supersaturation and phase behavior of ezetimibe amorphous solid dispersions upon dissolution in different biorelevant media. Mol Pharm. 2018;15(1):193–206. https://doi.org/10.1021/acs.molpharmaceut.7b00814.
Ashwathy P, Anto AT, Sudheesh MS. A mechanistic review on the dissolution phase behavior and supersaturation stabilization of amorphous solid dispersions. Drug Dev Ind Pharm. 2021;47(1):1–11. https://doi.org/10.1080/03639045.2021.1879843.
Puppolo MM, Hughey JR, Dillon T, Storey D, Jansen-Varnum S. Biomimetic dissolution: a tool to predict amorphous solid dispersion performance. AAPS PharmSciTech. 2017;18(8):2841–53. https://doi.org/10.1208/s12249-017-0783-4.
Litou C, Turner DB, Holmstock N, Ceulemans J, Box KJ, Kostewicz E, et al. Combining biorelevant in vitro and in silico tools to investigate the in vivo performance of the amorphous solid dispersion formulation of etravirine in the fed state. Eur J Pharm Sci. 2020;149:105297. https://doi.org/10.1016/j.ejps.2020.105297.
Kambayashi A, Kiyota T, Fujiwara M, Dressman JB. PBPK modeling coupled with biorelevant dissolution to forecast the oral performance of amorphous solid dispersion formulations. Eur J Pharm Sci. 2019;135:83–90. https://doi.org/10.1016/j.ejps.2019.05.013.
Boyd BJ, Bergström CAS, Vinarov Z, Kuentz M, Brouwers J, Augustijns P, et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur J Pharm Sci. 2019;137:104967. https://doi.org/10.1016/j.ejps.2019.104967.
Keen JM, McGinity JW, Williams RO 3rd. Enhancing bioavailability through thermal processing. Int J Pharm. 2013;450(1-2):185–96. https://doi.org/10.1016/j.ijpharm.2013.04.042.
Seibert KD, Collins PC, Fisher E. Milling operations in the pharmaceutical industry. In: Ende DJ, editor. Chemical engineering in the pharmaceutical industry. New Jersey: John Wiley & Sons, Inc.; 2010. p. 365–78.
Agrawal A, Dudhedia M, Deng W, Shepard K, Zhong L, Povilaitis E, et al. Development of tablet formulation of amorphous solid dispersions prepared by hot melt extrusion using quality by design approach. AAPS PharmSciTech. 2016;17(1):214–32. https://doi.org/10.1208/s12249-015-0472-0.
Kolter K, Karl M, Gryczke A. Hot-melt extrusion with BASF pharma polymers: extrusion compendium. Germany: BASF SE - Pharma Ingredients and Services; 2012.
Iyer R, Hegde S, Zhang YE, Dinunzio J, Singhal D, Malick A, et al. The impact of hot melt extrusion and spray drying on mechanical properties and tableting indices of materials used in pharmaceutical development. J Pharm Sci. 2013;102(10):3604–13. https://doi.org/10.1002/jps.23661.
Carley JF, McKelvey JM. Extruder scale-up theory and experiments. Ind Eng Chem. 1953;45(5):989–92. https://doi.org/10.1021/ie50521a036.
Nakatani M. Short communication: scale-up theory for twin-screw extruder, keeping the resin temperature unchanged. Adv Polym Technol. 1998;17(1):19–22. https://doi.org/10.1002/(sici)1098-2329(199821).
Chung CI. On the scale-up of plasticating extruder screws. Polym Eng Sci. 1984;24(9):626–32. https://doi.org/10.1002/pen.760240904.
Meijer HEH, Elemans PHM. The modeling of continuous mixers. Part I: The corotating twin-screw extruder. Polym Eng Sci. 1988;28(5):275–90. https://doi.org/10.1002/pen.760280504.
Agur EE. Extruder scale-up in a corotating twin-screw extrusion compounding process. Adv Polym Technol. 1986;6(2):225–31. https://doi.org/10.1002/adv.1986.060060208.
Bigio D, Wang K. Scale-up rules for mixing in a non-intermeshing twin-screw extruder. Polym Eng Sci. 1996;36(23):2832–9. https://doi.org/10.1002/pen.10684.
Hughey J. A practical guide to hot-melt extrusion scale-up for pharmaceutical applications. Pharm Technol. 2014;Solid Dosage & Excipients:25-9.
Ghosh I, Vippagunta R, Li S, Vippagunta S. Key considerations for optimization of formulation and melt-extrusion process parameters for developing thermosensitive compound. Pharm Dev Technol. 2012;17(4):502–10. https://doi.org/10.3109/10837450.2010.550624.
Agrawal AM, Dudhedia MS, Zimny E. Hot melt extrusion: development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSciTech. 2016;17(1):133–47. https://doi.org/10.1208/s12249-015-0425-7.
Bochmann ES, Steffens KE, Gryczke A, Wagner KG. Numerical simulation of hot-melt extrusion processes for amorphous solid dispersions using model-based melt viscosity. Eur J Pharm Biopharm. 2018;124:34–42. https://doi.org/10.1016/j.ejpb.2017.12.001.
Dubey SP, Abhyankar HA, Marchante V, Brighton JL, Blackburn K, Temple C, et al. Modelling and validation of synthesis of poly lactic acid using an alternative energy source through a continuous reactive extrusion process. Polymers. 2016;8(4):164. https://doi.org/10.3390/polym8040164.
Zecevic DE, Evans RC, Paulsen K, Wagner KG. From benchtop to pilot scale-experimental study and computational assessment of a hot-melt extrusion scale-up of a solid dispersion of dipyridamole and copovidone. Int J Pharm. 2018;537(1-2):132–9. https://doi.org/10.1016/j.ijpharm.2017.12.033.
Kalepu S, Nekkanti V. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sin B. 2015;5(5):442–53. https://doi.org/10.1016/j.apsb.2015.07.003.
Loreti G, Maroni A, Del Curto MD, Melocchi A, Gazzaniga A, Zema L. Evaluation of hot-melt extrusion technique in the preparation of HPC matrices for prolonged release. Eur J Pharm Sci. 2014;52:77–85. https://doi.org/10.1016/j.ejps.2013.10.014.
Wilson MR, Jones DS, Andrews GP. The development of sustained release drug delivery platforms using melt-extruded cellulose-based polymer blends. J Pharm Pharmacol. 2017;69(1):32–42. https://doi.org/10.1111/jphp.12656.
Yi S, Wang J, Lu Y, Ma R, Gao Q, Liu S, et al. Novel hot melt extruded matrices of hydroxypropyl cellulose and amorphous felodipine-plasticized hydroxypropyl methylcellulose as controlled release systems. AAPS PharmSciTech. 2019;20(6):219. https://doi.org/10.1208/s12249-019-1435-7.
Almutairi M, Almutairy B, Sarabu S, Almotairy A, Ashour E, Bandari S, et al. Processability of AquaSolve LG polymer by hot-melt extrusion: effects of pressurized CO2 on physicomechanical properties and API stability. J Drug Deliv Sci Technol. 2019;52:165–76. https://doi.org/10.1016/j.jddst.2019.04.029.
Xue X, Chen G, Xu X, Wang J, Wang J, Ren L. A combined utilization of Plasdone-S630 and HPMCAS-HF in ziprasidone hydrochloride solid dispersion by hot-melt extrusion to enhance the oral bioavailability and no food effect. AAPS PharmSciTech. 2019;20(1):37. https://doi.org/10.1208/s12249-018-1216-8.
Wu CY, Lui WB, Peng J. Optimization of extrusion variables and maleic anhydride content on biopolymer blends based on poly(hydroxybutyrate-co-hydroxyvalerate)/poly(vinyl acetate) with tapioca starch. Polymers. 2018;10(8):827. https://doi.org/10.3390/polym10080827.
Pimparade MB, Vo A, Maurya AS, Bae J, Morott JT, Feng X, et al. Development and evaluation of an oral fast disintegrating anti-allergic film using hot-melt extrusion technology. Eur J Pharm Biopharm. 2017;119:81–90. https://doi.org/10.1016/j.ejpb.2017.06.004.
Fukuda M, Peppas NA, McGinity JW. Properties of sustained release hot-melt extruded tablets containing chitosan and xanthan gum. Int J Pharm. 2006;310(1-2):90–100. https://doi.org/10.1016/j.ijpharm.2005.11.042.
Verhoeven E, Vervaet C, Remon JP. Xanthan gum to tailor drug release of sustained-release ethylcellulose mini-matrices prepared via hot-melt extrusion: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2006;63(3):320–30. https://doi.org/10.1016/j.ejpb.2005.12.004.
Leelakanok N, Geary SM, Salem AK. Antitumor efficacy and toxicity of 5-fluorouracil-loaded poly(lactide co-glycolide) pellets. J Pharm Sci. 2018;107(2):690–7. https://doi.org/10.1016/j.xphs.2017.10.005.
Bode C, Kranz H, Fivez A, Siepmann F, Siepmann J. Often neglected: PLGA/PLA swelling orchestrates drug release: HME implants. J Control Release. 2019;306:97–107. https://doi.org/10.1016/j.jconrel.2019.05.039.
Kamel R, Abbas H. PLGA-based monolithic filaments prepared by hot-melt extrusion: In-vitro comparative study. Ann Pharm Fr. 2018;76(2):97–106. https://doi.org/10.1016/j.pharma.2017.09.002.
Heller J, Barr J, Ng S, Shen HR, Gurny R, Schwach-Abdelaoui K, et al. Development of poly(ortho esters) and their application for bovine serum albumin and bupivacaine delivery. J Control Release. 2002;78(1-3):133–41. https://doi.org/10.1016/s0168-3659(01)00482-5.
Heller J, Barr J, Ng SY, Shen HR, Schwach-Abdellaoui K, Einmahl S, et al. Poly(ortho esters)—their development and some recent applications. Eur J Pharm Biopharm. 2000;50(1):121–8. https://doi.org/10.1016/s0939-6411(00)00085-0.
Verstraete G, Vandenbussche L, Kasmi S, Nuhn L, Brouckaert D, Van Renterghem J, et al. Thermoplastic polyurethane-based intravaginal rings for prophylaxis and treatment of (recurrent) bacterial vaginosis. Int J Pharm. 2017;529(1-2):218–26. https://doi.org/10.1016/j.ijpharm.2017.06.076.
Verstraete G, Mertens P, Grymonpre W, Van Bockstal PJ, De Beer T, Boone MN, et al. A comparative study between melt granulation/compression and hot melt extrusion/injection molding for the manufacturing of oral sustained release thermoplastic polyurethane matrices. Int J Pharm. 2016;513(1-2):602–11. https://doi.org/10.1016/j.ijpharm.2016.09.072.
Verstraete G, Van Renterghem J, Van Bockstal PJ, Kasmi S, De Geest BG, De Beer T, et al. Hydrophilic thermoplastic polyurethanes for the manufacturing of highly dosed oral sustained release matrices via hot melt extrusion and injection molding. Int J Pharm. 2016;506(1-2):214–21. https://doi.org/10.1016/j.ijpharm.2016.04.057.
Li LC, Deng J, Stephens D. Polyanhydride implant for antibiotic delivery—from the bench to the clinic. Adv Drug Deliv Rev. 2002;54(7):963–86. https://doi.org/10.1016/s0169-409x(02)00053-4.
Deng JS, Meisters M, Li L, Setesak J, Claycomb L, Tian Y, et al. The development of an injection-molding process for a polyanhydride implant containing gentamicin sulfate. PDA J Pharm Sci Technol. 2002;56(2):65–77.
Simões MF, Pereira A, Cardoso S, Cadonau S, Werner K, Pinto RMA, et al. A 5-stage approach for a systematic screening and development of etravirine amorphous solid dispersions by hot-melt extrusion. Mol Pharm. 2019. https://doi.org/10.1021/acs.molpharmaceut.9b00996.
Evans RC, Bochmann ES, Kyeremateng SO, Gryczke A, Wagner KG. Holistic QbD approach for hot-melt extrusion process design space evaluation: linking materials science, experimentation and process modeling. Eur J Pharm Biopharm. 2019;141:149–60. https://doi.org/10.1016/j.ejpb.2019.05.021.
Ma X, Huang S, Lowinger MB, Liu X, Lu X, Su Y, et al. Influence of mechanical and thermal energy on nifedipine amorphous solid dispersions prepared by hot melt extrusion: preparation and physical stability. Int J Pharm. 2019;561:324–34. https://doi.org/10.1016/j.ijpharm.2019.03.014.
Zi P, Zhang C, Ju C, Su Z, Bao Y, Gao J, et al. Solubility and bioavailability enhancement study of lopinavir solid dispersion matrixed with a polymeric surfactant—Soluplus. Eur J Pharm Sci. 2019;134:233–45. https://doi.org/10.1016/j.ejps.2019.04.022.
Simões MF, Nogueira BA, Tabanez AM, Fausto R, Pinto RMA, Simões S. Enhanced solid-state stability of amorphous ibrutinib formulations prepared by hot-melt extrusion. Int J Pharm. 2020;579:119156. https://doi.org/10.1016/j.ijpharm.2020.119156.
Ugaonkar SR, Wesenberg A, Wilk J, Seidor S, Mizenina O, Kizima L, et al. A novel intravaginal ring to prevent HIV-1, HSV-2, HPV, and unintended pregnancy. J Control Release. 2015;213:57–68. https://doi.org/10.1016/j.jconrel.2015.06.018.
Almeida A, Possemiers S, Boone MN, De Beer T, Quinten T, Van Hoorebeke L, et al. Ethylene vinyl acetate as matrix for oral sustained release dosage forms produced via hot-melt extrusion. Eur J Pharm Biopharm. 2011;77(2):297–305. https://doi.org/10.1016/j.ejpb.2010.12.004.
Feng Z, Li M, Wang W. Improvement of dissolution and tabletability of carbamazepine solid dispersions with high drug loading prepared by hot-melt extrusion. Pharmazie. 2019;74(9):523–8. https://doi.org/10.1691/ph.2019.9008.
Meng F, Meckel J, Zhang F. Investigation of itraconazole ternary amorphous solid dispersions based on povidone and Carbopol. Eur J Pharm Sci. 2017;106:413–21. https://doi.org/10.1016/j.ejps.2017.06.019.
Bhagurkar AM, Darji M, Lakhani P, Thipsay P, Bandari S, Repka MA. Effects of formulation composition on the characteristics of mucoadhesive films prepared by hot-melt extrusion technology. J Pharm Pharmacol. 2019;71(3):293–305. https://doi.org/10.1111/jphp.13046.
Palazi E, Karavas E, Barmpalexis P, Kostoglou M, Nanaki S, Christodoulou E, et al. Melt extrusion process for adjusting drug release of poorly water soluble drug felodipine using different polymer matrices. Eur J Pharm Sci. 2018;114:332–45. https://doi.org/10.1016/j.ejps.2018.01.004.
Xu X, Siddiqui A, Srinivasan C, Mohammad A, Rahman Z, Korang-Yeboah M, et al. Evaluation of abuse-deterrent characteristics of tablets prepared via hot-melt extrusion. AAPS PharmSciTech. 2019;20(6):230. https://doi.org/10.1208/s12249-019-1448-2.
Acknowledgements
The authors thank Teresa Filipe for her support on illustrations.
Funding
This work was supported by Bluepharma, Coimbra, Portugal, and by Fundação para a Ciência e Tecnologia (FCT), Portugal (Ph.D. fellowship PD/BDE/135149/2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Simões, M.F., Pinto, R.M.A. & Simões, S. Hot-Melt Extrusion: a Roadmap for Product Development. AAPS PharmSciTech 22, 184 (2021). https://doi.org/10.1208/s12249-021-02017-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-021-02017-7