
Abstract
Chimeric/mixed stimuli-responsive nanocarriers are promising agents for therapeutic and diagnostic applications, as well as in the combinatorial field of theranostics. Herein, we designed chimeric nanosystems, composed of natural phospholipid and pH-sensitive amphiphilic diblock copolymer, in different molar ratios and assessed the polymer lyotropic effect on their properties. Initially, polymer-grafted bilayers were evaluated for their thermotropic behavior by thermal analysis. Chimeric liposomes were prepared through thin-film hydration and the obtained vesicles were studied by light scattering techniques, to measure their physicochemical characteristics and colloidal stability, as well as by imaging techniques, to elucidate their global and membrane morphology. Finally, in vitro screening of the systems’ toxicity was held. The copolymer effect on the membrane phase transition strongly depended on the pH of the surrounding environment. Chimeric nanoparticles were around and above 100 nm, while electron microscopy revealed occasional morphology diversity, probably affecting the toxicity of the systems. The latter was assessed to be tolerable, while dependent on the nanosystems’ material concentration, polymer concentration, and polymer composition. All experiments suggested that the thermodynamic and biophysical properties of the nanosystems are copolymer-composition- and concentration-dependent, since different amounts of incorporated polymer would produce divergent effects on the lyotropic liquid crystal membrane. Certain chimeric systems can be exploited as advanced drug delivery nanosystems, based on their overall promising profiles.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The therapeutic approach on many complex human diseases has lately been in the transit from conventional to more sophisticated methods. One aspect of this transition relates to the utilization of innovative medicines that are developed and function in the nanoscale, based on drug delivery nanosystems (DDnSs). Liposomes, dendrimers, and other types of nanoparticles belong to this class of nanosystems and may be characterized as conventional (cDDnSs) or advanced (aDDnSs), depending on their composition. Nevertheless, their utility remains the same and involves the control and modulation of drug release, which brings about a number of therapeutic benefits. Briefly, pharmaceutical nanotechnology has offered solutions in fields like imaging and diagnosis of tumors, real-time monitoring of treatments and therapy, through delivery systems that provide spatiotemporal drug release. Concerning DDnSs, specific tissue targeting, long circulation, biodegradability, as well as physical and biological stability of drugs are only some of the advantages provided. The delivery of genes for efficient cancer therapy has also been facilitated through such systems (1,2).
Recent advances in synthetic chemistry offer new possibilities for controlling the in vivo behavior and consequent absorption, distribution, metabolism, and excretion (ADME) profile of aDDnSs. Namely, stimuli-sensitive/responsive biomaterials (e.g., polymers) have been widely discussed and studied as components of self-regulated nanostructures, either through their own self-assembly or by incorporating them into previously developed systems. Their utility in targeted drug delivery is based on their tailored and precise composition, architecture, and hydrophilic-to-hydrophobic balance, which is altered under defined physiological conditions, inducing structural rearrangements and perturbations on various levels of the nanocarrier and, eventually, promoting drug release. The hydrophilic-to-hydrophobic balance of biomaterials has been generally shown to determine the thermotropic phase behavior and because of that, the surface morphology of the nanostructures, which plays a major role in their physical and biological stability. Stimuli-responsive nanosystems allow spatially and temporally controlled drug release, improving drug pharmacokinetic/pharmacodynamic (PK/PD) profile and biodistribution and eliminating the toxicity of otherwise toxic dosages (3,4).
The diverse physiology presented in certain pathological states, like cancer or inflammation, compared with normal ones, facilitates the utilization of biomaterials that undergo functional alterations under such conditions. In the effort to exploit the various phenomena that cause tissue and intracellular acidosis for more effective diagnosis or treatment, pH-responsive nanosystems were envisaged (5,6). There are a number of ways to build pH-responsive liposomes, for example by using pH-sensitive lipids and fusogenic peptides/proteins or by anchoring pH-sensitive polymers on the lipid bilayer, usually through hydrophobic alkyl chains (7). The pH-sensitive polymer is usually composed of protonable/deprotonable groups, which can be either anionic, like in the case of poly(acrylic acid) (PAA) or cationic, like in poly(2-(dimethylamino) ethyl methacrylate) (PDMAEMA) (8). Another interesting application of PAA, combined with N-isopropylacrylamide (NIPAAm), which is thermosensitive, and other copolymers regards a series of dual-sensitive hydrogels, utilized for encapsulation of cardiosphere derived cells (CDCs) and stem cell cardiac therapy. These hydrogels remain liquid at blood pH, while they solidify at the pH of the infracted heart, facilitating the efficient delivery of cells (9).
Besides pH-responsive, thermoresponsive or thermosensitive (nano)systems have been widely studied, since most pathological sites present hyperthermia, but also because of the elevated temperature of the physiological environment. As a result, many applications on the aforementioned myocardial infarction have arisen (10,11). Other types of stimuli include endogenous ones, such as redox, ions, enzymes, and biomolecules, while exogenous are ultrasound, magnetic field, electric voltage, light. Shear stress, as it is with heat, can result from both endogenous and exogenous sources (12).
However, in order to elucidate and regulate the morphology of such nonequilibrium systems, the thermodynamic contribution and energy content of the surfaces resulting from self-assembly must be clarified, and nanoscale-related properties should be taken into account (13). Such properties are the chemical potential, number of surface molecules and atoms, interfacial free energy, and surface properties. Surface thermodynamics and subdivision potential govern self-assembly, and the result is always a heterogeneous system, rich in metastable and morphologically diverse phases (14,15,16). By combining thermal analysis and imaging techniques, it is possible to link the nature of biomolecules with the thermodynamics and morphology of the emerging surfaces, which constitute a roadmap for the biophysical behavior of nanoparticles (17).
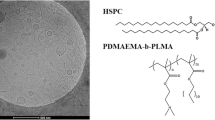
The aim of this research work was to design, develop, and study pH-responsive chimeric/mixed liposomal nanoplatforms, attempting to delineate their properties and behavior, so as to enable their future application as innovative excipients of advanced therapeutics. In particular, L-a-phosphatidylcholine, hydrogenated (soy) (HSPC) phospholipid (Fig. 1a) and poly(2-(dimethylamino)ethyl methacrylate)-b-poly(lauryl methacrylate) (PDMAEMA-b-PLMA) amphiphilic diblock copolymers (Fig. 1b) were mixed at different molar ratios, in order to obtain nanoassemblies in aqueous medium. Initially, thermal analysis was applied on lipid:block copolymer bilayers, to evaluate the biomaterial cooperativity, based on the system thermotropic behavior. Based on that, specific molar ratios were chosen to develop several nanosystems, afterwards studied by a series of light scattering and imaging techniques, to elucidate their physicochemical characteristics and morphology, respectively. The systems were also evaluated for their in vitro toxicity, to assess their safety and biocompatibility for future in vivo applications. Two PDMAEMA-b-PLMA copolymers, denoted as 1 and 2, were used, which differed in their composition and, consequently, hydrophilic-to-hydrophobic balance. The hydrophobic segment, that is PLMA, serves as a hydrophobic anchor for the attachment of the copolymer onto the liposomal membrane, through hydrophobic interactions with the phospholipid tails (18). A special feature of the copolymer is the stimuli responsivity that it exhibits in pH fluctuations. PDMAEMA is a polyamine, with each individual amino group having a pKa of 7.5–8, according to the literature (19,20). HSPC is a natural phospholipid with a relatively high main transition temperature Tm = 51 ± 2°C (21). It is a major component of liposomal products, with significant presence in the market (22).

Chemical structures of a HSPC phospholipid and b PDMAEMA-b-PLMA block copolymer. The segments’ molar ratio (n:m) of the copolymers is 70–30 for the PDMAEMA-b-PLMA 1 and 58–42 for the PDMAEMA-b-PLMA 2
MATERIALS AND METHODS
Materials
The natural phospholipid HSPC was purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA) and used without further purification (Fig. 1a). It has a molecular weight (Mw) of 783.774, a fatty acid distribution of lipid chains 16:0 11.4 and 18:0 88:6%, and its main transition temperature Tm is around 51 ± 2°C. It is also a component of liposomal products. Chloroform and other reagents were of analytical grade and purchased from Sigma-Aldrich Chemical Co. The PDMAEMA-b-PLMA amphiphilic diblock copolymers were synthesized by RAFT polymerization methodologies, in two different molar and weight compositions, 70–30 and 59–41 for PDMAEMA-b-PLMA 1 and 58–42 and 46–54 for PDMAEMA-b-PLMA 2 (Fig. 1b). The Mw of copolymers, determined by size exclusion chromatography (SEC), equals 8900 and 10,800, respectively, while the calculated length of extended chain was L = 12.5 nm for PDMAEMA-b-PLMA 1 and L = 14.0 nm for PDMAEMA-b-PLMA 2.
Synthesis of pH-Responsive Amphiphilic Block Copolymers
The PDMAEMA block was prepared first and then used as a macromolecular chain transfer agent for the synthesis of the second PLMA block. A typical RAFT polymerization scheme for the synthesis of PDMAEMA homopolymer is described in the following: to a round bottom flask (25 mL), 2-(dimethylamino)ethyl methacrylate (DMAEMA) (3 g, 19.1 mmol), 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (0.24 g, 0.6 mmol), AIBN (0.0098 g, 0.06 mmol), and 7 mL of dioxane were added. The final solution was degassed by nitrogen gas bubbling for 15 min and subsequently left for polymerization at 65°C for 18 h. The Mw of the PDMAEMA block was 4900 and 5000 for PDMAEMA-b-PLMA 1 and PDMAEMA-b-PLMA 2, while the molecular weight polydispersity index (Mw/Mn) was 1.15 and 1.14, respectively. Both values were obtained by SEC. The RAFT polymerization procedure for the synthesis of the PDMAEMA-b-PLMA block copolymers is as follows: to a round bottom flask (25 mL), lauryl methacrylate (LMA) (0.5/1.0 g, 1.96/3.93 mmol), poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA 1/PDMAEMA 2) (1.1/1.0 g, 0.224/0.198 mmol), AIBN (0.0184/0.0163 g, 0.1121/0.0995 mmol), and 4.8/6.0 mL of benzene were added. The final solution was degassed by nitrogen gas bubbling for 15 min and left for polymerization at 65°C for 24 h. The Mw values of the diblock copolymers were 8900 and 10,800 and the Mw/Mn values were 1.16 and 1.19 for PDMAEMA-b-PLMA 1 and PDMAEMA-b-PLMA 2, respectively. Both values were obtained by SEC. The copolymers contained 41.09/53.61 wt% PLMA, as determined by 1H NMR spectroscopy (23). Finally, the chain length was calculated from the number of segments and the known length of each monomeric unit for methacrylate-based polymers (0.254 nm).
Preparation of Pure and Chimeric Bilayers
Pure lipid and chimeric bilayers were prepared by mixing the appropriate amounts of HSPC and PDMEMA-b-PLMA 1/2 in chloroform/methanol (9:1 v/v) solutions and the subsequent evaporation of the solvents under vacuum and heat. Briefly, stock solutions were prepared by dissolving the copolymers PDMEMA-b-PLMA 1/2 in chloroform/methanol (9:1 v/v). Appropriate amounts of these solutions were mixed with 30.0 mg of HSPC, in order to obtain the desirable molar ratios (9:0.02, 9:0.05, 9:0.1, 9:0.2, and 9:0.5), and the solutions were transferred into vials. Then, the vials were placed into a vacuum machine (TechneDri-Block DB-3 Thermostat Teche Sample Concentrator). Chimeric phospholipids/block copolymer films were formed by removing the solvent at 50°C. The films were maintained under vacuum for 2 h and then in a desiccator for at least 24 h, in order to remove traces of solvent. The obtained laminated bilayers were hydrated into the appropriate aqueous medium and then studied by differential scanning calorimetry (DSC).
Differential Scanning Calorimetry
DSC experiments were carried out using an 822eMettler-Toledo (Schwerzenbach, Switzerland) calorimeter calibrated with pure indium (Tm = 156.6°C). Sealed aluminum 40-μL crucibles were used as sample holders. Conventional HSPC and chimeric HSPC:PDMAEMA-b-PLMA 1/2 (9:0.02, 9:0.05, 9:0.1, 9:0.2, and 9:0.5 molar ratios) fully hydrated bilayers were investigated. Thirty minutes prior to measurements, 3.0 mg of each mixture, after placed in the crucible, was hydrated using 20 μL of medium (phosphate buffer saline (PBS), pH = 7.4 or citrate buffer, pH = 4.5). Then, the crucible was sealed. Two heating-cooling cycles and a heating scan were performed, in order to ensure good reproducibility of the data. The temperature range was from 20 to 60°C and the scanning rate 5°C/min. Before each cycle, the samples were subjected to a constant temperature of 20°C, to ensure the equilibration, while an empty aluminum crucible was used as reference. The second heating and cooling runs were taken into account, and the calorimetric data obtained (characteristic transition temperatures Tonset,m/s and Tm/s, enthalpy changes ΔHm/s and widths at half peak height of the Cp profiles ΔT1/2,m/s) were analyzed using Mettler-Toledo STARe software. Furthermore, the linear correlation between the polymer concentration and the transition specific enthalpy values for each polymer in each buffer medium was investigated through scatter analysis, using “EXCELL.” For this purpose, the R-squared values were assessed.
Preparation of Pure and Chimeric Vesicles
Different chimeric formulations of HSPC:PDMAEMA-b-PLMA 1 and 2 have been prepared, using the thin-film hydration method. Briefly, appropriate amounts of HSPC and PDMAEMA-b-PLMA 1/2 (9:0.02, 9:0.1, and 9:0.5 molar ratios) were dissolved in chloroform/methanol (9:1 v/v) and then transferred into a round flask, connected to a rotary evaporator (Rotavapor R-114, Buchi, Switzerland). Vacuum was applied and the chimeric phospholipid/block copolymer thin film was formed by slow removal of the solvent at 41°C. The mixed film was maintained under vacuum for at least 24 h in a desiccator to remove traces of solvent. Subsequently, it was hydrated with PBS (pH = 7.4), by slowly stirring for 1 h in a water bath, above the phase transition temperature of the lipid (52°C for HSPC), for a total concentration of 5 mg/mL. The resultant structures (apparently multilamellar vesicles, MLVs) were subjected to two 5-min sonication cycles (amplitude 70%, cycle 0.5 s) interrupted by a 5-min resting period, using a probe sonicator (UP 200S, dr. Hielsher GmbH, Berlin, Germany). The resultant chimeric nanostructures (tentatively assigned as small unilamellar vesicles, SUVs) were allowed to anneal for 30 min.
Light Scattering Techniques
The size, size distribution, ζ-potential, and morphology of the obtained structures were investigated by dynamic and electrophoretic light scattering (DLS and ELS). The physicochemical characteristics were measured immediately after preparation (t = 0 days), as well as over time (t = 30 days for neat liposomes and t = 60 days for chimeric systems), for the monitoring of the system’s physical stability. For DLS and ELS, 100 μL aliquots were 30-fold diluted in HPLC-grade water. In addition, acidic protocol was performed, by 10-fold diluting 100 μL of the samples in citrate buffer (pH = 4.5), allowing them to anneal for 15 to 20 min and then 3-fold diluting them in HPLC-grade water. Measurements were performed at a detection angle of 90° and at 25°C, in a photon correlation spectrometer (Zetasizer 3000 HSA, Malvern, UK) and analyzed by CONTIN method (MALVERN software). Details on the methods have been published elsewhere (24).
Atomic Force Microscopy
For atomic force microscopy (AFM) measurements, aqueous dispersions were spin coated onto mica surface using a standard spin coater model SPIN150, SPS-Europe B.V. (the Netherlands) with 400 rpm for 600 min, and then samples were dried at room temperature for 24 h. AFM images were obtained using a MultiMode with Nanoscope IIId controller, Veeco (USA) AFM equipped with a piezoelectric scanner with a scan range of 10 × 10 μm2. The imaging of samples was conducted in the tapping mode in ambient air conditions at a scan rate of 1 Hz using etched silicon probes (TESP, BRUKER) of nominal spring constant 42 N/m and operating at a resonant frequency of 320 kHz. All samples were imaged at room temperature. The Veeco NanoScope V531r1 program was employed to analyze the recorded images.
Cryogenic Transmission Electron Microscopy
Cryogenic transmission electron microscopy (cryo-TEM) micrographs were obtained using a Tecnai F20 TWIN microscope (FEI Company, USA), equipped with field emission gun, operating at an acceleration voltage of 200 kV. Images were recorded on the Eagle 4k HS camera (FEI Company, USA) and processed with TIA software (FEI Company, USA). Specimen preparation was done by vitrification of the aqueous (HPLC-grade water) suspensions on grids with holey carbon film (Quantifoil R 2/2; Quantifoil Micro Tools GmbH, Germany). Prior to use, the grids were activated for 30 s in oxygen plasma using a Femto plasma cleaner (Diener Electronic, Germany). Cryo-samples were prepared by applying a droplet (2.1 μL) of the suspension to the grid, blotting with filter paper and immediate freezing in liquid ethane using a fully automated blotting device Vitrobot Mark IV (FEI Company, USA). After preparation, the vitrified specimens were kept under liquid nitrogen until they were inserted into a cryo-TEM holder Gatan 626 (Gatan Inc., USA) and analyzed in the TEM at − 178°C. Pictures were processed using ImageJ software.
In Vitro Screening
The MCF 10A cell line was used for all the in vitro experimental procedures. The cells were cultured in a 5% CO2 at 37°C. At confluence, they were harvested and reseeded in a 96-well plate at a density of 10,000 cells per well. The chimeric systems were inoculated for 3 days with the cells. Then, the cell viability was measured, using an MTT assay described in previous publications (25).
Statistical Analysis
DLS and ELS results are shown as mean value ± standard deviation (SD) of three independent measurements. Statistical analysis was performed using Student’s t test and multiple comparisons were done using one-way ANOVA. P values < 0.05 were considered statistically significant. All statistical analyses were performed using “EXCELL.” Concerning the in vitro experiments, we utilized logit-log plot for linear regression of the cell viability data (average values from 3 independent experiments), in order to calculate the IC50 curves for the studied systems. The half-maximal inhibitory concentrations (IC50) were determined using a logit-log plot and linear regression of the data, using SigmaPlot software (Systat Software, Inc., San Jose, CA, USA).
RESULTS AND DISCUSSION
Thermotropic Behavior Investigation of Chimeric Bilayers
The DSC profiles of the prepared pure HSPC bilayers, as well as of the PDMAEMA-b-PLMA 1 and PDMAEMA-b-PLMA 2-grafted HSPC bilayers, in PBS (pH = 7.0) and citrate buffer (pH = 4.5) are presented in Figs. 2, 3, 4, and 5. Both heating and cooling scans are included. The values of the corresponding thermodynamic parameters for the system phase transitions are shown in Tables I, II, III, and IV. The effect of the presence and amount of the block copolymers upon the lipid membrane’s thermotropic/phase behavior is discussed, in terms of self-assembled organization, fluidity, and cooperativity.

DSC heating scans of HSPC:PDMAEMA-b-PLMA 1 chimeric bilayers in a PBS (pH = 7.4) and b citrate buffer (pH = 4.5). Molar ratios for the two components are a. 9:0, b. 9:0.02, c. 9:0.05, d. 9:0.1, e. 9:0.2, and f. 9:0.5

DSC heating scans of HSPC:PDMAEMA-b-PLMA 2 chimeric bilayers in a PBS (pH = 7.4) and b citrate buffer (pH = 4.5). Molar ratios for the two components are a. 9:0, b. 9:0.02, c. 9:0.05, d. 9:0.1, e. 9:0.2, and f. 9:0.5

DSC cooling scans of HSPC:PDMAEMA-b-PLMA 1 chimeric bilayers in a PBS (pH = 7.4) and b citrate buffer (pH = 4.5). Molar ratios for the two components are a. 9:0, b. 9:0.02, c. 9:0.05, d. 9:0.1, e. 9:0.2, and f. 9:0.5

DSC cooling scans of HSPC:PDMAEMA-b-PLMA 2 chimeric bilayers in a PBS (pH = 7.4) and b citrate buffer (pH = 4.5). Molar ratios for the two components are a. 9:0, b. 9:0.02, c. 9:0.05, d. 9:0.1, e. 9:0.2, and f. 9:0.5
The neat HSPC bilayers exhibit a major sharp endothermic phase transition, from gel (Lβ′) to liquid crystalline phase (Lα), centered at 54.3°C, when hydrated in PBS medium (pH = 7.4) (Fig. 2a). The calculated values for the thermodynamic parameters of the main transition are in line with past studies (26). After the hydration of the bilayers in citrate buffer (pH = 4.5), the main transition becomes sharper and more symmetric (Fig. 2b). The onset (Tonset,m) and peak (Tm) temperatures of the main transition remained almost unaffected, while the half width at half peak height (ΔT1/2,m) underwent a significant reduction, of 1.5°C, and the transition specific enthalpy (ΔHm) was increased by 9.2 J/g. These alterations in acidic pH indicate increased chain cooperativity inside the phospholipid bilayer. Finally, a secondary broad low-enthalpy pretransition, from gel (Lβ′) to rippled (Pβ′) phase, was only observed in acidic pH, during heating and in PBS, during the cooling process. Both are broad and low-enthalpy transitions that occur at temperature lower than the main transition event. It should be noted that the observation of the pretransition and calculation of its specific enthalpy are not easy in the case of HSPC (27).
The grafting of the HSPC bilayers with the PDMAEMA-b-PLMA 1 block copolymer made the main transition sharper and more symmetric for the chimeric membranes, after hydration in PBS (Fig. 2a). Concerning the thermodynamic parameters, the onset temperature of the main transition remained almost unaffected. Conversely, the peak temperature underwent a downshift of around 1°C, for all the polymer ratios, as did the half width at half peak height, up to 1.4°C, which is almost 50%, leading to sharper transitions. An exception to the last is the HSPC:PDMAEMA-b-PLMA 1 9:0.02 system, where the half width increased by 0.6°C, because of the appearance of a shoulder on the main transition. These phenomena indicate slight fluidization, induced by the insertion of the block copolymer, as well as increased biomaterial transition cooperativity, at the same degree for all molar ratios (28). At the same time, the transition specific enthalpy presents a gradual reduction for the increasing polymer ratio, for a maximum difference of 19.4 J/g between 9:0.5 and HSPC systems, which comes hand in hand with the sharpening of the transition.
The effect of the HSPC:PDMAEMA-b-PLMA 1 on the HSPC membranes was stronger in the acidic environment of the citrate buffer (Fig. 2b). Specifically, the main transition exhibits a gradual broadening, asymmetry, and shortening, as the polymer ratio increases, with respective change of the thermodynamic parameters. The onset and peak temperatures exhibit an escalated reduction, of 7.3 and 2.8°C correspondingly, while half width at half pick dramatically increases, for a final 3.4°C, leading to broader transitions. The different rates of these changes result in the transition being more asymmetric, as more copolymer is incorporated inside the bilayer. Finally, a concurrent escalated reduction for the specific enthalpy is observed, for a maximum difference of 35.3 J/g between 9:0.5 and HSPC systems, accompanying the great shortening of the transition, as the polymer ratio increases. In this case, the concentration-dependent fluidization of the membrane is obvious, explained by the electrostatic repulsion between the practically fully charged PDMAEMA amino groups, which mobilizes the incorporated PLMA groups and leads to bilayer perturbation and probable disruption/pore creation. What we can also observe is the disappearance of the pretransition for the chimeric systems in citrate buffer. This finding is attributed to the PDMAEMA segment of the polymer interacting with the polar head groups of the phospholipid, which reside on the exterior of the membrane, affecting their mobility and stabilizing the gel phase against the ripple phase (16).
Similar to PDMAEMA-b-PLMA 1, the insertion of PDMAEMA-b-PLMA 2 in the HSPC membranes made the main transition sharper and more symmetric, in the PBS medium (Fig. 3a). The thermodynamic parameter variations also showed the same trends, with the onset temperature remaining almost the same, the peak temperature exhibiting a small downshift of 1°C, the half width at half peak height significantly declining, with a difference up to 1.5°C, and the transition specific enthalpy gradually being reduced, for a maximum difference of 19.9 J/g. Slight fluidization and increased cooperativity are also the case for this lipid/copolymer system (28). What distinguishes these systems from the previous ones is that, for the 0.02, 0.05, and 0.1 copolymer ratios, a pretransition appeared but was not observed when more copolymer was incorporated inside the lipid bilayer. These events are considered as metastable and are expected to influence the liposomal membrane biophysical properties, including stability and drug release (16).
As for the citrate buffer, the PDMAEMA-b-PLMA 2-grafted bilayers exhibit similar thermodynamic behavior with the PDMAEMA-b-PLMA 1-grafted ones (Fig. 3b). However, the effects of the second polymer on the HSPC membrane were not as intensive. As expected, the PDMAEMA-b-PLMA 2 polymer consists of fewer pH-sensitive DMAEMA segments and, consequently, has fewer ionized groups in acidic pH, thus affecting the lipid bilayer to a lesser degree. Finally, as with PDMAEMA-b-PLMA 1, the pretransition of the HSPC bilayer disappears in citrate buffer, probably because of the interaction of the PDMAEMA segment of the PDMAEMA-b-PLMA 2 with the polar head groups and the stabilization of the gel phase (16).
Finally, the long hysteresis in the cooling diagrams of all systems, conventional and chimeric, possibly suggests the formation of interdigitated phase (Figs. 4 and 5). Especially in the case of HSPC, the appearance of a low-enthalpy pretransition event during the cooling process, combined with the hysteresis, strengthens this assumption (29).
The plot of the transition enthalpy as a function of polymer concentration exhibits a rather linear character in most cases of heating the samples, which declares that the lyotropism of the systems is altered proportionally to the polymer-guest amount. In the first case of HSPC:PDMAEMA-b-PLMA 1 in PBS, the regression line approximates the data very well, with regression coefficient r2 = 0.9651 (Fig. 6a). This effect is rather surprising, since lipid membranes are considered dynamic/nonlinear and this finding suggests that certain features of their behavior are the resultant of individual linear relationships. The enthalpy-concentration relationship approaches linearity in the second and third cases as well, with r2 = 0.9681 and 0.9702, respectively (Fig. 6b). In the fourth case, the r2 coefficient was lower and equal to 0.9187 (Fig. 6d).

Linear regression analysis plots of transition specific enthalpy (ΔΗm) versus the concentration of the polymer inside the system for PDMAEMA-b-PLMA 1 in a PBS and b acidic pH and PDMAEMA-b-PLMA 2 in c PBS and d acidic pH
Overall, the thermotropic effect of the block copolymer guest on the bilayer in both PBS and acidic environment suggests a homogeneous distribution among the phospholipids, which in PBS does not affect the fluidity, enhances cooperativity, and reduces the total transition enthalpy, while in acidic pH perturbates the membrane, affecting all thermodynamic parameters, in a concentration and composition-dependent manner.
Physicochemical Characterization, Stability Evaluation, and pH-Responsiveness of Chimeric Vesicles
The physicochemical characteristics of conventional HSPC and chimeric HSPC:PDMAEMA-b-PLMA 1 and 2 systems (9:0.02, 0.1, and 0.5 molar ratios) in PBS and acidic conditions (pH = 4.5) are presented in Table V. Their respective size distribution curves are provided in our previous publication (30). The PBS buffer is chosen as a physiology simulator, attributed to its pH being around 7.4 and its ionic strength I = 0.154 M. In this case, it is of interest to study the various effects of the buffer ions (e.g., sodium and phosphate) on the solvation effects occurring on the bilayer surface (i.e., phospholipid head groups) and how those affect the properties of the systems. In addition, the acidic pH simulates the environment existing within late endosomes and lysosomes. The pH transition from 7.4 to 4.5 helps us determine the pH-responsive behavior of these pH-sensitive block copolymer-grafted vesicles.
The size of HSPC liposomes in PBS was around 100 nm, which is in agreement with previous publications. Their ζ-potential was close to 0, because of the absence of net charge on the nanocarrier surface (4,31,32).
Concerning size, the incorporation of either block copolymer led to particles of larger diameter, with the largest being that of low hydrophobic ratio. In particular, among the chimeric systems, for each copolymer separately, the largest particles consisted of low polymer ratio, the smallest had medium amount, and the high polymer ratio resulted in intermediate diameter values. The polydispersity of the chimeric systems, compared with conventional liposomes, for both copolymers, was higher for the lowest polymer ratio, lower for medium and high amounts of PDMAEMA-b-PLMA 1, indicating more homogeneous colloidal dispersions and almost the same for medium and high amounts of PDMAEMA-b-PLMA 2. The ζ-potential of the chimeric liposomes was found to be higher than that of conventional ones, ranging from around 10 mV to a bit higher than 20 mV. This was expected, since the pH of PBS medium (7.4) is slightly lower than the pKa value of the amino groups in the PDMAEMA block (7.5 to 8.0) and the outer hydrophilic PDMAEMA layer should be positively charged under such conditions (19,20). It is notable that even low polymer molar ratios (0.02, 0.1, or 0.5) raise the particle charge substantially, since the copolymer chains extend around the liposomal membrane.
A study of the chimeric liposomes’ physicochemical behavior in acidic pH was also conducted. The buffer was citrate, with pH = 4.5. In those conditions, HSPC liposomes more or less retained their size, polydispersity, and ζ-potential.
By assessing the particle hydrodynamic diameter difference for each system, we observed that all of them exhibited size reduction, with most being around 10 to 20 nm smaller in acidic pH, except for the HSPC:PDMAEMA-b-PLMA 2 9:0.02, which was over 100 nm smaller. Furthermore, aside from those with the highest lipid:copolymer ratio, most colloidal systems were found to be more homogeneous, regarding size, as indicated by their reduced PDI values from DLS. Potentially, the liposomes approach one another more easily when PDMAEMA chains are only half-protonated, that is in physiological pH, in comparison with the case of full protonation. This may lead to increased size extracted by DLS, while polydispersity might also vary. On the other hand, when PDMAEMA segments are 100% charged and extended, vesicles repulse each other, size drops, and heterogeneity meliorates (33). Changes in size may also be a result of the brush conformation of PDMAEMA chains due to protonation. Such a conformation increases the spatial requirements of the PDMAEMA-grafted chains, leading on a change of the lipid membrane structure and to a lower overall size of the nanostructure. Finally, ζ-potential values were generally higher in acidic pH, which is attributed to the protonable amino groups of the PDMAEMA chains being fully protonated in such conditions.
The colloidal stability, in terms of size and polydispersity, of the chimeric systems in PBS was also assessed, for a 2-month period (Fig. 7 and Fig. S1). HSPC conventional liposomes, which were studied for 34 days, underwent size increase, reaching ca. 152 nm and polydispersity decrease, reaching 0.288 (32).

Stability assessment of a HSPC conventional and HSPC:PDMAEMA-b-PLMA 1 and b HSPC conventional and HSPC:PDMAEMA-b-PLMA 2 chimeric structure size in PBS. Standard deviation is provided on each measurement
Size significantly increased for the systems with low copolymer ratios, to 444 nm for the first copolymer and to 551 nm for the second, after 2 months. Their respective PDI values were 0.968 and 1.000, indicating total heterogeneity. The incorporation of higher amounts of copolymers into the liposomal bilayer led all the other four chimeric systems to be practically stable, concerning size and polydispersity. This is mechanistically explained by the pH-sensitive character of the PDMAEMA chains, which at pH values below their pKa have their amino groups in charged protonated form, and thus, the segments adopt a fully stretched conformation, due to electrostatic repulsion between them. The result is repulsion and colloidal stability, due to steric interactions, osmotic effects, because of increased macromolecule concentration in the overlap region and enthalpic stabilization, which arises from the ejection of surface-adsorbed water molecules and consequent enthalpy increase (34).
Overall Morphology and Membrane Structure Investigation of Chimeric Systems
The developed vesicles with molar ratios 9:0.1 and 9:0.5 were further studied by AFM, along with cryo-TEM, for the parallel elucidation of the morphological characteristics of the chimeric systems in PBS. AFM and cryo-TEM, combined with light scattering results, provide a more comprehensive understanding of the self-assembly behavior of such combinatorial systems, consisting of more than one biomaterials and have been previously employed for this purpose (4,30). Representative AFM and cryo-TEM micrographs are given for every one of the four systems in Figs. 8 and 9, respectively.

AFM images of HSPC:PDMAEMA-b-PLMA 1 a 9:0.1 and b 9:0.5 chimeric systems

Cryo-TEM images of HSPC:PDMAEMA-b-PLMA 1 a 9:0.1 and b 9:0.5 and HSPC:PDMAEMA-b-PLMA 2 c 9:0.1 and d 9:0.5 chimeric systems. The scale bar in all micrographs is 500 nm
Concerning AFM, the diameter of sphere-like structures is calculated, through measurement of the horizontal distance of the cap-like structures formed by adsorption of the prepared vesicles on the surface coating (Fig. 8) (35,36). All chimeric system objects revealed on the micrographs were differentiated into two fractions. Τhe average diameters and heights of small and large vesicles, extracted by calculation of the horizontal and vertical distances of the cap-like structures, are provided in our previous work (30).
The average diameter and height of small fraction particles was 100–130 and 20–28 nm approximately, while large objects were 130–280 nm and with 30–80 nm height. The small object diameters are in line with DLS results, except for HSPC:PDMAEMA-b-PLMA 2 9:0.5. This is explained by the bimodal distribution observed in its DLS curve, which is more or less expressed by the two AFM fractions, with the DLS diameter being an approximate average of them. As a result, we could say that the two object populations are almost equal in number. For the other two chimeric systems exhibiting bimodal curves, the AFM small fraction is close to the first DLS curve peak. The first system is morphologically the most homogeneous, as the DLS diameter, DLS curve, and AFM small fraction are in very good correspondence. Large fraction particles are present there too, but probably in small quantity.
Chimeric systems of molar ratios 9:0.1 and 9:0.5 were also analyzed by cryo-TEM (Fig. 9). Vitrification by plunge freezing during cryo-preparation is utilized to preserve the hydrated state of the various dispersed structures and aggregates. This process is performed under controlled conditions, to avoid concentration alterations. The transparent film, formed by the amorphous ice, allows observation of the objects contained in the film. Cryo-TEM provides information about species morphology, size, and wall thickness. Black dots of size around 15 nm are also occasionally visible. These probably originate from ice contamination appearing during cryo-preparation but could also be the result of polymer self-assembly. All species observed in the cryo-TEM micrographs were categorized based on their conformation, size, and membrane profile, while the diameter and percentage quantity of objects were calculated and are provided in Table S1. Information on the wall thickness for vesicles and core diameter for worms have been previously presented (30).
The majority of objects identified by cryo-TEM for all chimeric systems are vesicular forms, with a close to 6-nm-thick membrane and sizes in the range of 69–80 nm. These morphologies are close to neat HSPC liposomes, considering both the diameter results from DLS and the known liposome membrane thickness documented in the literature, which is around 4–5 nm (35,37). However, their bilayer is thicker, probably indicating the incorporation of a small and nonobservable amount of polymer, through anchoring of the PLMA hydrophobic block. DSC results also confirm this assumption, where the alteration of the membrane phase transition thermodynamic parameters indicates distribution of the copolymer inside the lipid bilayer. These liposomes exhibit faceting, which means that their membrane is not smoothly round, but rather angularly shaped. This phenomenon has been related with the cryo-TEM preparation conditions, where the liposomal membrane is in the gel phase, due to submission to temperatures below the main transition point Tm (14). However, a study suggested that HSPC liposomes above 40 nm do not adopt faceted conformation and another that the same applies to liposomes without cholesterol above 100 nm (38,39). As a result, the observed faceting could be induced by the insertion of the polymer inside the membrane, which plays the role of cholesterol and is due to nanodomain formation, which could not be confirmed by classic DSC (7,40).
Other morphologies observed in cryo-TEM images include polymersomes, disc-like objects, and worm-like micelles, all of them being responsible for the bimodal distribution in most DLS curves. Polymersomes are larger (116–154 nm) and with membrane that is thicker (13–15 nm) and with lower electron beam contrast than liposomes. The self-assembly of the PDMAEMA-b-PLMA 2 copolymer resulted in vesicles of larger diameter, with thicker membrane, attributed to its relatively higher molecular weight (41). They might also be chimeric, containing a small and nonobservable amount of lipid, not high enough to induce faceting, because polymers prevent the gel phase rigidity of lipids (8). Furthermore, some vesicles seem to have a visually heterogeneous “patchy” membrane. Literature suggests that this visual finding occurs when the electron beam ends up on differently oriented facets (42). The formation of polymer “raft-like” nanodomains is also probable in these systems, as discussed for the faceting phenomenon (4). Besides unilamellar structures, multilamellar and multivesicular structures were observed, which consist of polymersomes, similar with the above described. A certain number of disc-like objects are also observable in the images, probably coming from compressed polymersomes, as they exhibit thickness which is close to twice the polymersome membrane (32). Finally, worm-like micelles are present in the chimeric systems, mainly for 9:0.5 molar ratio cases, occasionally for HSPC:PDMAEMA-b-PLMA 2 9:0.1 and not at all for HSPC:PDMAEMA-b-PLMA 1 9:0.1. Their core diameter corresponds well with polymersome membrane thickness and their length begins from 800 nm and reaches over 1000 nm. This kind of structures has been mentioned for systems composed of lipid:PEGylated lipid and, recently, for chimeric/mixed systems, composed of lipid and polymer (43,44).
Concluding, by assessing the overall statistics of each structure in every chimeric system (Table S1), the formation of nonliposomal morphologies was found to be both copolymer concentration- and composition-dependent. Specifically, all these objects appear more often in systems of 9:0.5 molar ratio, while also the self-assembly of HSPC:PDMAEMA-b-PLMA 2 into polymersomes, discs, and worms is more frequent and is attributed to its relatively higher hydrophobic character. The most noteworthy polymersome (25%) and worm (21%) populations are observed in the HSPC:PDMAEMA-b-PLMA 2 9:0.5 system, where liposomes are only 51%. Overall, the different morphologies, as well as the various membrane phenomena, highlight the lyotropic effect of amphiphilic molecules of different hydrophilic-to-hydrophobic balance on lipid membranes, even in small amounts, which arises from the gap between the biomaterials’ properties and leads to various paths of self-assembly and final polymorphic structures.
In Vitro Screening of the Cytotoxicity of Chimeric Systems
Having a complete picture about the thermotropic, physicochemical, and morphological properties of the chimeric systems, we decided to move on to assessing their in vitro cytotoxicity, evaluating their safety and biocompatibility for future utilization in drug delivery applications. To the best of the authors’ knowledge, there are limited publications regarding the nanotoxicity of pure newly designed nanocarriers in the literature, though the growth-inhibiting effect of lipid vesicles on cells was investigated a long time ago (45). In our opinion, this is a very important issue, because the safety of nanocarriers is strongly related to the future effectiveness of encapsulated/incorporated drug molecules. An example for that is a previously established cell growth inhibition on murine cells of drug-free liposomes, compared with amphotericin C-loaded ones (46). What is more, cationic liposomes have been previously reported to exhibit elevated cytotoxicity on certain types of cells and in vitro screening is a fast way to assess their effect on cellular growth. In addition, it has been previously reported that aside from dose and charge, factors like size, composition, shape, and solubility play an important role in the toxicity of nanoparticles (47).
The toxicity profiles of all chimeric systems are presented in Fig. 10, for each system separately. For this evaluation, human mammary fibroblasts (MCF 10A) were inoculated with the various systems, at the following concentrations: 30, 60, 125, 250, and 500 μM, and the resultant toxicity, in terms of percentage cell survival, was recorded, for each case. In every one of the six cases, the survival was inversely proportional to the concentration of the system, occasionally beginning from above 97%, for very low chimeric system concentrations and reaching close to 10%, for very high ones. Another observation, based on the half-maximal inhibitory concentration (IC50) values, suggests that the molar ratio of the amphiphilic copolymer in the system affects the toxicity of the whole system, for both types of copolymer utilized. To be more specific, the in vitro toxicity of the chimeric systems is copolymer concentration-dependent; higher PDMAEMA-b-PLMA amounts result in less cell survival. Moreover, by comparing the two different amphiphilic block copolymers, PDMAEMA-b-PLMA 1 and PDMAEMA-b-PLMA 2, which have a different composition and hydrophilic-to-hydrophobic balance, one can observe varying IC50 values for the same concentrations. In this case, the lowest concentration of PDMAEMA-b-PLMA 1 led to higher toxicity, compared with PDMAEMA-b-PLMA 2. In general, the toxicity of the chimeric mixture also depends on the composition of the block copolymer. Namely, concentrations of chimeric systems with molar ratio 9:0.02 or 9:0.1 up to 125 μM ensure the survival of over 80% of the cell population. In both cases of 9:0.1, the IC50 drops dramatically, because of the high toxicity of the two highest nanosystem concentrations (250 and 500 μM).

The cell survival in conjunction with the concentration of each chimeric system is depicted. The Y axis is the % cell viability (logit scale) and the X axis is the concentration in micromolars (log scale)
To sum up, the overall toxicity of the novel formulations is higher, in comparison to our previous investigation of other chimeric nanosystems (4). In that study, conventional liposomes composed of synthetic phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) were found to be more toxic than those of natural phospholipid HSPC (IC50 578 and > 970 μM, respectively). However, those biomaterials are considered safe and biocompatible and are ingredients of liposomal products that are present in the market (48). The chimeric systems exhibited very high IC50 values (> 970 μM), due to the “stealth” properties of the utilized copolymer. Compared to those results, the chimeric liposomes herein are a bit more toxic, however are still in the range of 100–1000 μM, and consequently, their toxicity is considered as acceptable and the formulations are safe to forward toward in vivo studies. The toxicity is also comparable with other liposomal formulations from the literature, where polymeric guest was not utilized (46,47).
Maybe the polymer concentration-dependent toxicity is due to the high pKa value of the amino groups leading to partially positively charged PDMAEMA chains, which act as “surfactants” on the negatively charged physiological cell membranes. Positive charge-related toxicity has been previously attributed to cationic lipids. As a solution to this, it has been reported that the cross-linking of membrane-incorporated polymers contributes to shield this charge and prevents its toxic effects (49). Another reason could be that the heterogeneity of the chimeric systems, in terms of structure and morphology, as presented in the cryo-TEM, is the cause of this behavior. The 9:0.1 molar ratio resulted in primarily vesicular forms (lipidic or polymeric), for both types of polymer, while 9:0.5 led to the appearance of worm-like structures, which may be responsible for the observed elevated cytotoxicity. Other studies have previously demonstrated the shape dependence of cytotoxic effects and cell viability, including apoptosis and necrosis, reactive oxygen species (ROS) generation, and proinflammatory response of macrophages (50). Furthermore, filamentous morphologies have been associated with increased circulation time and organ accumulation, which are discouraging for biomedical applications, as they are likely to cause severe side effects (51).
CONCLUSIONS
The utilization of “smart”/functional biomaterials in imaging and treatment methods for complex diseases is very promising. Liposomes are present in the market for many years now, offering many advantages in these fields, while stimuli-responsive nanosystems are a recently emerging innovation, with the first thermosensitive formulation having reached phase 3 in the clinic. However, in order for aDDnSs to reach the clinical stage, it is essential that they initially exhibit promising toxicity profiles and biological effectiveness. Especially for dynamic/nonequilibrium nanosystems, such as liposomes and their derivatives, this can only be guaranteed if their contained units are well-defined, in terms of composition, hydrophilic-to-hydrophobic balance, and self-assembly behavior, and the resulting nanostructures are thoroughly studied with physicochemical, thermodynamic, and imaging techniques. In the present study, chimeric liposomes, comprised of the natural phospholipid HSPC and the amphiphilic diblock copolymer PDMAEMA-b-PLMA, were designed, developed, and investigated. The distinctive feature of the utilized copolymers is their pH responsivity, which comes from the PDMAEMA amino groups (extensively protonable at pH values below 7.5–8.0) and renders the liposomes pH-responsive. The different number of monomers leads to different segment lengths and, thus, different copolymer composition, which defines its architecture, hydrophilic-to-hydrophobic balance, and consequently, its pH responsivity, as evaluated by the experimental results.
At first, DSC measurements were performed on chimeric bilayers, hydrated with physiological or acidic medium. The PLMA group penetrates the lipid bilayer, affecting its packing, organization, and fluidity. The results denoted the slight fluidization and concentration-dependent cooperativity induced by the copolymer insertion in PBS, while in citrate buffer, fluidization was more intense, especially in the case of the copolymer with more pH-sensitive PDMAEMA monomers. Concerning liposomes, the physicochemical study showed that the incorporation of the copolymers, in all cases, led to positively charged particles of larger diameter, but higher lipid:copolymer ratios would lead to better properties, including homogeneity and colloidal/physical stability, the latter due to steric repulsions. By performing an acidic protocol, we could also ascertain the pH responsivity of the nanosystems, which altered their physicochemical characteristics. The AFM and cryo-TEM studies offered an insight on the membrane morphology and structure heterogeneity generated in these chimeric systems, because of mismatch between the two kinds of biomaterials (e.g., phospholipid and block copolymer) and structural polymorphism arising from the interfacial phenomena during the self-assembly. These phenomena could not be verified through classic DSC, either because their membrane thermodynamic contribution is not important and lies beneath the method’s limit of detection or because they are somehow hidden inside the main transition, as parts of its nanothermodynamics. The hydrophobicity and amount of block copolymer in lyotropic systems are two determinant factors that induce the formation of nonvesicular assemblies, including worm-like micelles, while the copolymer molar ratio is generally very low (0.1 or 0.5). Finally, the in vitro screening of the chimeric nanosystems correlated the composition and concentration of the copolymer with the cytotoxicity of the developed formulations. In this context, the probable relationship between morphologically heterogeneous systems and increased toxicity is suggested. However, all chimeric systems are considered as nontoxic in vitro, due to the IC50 values being in the range of 100–1000 μM and, as a result, probable candidates for in vivo applications.
In summary, the most suitable pH-responsive chimeric nanosystems for further applications in drug delivery or other fields are the HSPC:PDMAEMA-b-PLMA 1/2 9:0.1 ones. Those exhibited the most promising properties, among which was morphological homogeneity, which is considered a prerequisite of paramount importance for biological stability and effectiveness. The control of the liposome membrane properties can offer promising aDDnSs, while the observed structural polymorphism and morphogenesis may provide explanation on how macromolecule insertion affects the biophysics of biological membranes.
References
Demetzos C, Pippa N. Advanced drug delivery nanosystems (aDDnSs): a mini-review. Drug Deliv. 2014;21(4):250–7.
Wang K, Huang Q, Qiu F, Sui M. Non-viral delivery systems for the application in p53 cancer gene therapy. Curr Med Chem. 2015;22(35):4118–36.
Pippa N, Stellas D, Skandalis A, Pispas S, Demetzos C, Libera M, et al. Chimeric lipid/block copolymer nanovesicles: physico-chemical and bio-compatibility evaluation. Eur J Pharm Biopharm. 2016;107:295–309.
Pippa N, Gardikis K, Pispas S, Demetzos C. The physicochemical/thermodynamic balance of advanced drug liposomal delivery systems. J Therm Anal Calorim. 2014;116(1):99–105.
Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance. Br J Pharmacol. 2016;173(6):970–9.
Xu H, Paxton JW, Wu Z. Enhanced pH-responsiveness, cellular trafficking, cytotoxicity and long-circulation of PEGylated liposomes with post-insertion technique using gemcitabine as a model drug. Pharm Res. 2015;32(7):2428–38.
Felber AE, Dufresne MH, Leroux JC. pH-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates. Adv Drug Deliv Rev. 2012;64(11):979–92.
Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials. 2016;85:152–67.
Li Z, Fan Z, Xu Y, Lo W, Wang X, Niu H, et al. pH and thermal sensitive hydrogels as stem cell carriers for cardiac therapy. ACS Appl Mater Interfaces. 2016;8(17):10752–60.
Fan Z, Fu M, Xu Z, Zhang B, Li Z, Li H, et al. Sustained release of a peptide-based matrix metalloproteinase-2 inhibitor to attenuate adverse cardiac remodeling and improve cardiac function following myocardial infarction. Biomacromolecules. 2017;18(9):2820–9.
Fan Z, Xu Z, Niu H, Gao N, Guan Y, Li C, et al. An injectable oxygen release system to augment cell survival and promote cardiac repair following myocardial infarction. Sci Rep. 2018;8(1):1371.
Naziris N, Pippa N, Pispas S, Demetzos C. Stimuli-responsive drug delivery nanosystems: from bench to clinic. Curr Nanomed. 2016;6(3):1–20.
Delogu F. Thermodynamics on the nanoscale. J Phys Chem B. 2005;109:21938–41.
Dao TPT, Fernandes F, Er-Rafik M, Salva R, Schmutz M, Brûlet A, et al. Phase separation and nanodomain formation in hybrid polymer/lipid vesicles. ACS Macro Lett. 2015;4:182–6.
Samsonov VM, Sdobnyakov NY, Bazulev AN. On thermodynamic stability conditions for nanosized particles. Surf Sci. 2003;532–535:526–30.
Pippa N, Pispas S, Demetzos C. The metastable phases as modulators of biophysical behavior of liposomal membranes. J Therm Anal Calorim. 2015;120(1):937–45.
Demetzos C. Biophysics and thermodynamics: the scientific building blocks of bio-inspired drug delivery nano systems. AAPS PharmSciTech. 2015;16(3):491–5.
Tribet C, Vial F. Flexible macromolecules attached to lipid bilayers: impact on fluidity, curvature, permeability and stability of the membranes. Soft Matter. 2008;4:68–81.
Samsonova O, Pfeiffer C, Hellmund M, Merkel OM, Kissel T. Low molecular weight pDMAEMA-block-pHEMA block-copolymers synthesized via RAFT-polymerization: potential non-viral gene delivery agents? Polymer. 2011;3(2):693–718.
Zengin A, Karakose G, Caykara T. Poly(2-(dimethylamino)ethyl methacrylate) brushes fabricated by surface-mediated RAFT polymerization and their response to pH. Eur Polym J. 2013;49(10):3350–8.
Wei X, Cohen R, Barenholz Y. Insights into composition/structure/function relationships of Doxil® gained from “high-sensitivity” differential scanning calorimetry. Eur J Pharm Biopharm. 2016;104:260–70.
Cheng Z, Elias DR, Kamat NP, Johnston ED, Poloukhtine A, Popik V, et al. Improved tumor targeting of polymer-based nanovesicles using polymer-lipid blends. Bioconjug Chem. 2011;22(10):2021–9.
Chrysostomou V, Pispas S. Stimuli-responsive amphiphilic PDMAEMA-b-PLMA copolymers and their cationic and zwitterionic analogs. J Polym Sci A Polym Chem. 2018;56(6):598–610.
Pippa N, Deli E, Mentzali E, Pispas S, Demetzos C. PEO-b-PCL grafted DPPC liposomes: physicochemical characterization and stability studies of novel bio-inspired advanced drug delivery nano systems (aDDnSs). J Nanosci Nanotechnol. 2014;14:5676–81.
Galani A, Tsitsias V, Stellas D, Psycharis V, Raptopoulou CP, Karaliota A. Two novel compounds of vanadium and molybdenum with carnitine exhibiting potential pharmacological use. J Inorg Biochem. 2014;142(C):109–17.
Koynova R, Caffrey M. Phases and phase transitions of the phosphatidylcholines. Biochim Biophys Acta. 1998;1376:91–145.
Kitayama H, Takechi Y, Tamai N, Matsuki H, Yomota C, Saito H. Thermotropic phase behavior of hydrogenated soybean phosphatidylcholine-cholesterol binary liposome membrane. Chem Pharm Bull (Tokyo). 2014;62(1):58–63.
Matsingou C, Hatziantoniou S, Georgopoulos A, Dimas K, Terzis A, Demetzos C. Labdane-type diterpenes: thermal effects on phospholipid bilayers, incorporation into liposomes and biological activity. Chem Phys Lipids. 2005;138(1–2):1–11.
Matsingou C, Demetzos C. Calorimetric study on the induction of interdigitated phase in hydrated DPPC bilayers by bioactive labdanes and correlation to their liposome stability: the role of chemical structure. Chem Phys Lipids. 2007;145(1):45–62.
Naziris N, Pippa N, Chrysostomou V, Pispas S, Demetzos C, Libera M, et al. Morphological diversity of block copolymer/lipid chimeric nanostructures. J Nanopart Res. 2017;19(10):347–57.
Takechi-Haraya Y, Sakai-Kato K, Goda Y. Membrane rigidity determined by atomic force microscopy is a parameter of the permeability of liposomal membranes to the hydrophilic compound calcein. AAPS PharmSciTech. 2017;18(5):1887–93.
Naziris N, Pippa N, Meristoudi A, Pispas S, Demetzos C. Design and development of pH-responsive HSPC:C12H25-PAA chimeric liposomes. J Liposome Res. 2017;27(2):108–17.
Sun X, Jiang G, Wang Y, Xu Y. Synthesis and drug release properties of novel pH- and temperature-sensitive copolymers based on a hyperbranched polyether core. Colloid Polym Sci. 2011;289:677–84.
Attwood D, Florence AT. Physical pharmacy. London: Pharmaceutical Press; 2008.
Winzen S, Bernhardt M, Schaeffel D, Koch A, Kappl M, Koynov K, et al. Submicron hybrid vesicles consisting of polymer-lipid and polymer-cholesterol blends. Soft Matter. 2013;9(25):5883–90.
Kuntsche J, Horst JC, Bunjes H. Cryogenic transmission electron microscopy (cryo-TEM) for studying the morphology of colloidal drug delivery systems. Int J Pharm. 2011;417(1–2):120–37.
Schulz M, Binder WH. Mixed hybrid lipid/polymer vesicles as a novel membrane platform. Macromol Rapid Commun. 2015;36(23):2031–41.
Kaiser N, Kimpfler A, Massing U, Burger AM, Fiebig HH, Brandl M, et al. 5-Fluorouracil in vesicular phospholipid gels for anticancer treatment: entrapment and release properties. Int J Pharm. 2003;256(1–2):123–31.
Ickenstein LM, Sandström MC, Mayer LD, Edwards K. Effects of phospholipid hydrolysis on the aggregate structure in DPPC/DSPE-PEG2000 liposome preparations after gel to liquid crystalline phase transition. Biochim Biophys Acta. 2006;1758(2):171–80.
Bowick MJ, Sknepnek R. Pathways to faceting of vesicles. Soft Matter. 2013;9(34):8088–95.
Itel F, Chami M, Najer A, Lörcher S, Wu D, Dinu IA, et al. Molecular organization and dynamics in polymersome membranes: a lateral diffusion study. Macromolecules. 2014;47(21):7588–96.
Andersson M, Hammarstroem L, Edwards K. Effect of bilayer phase transitions on vesicle structure, and its influence on the kinetics of viologen reduction. J Phys Chem. 1995;99(39):14531–8.
Johnsson M, Edwards K. Liposomes, disks, and spherical micelles: aggregate structure in mixtures of gel phase phosphatidylcholines and poly(ethylene glycol)-phospholipids. Biophys J. 2003;85(6):3839–47.
Dao TPT, Brûlet A, Fernandes F, Er-Rafik M, Ferji K, Schweins R, et al. Mixing block copolymers with phospholipids at the nanoscale: from hybrid polymer/lipid wormlike micelles to vesicles presenting lipid nanodomains. Langmuir. 2017;33(7):1705–15.
Campbell PI. Toxicity of some charged lipids used in liposome preparations. Cytobios. 1983;37(145):21–6.
Szoka FC Jr, Milholland D, Barza M. Effect of lipid composition and liposome size on toxicity and in vitro fungicidal activity of liposome-intercalated amphotericin B. Antimicrob Agents Chemother. 1987;31(3):421–9.
Roursgaard M, Knudsen KB, Northeved H, Persson M, Christensen T, Kumar PEK, et al. In vitro toxicity of cationic micelles and liposomes in cultured human hepatocyte (HepG2) and lung epithelial (A549) cell lines. Toxicol in Vitro. 2016;36:164–71.
Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2011;7:49–60.
Simões MG, Alves P, Carvalheiro M, Simões PN. Stability effect of cholesterol-poly(acrylic acid) in a stimuli-responsive polymer-liposome complex obtained from soybean lecithin for controlled drug delivery. Colloids Surf B: Biointerfaces. 2017;152:103–13.
Oh WK, Kim S, Yoon H, Jang J. Shape-dependent cytotoxicity and proinflammatory response of poly(3,4-ethylenedioxythiophene) nanomaterials. Small. 2010;6(7):872–9.
Eliezar J, Scarano W, Boase NRB, Thurecht KJ, Stenzel MH. In vivo evaluation of folate decorated cross-linked micelles for the delivery of platinum anticancer drugs. Biomacromolecules. 2015;16(2):515–23.
Funding
This research has been financially supported by the General Secretariat for Research and Technology (GSRT) and the Hellenic Foundation for Research and Innovation (HFRI) (Scholarship Code: 392).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
ESM 1
The stability evaluation of polydispersity, as well as diameters and wall dimensions extracted by cryo-TEM for the prepared chimeric nanosystems are provided in the supplementary material section. (DOCX 201 kb)
Rights and permissions
About this article

Cite this article
Naziris, N., Pippa, N., Stellas, D. et al. Development and Evaluation of Stimuli-Responsive Chimeric Nanostructures. AAPS PharmSciTech 19, 2971–2989 (2018). https://doi.org/10.1208/s12249-018-1112-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-018-1112-2