
Abstract
Purpose
The in vitro and in vivo properties of PEGylated pH-sensitive liposomes (PSL) prepared by pre- and post-insertion techniques were investigated.
Methods
A pre-insertion or post-insertion technique was used for PSL PEGylation. For the first time, confocal laser scanning microscopy coupled with a modified calcein self-quench assay was applied to evaluate the endosome escape capability. PSL cellular uptake was evaluated using macrophages and the cytotoxicity using a gemcitabine (model drug)-resistant MIA PaCa-2 cells. The pharmacokinetics of PSL encapsulated gemcitabine was investigated in rats.
Results
PEGylation reduced the pH-sensitivity in a concentration-dependent manner (0.5–5% mol). Both PEGylation methods reduced the uptake of PSL by macrophages by over 60%. Cytotoxicity was ranked in the order: post-inserted PSL ≥ pre-inserted PSL > non-PSL > gemcitabine solution, consistent with the confocal microscopic observation and pH-sensitivity. Both pre and post-inserted PSL resulted in significant reductions (p < 0.05) in plasma clearance (58.6 and 38.4 ml/h/kg), increases in the area-under-the-concentration-time curve (56.9 and 87.1 μM· h) and half-life (6.1 and 6.2 h) compared to gemcitabine solution (152.9 ml/h/kg, 22.2 μM· h and 1.4 h).
Conclusion
PEGylation by post-insertion offers advantages over pre-insertion to obtain PSL with enhanced pH-sensitivity, more effective intra-cytoplasmic delivery, and a superior pharmacokinetics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liposomes are considered to be one of the most biocompatible vesicular drug carriers with a capacity for accommodating both hydrophilic and hydrophobic compounds. Since their discovery in the 1960s by Bangham (1), in vitro studies have indicated that liposomes are taken into cells via endocytosis which may increase the efficiency of intracellular drug delivery (2,3). However, after endocytosis by cells, the conventional liposomes, entrapped in the endosome organelle, is destined for the lysosome, where the contents may be degraded by enzymes. This may limit the extent of intracellular drug available to its site of action, such as DNA (4–6). To address this limitation, pH-sensitive liposomes (PSL) may be used to enhance the intracellular delivery of drug content to cytosol via a process known as ‘endosome escape’ [11, 12]. PSL are designed to be stable at the physiological pH, but destabilize under acidic conditions by using pH-sensitive lipid components, such as 1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) in the liposomal membrane. PSL take advantage of the acidification of the endosomal lumen (as acidic as pH 5–5.5) (7,8) to allow protonation of the head group of DOPE, resulting in an inversion of DOPE into an unstable hexagonal phase. The latter is key factor for cytoplasmic delivery of the entrapped compounds and controlled release of its cargo in pathological tissues, such as tumours or inflamed and infected areas which exhibit an acidic environment as compared to normal tissues (9). By enhancing intracellular drug delivery and the endosome escape pathway, PSL may bypass multi-drug resistant efflux pumps and thus restore the cell’s sensitivity to some drugs (10). To date, PSL have been applied to macrophage cell target delivery (6), intracellular transport of antigens (9), efficient delivery of neoplastic drugs or recombinant proteins (11) and intracellular transport of genetic material for antisense therapies (5). However, in addition to the pH-sensitivity, the in vivo efficacy of PSL depends strongly on their interactions with serum components that influence their pharmacokinetics (6,9). After intravenous administration, PSL are cleared rapidly from blood circulation by the reticulo-endothelial system (RES), accumulating in the liver and spleen (12), which is an issue if the target site is not in these organs. To overcome this problem, surface coating with polyethylene glycol (PEG) polymer (termed PEGylation) provides steric inhibition of opsonin proteins adhering to the liposome surface, thus avoiding recognition and the subsequent rapid clearance by the RES, and leading to a prolonged circulation time which is key factor for PSL’s in vivo tumor accumulation (13).
Although PEGylation has been reported to prolong circulation time (14), in vitro studies have suggested that PEGylation may reduce the pH-sensitivity, cellular uptake (15) and endosome escape of the PSL (16). Currently, two techniques, pre-insertion (the traditional method) and post-insertion are used to prepare PEGylated liposomes. With the former, the PEGylated phospholipids are added to other lipids, such as DOPE and cholesteryl hemisuccinate (CHEMS), during liposome membrane preparation, allowing the PEG chains to link to both internal and external layers of the liposome. With the post-insertion method, the PEGylated lipid is grafted on the external layer of the preformed liposomes (17,18). The latter was demonstrated to have better retention of the entrapped drug than the classical non-pH-sensitive PEGylated liposomes (CL), and a superior pharmacokinetics in vivo, possible due to reduced leakage of the drug (17,18). To our knowledge, there have been no reports on whether the pre-insertion or post-insertion method can cause significant differences in the pH-sensitivity, cellular trafficking, cytotoxicity and pharmacokinetics of the resulting PSL. Furthermore, there are few pharmacokinetic data on PSL gemcitabine published in the literature to date.
Gemcitabine, a potent first-line drug commonly used for the treatment of pancreatic cancer (PC) was selected as a model drug to investigate the PSL effect on cytotoxicity and pharmacokinetics. Gemcitabine is phosphorylated to its active moiety in the cell, which inhibits DNA synthesis (2,19). However, after systemic admistration, rapid conversion into the inactive metabolite by cytidine deaminase and development of resistance has been reported to markedly reduce the clinical efficacy of gemcitabine (20–22).
Our aim was to develop a PEGylated PSL with improved pH-sensitivity, more effective intracellular delivery and superior in vivo pharmacokinetics for compounds encapsulated in the aqueous core. We have investigated the in vitro and in vivo properties of PEGylated PSL prepared by pre- and post-insertion techniques, including their cellular uptake and endosome escape capabilities, in vitro drug release and their pharmacokinetics in rats. PSL formulations containing a model drug, gemcitabine, were developed and their cytotoxicity towards a drug-resistant PC cell line (MIA PaCa-2) was compared. In addition, a high drug loading (DL) is desirable to increase the cytotoxicity to cancer cells in vitro, as well as to reduce possible side effects caused by liposomal phospholipids and cholesterol (23,24). Based on our previous study with CL, a ‘small volume incubation (SVI)’ method was developed to increase the loading of gemcitabine into the PSL when active loading cannot be applied (3).
Materials and Methods
Materials
Cholesteryl hemisuccinate (CHEMS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and N-(carbonyl-methoxypolyethylene-glycol-2000)-1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE-mPEG2000) were purchased from Avanti Polar Lipids (Alabama, USA), and 1,2-dipalmitoyl-sn-glycero-3-phospocholinemonohydrate (DPPC) and N-(carbonyl-methoxypolyethylene-glycol-2000)-1,2-distearoyl-sn-glycero-3-phospho-thanolamine (DSPE-mPEG2000) from Lipoid (Steinhausen, Switzerland). Cholesterol, gemcitabine (purity >98%), calcein and 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT, for cytotoxicity studies) were from Sigma (Auckland, New Zealand). Milli-Q water was prepared using a water purification system (Millipore Corp., Bedford, MA, USA). All the other materials for this study were of analytical grade.
For the pharmacokinetic study, Sprague–Dawley (SD) rats (195–205 g) were obtained from the Vernon Jansen Unit (The University of Auckland), and maintained according to the standards relating to the care and management of experimental animals of New Zealand.
All procedures were approved by the Committee on Animal Experiments of The University of Auckland (Ethics approval number 001228).
Preparation of PEGylated pH-Sensitive Liposomes
PEGylation by the Pre-Insertion Method
PSL (DOPE: CHEMS at a fix molar ratio of 6:4, with either DSPE-mPEG2000 or DOPE-mPEG2000), and CL (DPPC: cholesterol: DSPE-mPEG2000 = 6:3:0.28) were prepared using the thin-film hydration-extrusion (TFHE) method. The ratio of PEGylated lipid to total lipids were, 0 0.5, 1, 3 and 5%. Briefly, the mixture of lipids was dissolved in 1 ml of chloroform: methanol (3:1, v/v). The thin film of the lipid was then obtained by removal of the organic solvent using a rotary evaporator under vacuum (R-215, Büchi, Switzerland). Further traces of the organic solvent were removed by flowing nitrogen at 45°C for 40 min. After hydration with 1 ml PBS (pH 7.4, 30 mM, with the osmotic pressure adjusted to 100 mOsm using NaCl), liposomes were extruded 20 times through a 200 nm membrane. The liposomes pellet was obtained by ultracentrifugation of the above suspension at 186,000×g for 1 h at 4°C, and kept at 4°C for further study.
PEGylation by the Post-Insertion Method
Briefly, blank liposomes (DOPE: CHEMS: = 6:4) were prepared using the above protocol. Blank liposomes were then incubated with different amounts of DSPE-mPEG2000 aqueous solutions (possibly with micelles, prepared using TFHE method, extruded through 50 nm membrane) for 1 h to make the final molar % of DSPE-mPEG2000 at 0.5, 1, 3 and 5%. The liposomes were obtained by ultracentrifuge at 186,000×g for 1 h at 4°C.
Preparation of Calcein or Drug Containing PSL
To prepare liposomes for the pH-responsiveness study and confocal microscopic analysis, the hydrophilic fluorescent dye calcein in aqueous solution (80 mM, pH 7.4, adjusted to 320 mOsm with NaCl) was used as the hydration solution.
To prepare gemcitabine PSL for cytotoxicity and pharmacokinetic studies, the previously reported Small Volume Incubation (SVI) method was modified for incorporation of gemcitabine into preformed PSL (3). Briefly, gemcitabine hydrochloride (9 mg) was dissolved in 2 ml Milli-Q water. The pH was adjusted to 7.4 with 0.1 M NaOH and equal aliquots were added to 6 separate tubes. After removal of all the water using a vacuum concentrator, the gemcitabine powder was re-suspended with 20 μl of 50 mM PBS (7.4) with vortexing for 3 min with preformed empty liposome pellets (total lipids 10 mg, containing 3% DSPE-mPEG2000). The mixture was then incubated at 60°C for 3 h for drug loading.
The gemcitabine CL containing 3% DSPE-mPEG2000 used for the cytotoxicity study were prepared as previously (3). After loading with gemcitabine, the predicted osmotic pressure in the liposomal cores was approximately 300 mOsm for both PSL and CL which is close to the osmotic pressure of blood.
Determination of Attachment Efficiency (AE) of PEGylated Lipid
The liposomal AE of the PEGylated lipids was determined by a previously reported HPLC method with modification (18). After incubation of the PEGylated lipid (either DSPE-mPEG and DOPE-mPEG) with the preformed non-PEGylated liposomes at 60°C for 1 h, the free DSPE-mPEG and DOPE-mPEG micelles were separated by ultracentrifugation at 186,000×g (a previous study confirmed that under these conditions the liposomes were maintained in the pellet without micelle formation). The PSL pellet was then dissolved in the mobile phase (methanol: isopropanol, 95:5, at a flow rate of 1 ml/min), and the lipid contents (DOPE, CHEMS and DSPE-mPEG2000) analysed by HPLC using an Agilent 1100 system with a refractive index (RI) detector and a Phenomenex C18 column (250 × 4.6 mm i.d., 5 μm particle size) at 35°C. The AE was calculated using the following equation:
Where Mpellet is the mass of PEGylated lipids attached, and Mtotal the total mass of PEGylated lipid used in preparation.
Determination of Entrapment Efficiency (EE) and Drug Loading (DL)
To determine the EE and DL of gemcitabine in various liposomes, un-entrapped gemcitabine was removed by gel-filtration using a Sephadex G50 column (bead size, 20–80 μm). The liposome proportion was collected after gel-filtration. To obtain the concentration of the encapsulated gemcitabine (Min), 100 μl of the filtered liposome suspension was diluted with 900 μl of 10% Triton-X100 solution and votexed to dissolve the vesicles before analysis for gemcitabine using a validated reverse-phase HPLC method (3). EE and DL were calculated using the following equations:
Where Mtotal the total drug mass used in preparation and Mlip is the total mass of drug loaded liposomes.
Particle Size, Zeta Potential and Morphology
The particle size distribution and zeta potential of various liposomes were measured by a laser diffraction particle analyser (Nano-ZS Malvern Instruments Ltd, UK).
The morphology of the drug loaded liposomes was examined by cryo-transmission electron microscopy (TEM) (FEI Tecnai G2 Spirit Twin 120Kv). Briefly, a 3 mm 200-mesh copper grid was placed on top of one drop of liposome suspension (0.2 mg/ml) and incubated for 2 min. The surplus was then removed by filter paper, and the liposome sample on the mesh was immediately frozen using ethane in a liquid nitrogen system until TEM analysis.
pH-Sensitivity of Liposomes
To develop sterically stabilized (PEGylated) PSL, a number of formulations were investigated. The optimal molar ratio 6:4 for DOPE: CHEMS for pH-sensitivity was chosen based on the literature (9). DOPE-mPEG2000 or DSPE-mPEG2000 was compared, at different concentrations (0–5%). The pH-sensitivity of liposomes was evaluated using a calcein self-quenching assay with modification (6,25). Following the leakage of encapsulated calcein (80 mM, self-quenched) from PSL, calcein was diluted by the extra-liposomal medium, resulting in an increase in fluorescence of the whole liposome suspension. Free calcein was removed from liposomes by a Sephadex G50 column using 10 mM PBS (adjusted to isotonic with NaCl) after preparation and then 100 μl of liposome sample was added to 900 μl PBS (150 mM with 1 mM EDTA) with different pH values (5, 5.5, 6.0, 6.5 and 7.4). The final concentration of total phospholipids (DOPE, DPPC, DSPE-mPEG2000 and DOPE-mPEG2000) was controlled at 200 nM. After 10 min incubation at 37°C, the calcein fluorescence was measured at each pH by a plate reader (excitation, 470 nm and emission, 509 nm). The total fluorescence was obtained by destroying the liposome with 10% of Triton-X100, followed by dilution with PBS (pH 7.4, 100 mM isotonic adjusted with NaCl). The final concentration of Triton-X100 was 0.1%. To calibrate the pH effect on the fluorescence of calcein, different standard curves of calcein at each pH were prepared to quantify the amount of calcein. The pH-sensitivity of the liposomes was calculated using the following equation:
Where M7.4 is the mass of calcein released to medium at pH7.4, MpH is the mass released at different pH buffers, and M100% is the total mass of calcein entrapped in the liposomes.
Liposomal Uptake by Macrophage-Like Cells
To predict their capacity for prolonged circulation in vivo, a macrophage-like cell line (RAW 264.7 from ATCC, TIB-71) was used to evaluate the uptake of the different PSL formulations. A lower cellular uptake would indicate a possible stealth property, i.e., avoidance of the clearance by RES (26). Calcein (as a marker) was encapsulated into PSL with different molar ratios of DSPE-mPEG2000 (0, 1, 3 and 5%). The cells were seeded at a density of 2 × 105 cells/well in a 24-well plate in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS, 100 UI/ml penicillin and 100 μg/ml streptomycin (complete DMEM), and maintained at 37°C for 24 h in an incubator with 5% CO2/95% air. The cells were then treated with 1 ml complete DMEM with different liposome suspensions. After 3 h incubation, the cells were washed 3 times with PBS, followed by the addition of 1 ml Triton-X100 solution and sonicated for 30 min to extract the intracellular calcein. After centrifuge at 125×g for 5 min, the supernatant was obtained and measured using a fluorimeter (Molecular Devices, US) with excitation and emission wavelengths of 470 and 509 nm, respectively. A standard curve of calcein in 10% Triton-X100 (pH 7.4, adjusted with NaOH) was prepared for the calculation of calcein. The background absorbance of media-only controls was subtracted from the sample readings, and uptake was expressed as the percentage of the control liposomes (non-PEGylated). Experiments were conducted in triplicate.
Intracellular Delivery Efficiency and Endosome Escape
To investigate the endosome escape ability of the PSL formulations, a calcein self-quenching assay in combination with confocal laser scanning microscopy was firstly applied in this study. In contrast to the conventional assay, only the calcein released from the liposomes through the endosome escape pathway (e.g., when concentration <80 mM) to cytoplasm can be measured. There would be no interference by the fluorescence encapsulated in the liposomes even if they are internalized in the cells. MIA PaCa-2 cells were incubated with various calcein-containing liposomes for 2 h at 37°C under a humidified (95%) CO2 (5%) atmosphere. The cells were washed three times with PBS before fixing with 4% paraformaldehyde in PBS for 15 min at room temperature and rinsing three times with PBS. Cells were stained with a nucleus dye, 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) for 5 min, followed by 3 rinses with PBS. Mounting media was then added to the cells which were covered with a glass slide for microscopic observations. The location of intracellular fluorescence was validated using a confocal laser scanning microscope (Olympus Fluoview FV1000, Olympus Corporation, Japan) with excitation wavelengths of 405 nm for DAPI, and 488 nm for calcein. All the optical sections were recorded with the same settings for each colour detected.
In Vitro Gemcitabine Release
The pH-dependent drug release from selected PSL was investigated using a dialysis method. Cellulose acetate dialysis bags (12–14 kDa, molecular weight cut-off) containing 1 ml of the liposome suspension were placed in 50 ml release medium (PBS, pH 7.4 or 6.5, 125 mM, adjusted with NaCl to 320 mOsm) at 37°C with stirring throughout the experiment. At different time intervals, a 0.1 ml sample was withdrawn and replaced with the same volume of fresh medium. Samples were analysed by HPLC and the % of the drug released was calculated.
Release profiles were compared using a similarity factor (f2) model based on the sum-squared error of the differences in percentage of cumulative release between two formulations (Tj and Rj).
Where, n represents the total number of time points, and wj is an optional weight factor. The f2 values may range from 0 to 100, and if the f2 is between 50 and 100, the two profiles are considered to be similar (27).
Cytotoxicity to Drug Resistant Pancreatic Cells
MIA PaCa-2 cells (a gift from the Auckland Cancer Society Research Centre) were cultured in complete DMEM and maintained at 37°C in an incubator with 5% CO2/95% air. The MTT cell viability assay was used to evaluate the cytotoxic effects of various gemcitabine liposomes in MIA PaCa-2 cells. Gemcitabine solution and blank liposomes were used as references. Cells were seeded (5 × 103 cells/well) in a 96-well plate in phenol-red free DMEM and cultured for 24 h. Cells were treated with free gemcitabine solutions, gemcitabine PSL prepared by pre- and post-insertion methods, and empty PSL, respectively. At 24 and 48 h, the medium was removed and the cells were washed with PBS before the cell viability was measured using the routine protocol for MTT assay (3) Cell viability was expressed as a percentage of control (untreated cells). Independent experiments were conducted at least in duplicate (n = 3, for each experiment).
Pharmacokinetics in Rats
Sprague–Dawley (SD) rats were randomly divided into three formulation groups (n = 4): gemcitabine solution and the gemcitabine loaded PSL PEGylated by pre-insertion or post-insertion method (both PSL with DOPE: CHEMS: DSPE-mPEG2000 = 6:4:0.3, DL = 4.2%). Each formulation (1 mL) was injected via the tail vein at 1 mg/kg equivalent gemcitabine. Blood samples (100–200 μl) were collected from the tail vein at 0 (before injection), 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 24 h after drug administration. The plasma (50 μl) was obtained after centrifugation (3000 rpm) and added to 0.5 ml acetonitrile. After vortexing for 3 min, the mixture was centrifuged and the supernatant collected. A further 0.5 ml acetonitrile was added to the precipitate and the extraction process repeated. The combined supernatants were evaporated to dryness using a concentrator and the residue re-dissolved by vortexing for 3 min with 50 μl Triton-X100 solution (10%). The sample was centrifuged and the supernatant was collected for HPLC analysis of gemcitabine (3). The quantification of gemcitabine was calculated using an external standard curve (ranging from 0.1 to 10 μM, r 2 = 0.9997) prepared with spiked plasma samples subjected to the same extraction protocol. No interference was observed from plasma components and the recovery after extraction for each concentration level was all over 95%.
The pharmacokinetic profiles of the gemcitabine formulations were fitted to a non-compartmental model using WinNonlin® version 5.3 (Pharsight, Mountain View, CA, USA). The area under the concentration-time profile curve (AUC) was calculated by the log trapezoidal rule with extrapolation of the terminal slope to infinity by log-linear regression. The elimination half-life (T½) was calculated by the equation T½ = ln2/λ (λ is the rate constant associated with the terminal elimination phase). The model-independent pharmacokinetic parameters, clearance (Cl), volume of distribution (Vd), and mean residence time (MRT) were calculated by the following equations: Cl = Dose/AUC; Vd = (Dose x AUMC)/(AUC)2, and MRT = AUMC/AUC; where AUMC represents the total area under the first moment of the concentration-time curve.
Statistical Analysis
Data were compared using one-way ANOVA (Analysis of Variance) with Tukey’s multiple comparisons test using Origin 8.0 and the p value for significance was set at 0.05.
Results
PEGylated Liposomes and Attachment Efficiency
The HPLC results showed that the AE of DSPE-mPEG2000 to PSL was greater than 97% regardless of the concentration used (0.5–3%). A longer incubation time (>1 h) did not further increase the attachment efficiency significantly, indicating that 1 h incubation was sufficient to complete the post-insertion process.
Increasing the amount of DSPE-mPEG2000 using the pre-insertion method significantly reduced the liposome size (p < 0.05); whereas no significant difference was observed using the post-insertion method (Table I). In all cases, the Polydispersity Index ranged from 0.05 to 0.08, indicating a mono-dispersal profile.
With the addition of DSPE-mPEG2000 at 1%, the zeta potential of all the liposomes significantly reduced from −37 to around −8 mV. However, not further change was observed when PEG density was increased.
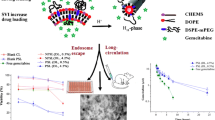
pH-Sensitivity
The addition of DSPE-mPEG2000 and particularly DOPE-MPEG2000 decreased the pH-sensitivity of the liposomes depending on the density of PEG (0.5–5%) (Fig. 1a).

pH-dependent leakage of calcein after 10 min from different PSL formulations with and without PEGylation: (a) pre-insertion with various PEGylated lipids, and (b) comparison of pre-insertion and post insertion of DSPE-mPEG2000. Data are mean ± SD (n = 3).
The post-insertion of PEGylated lipid also significantly (p < 0.05) increased the in vitro pH sensitivity of the liposomes (Fig. 1b); and with the same molar ratio of PEG, the release of calcein from at pH 5.0 almost double that of liposomes produced by the pre-insertion method.
Characterization of pH-sensitive Gemcitabine Liposomes
Using the modified SVI method, a high EE (28.2 ± 0.5%) and DL (4.2 ± 1% w/w) for gemcitabine were obtained (to our knowledge, the highest EE that have been reported for gemcitabine with a PSL size of approximately 160 nm). PEGylation (3% DSPE-mPEG2000) with post-insertion method resulted in larger vesicles than with pre-insertion (160 nm vs 140 nm). However, no difference was observed in their zeta potential (−8.1 ± 0.3) or morphology, as shown in the TEM (Fig. 2).

Cryo-TEM morphology of PEGylatd PSL prepared by pre-insertion (a) and post insertion (b).
Uptake by Macrophage-Like Cells (RAW 264.7)
The post-insertion of 0.5% DSPE-mPEG2000 effectively reduced the macrophage uptake of liposomes to less than 40% (p < 0.05, compared to non-PEGylated PSL) by RAW 264.7 cells (as measured by intracellular calcein accumulation) was observed (Fig. 3), suggesting stealth properties and the potential for long-circulation of the PEGylated PSL in vivo. There is no significant difference among all the PEGylated PSL (p > 0.05), indicating that increase the ratio of DSPE-mPEG2000 cannot further reduce the uptake.

Uptake by macrophage-like cells of different PSL formulations prepared by pre-insertion and post-insertion methods. Data are mean ± SD (n = 3).
Intracellular Delivery Efficiency and Endosome Escape
The strongest intracellular calcein fluorescence was observed with PSL without PEGylation (Fig. 4). With the addition of 3% DSPE-mPEG2000, the intracellular signal of calcein was remarkably decreased Furthermore, cells incubated with pre-insertion PSL showed more distinct ‘bright dots’ (probably endosomes) around the nucleus than in the cells treated with the PSL prepared by the post-insertion, suggesting a slightly better endosomal escape of the calcein from the latter formulation. With the CL, a very weak signal was observed in the MIA PaCa-2 cells.

Confocal microscopy analysis of MIA PaCa-2 cells treated with different liposome formulations encapsulating 80 mM calcein.
In vitro Drug Release
At pH 7.4, there was little release of gemcitabine over 24 h from CL, and this was not altered at the lower pH (f2 = 97.2). In contrast, the release rate of gemcitabine from PSL was significantly greater than the CL. Compared with pre-insertion, post-insertion resulted in PSL with more rapid release at acidic condition (f2 = 31.0), but not at the physiological pH (f2 = 78.7) (Fig. 5).

Cumulative release of gemcitabine from different liposomes (all containing 3% DSPE-mPEG2000) in PBS (pH 7.4 and 6.5). Data are mean ± SD (n = 3).
Cytotoxicity to Drug-Resistant Pancreatic Cancer Cells
Blank liposomes (diluted to lipid concentrations equivalent to the gemcitabine-loaded liposomes) were not cytotoxic to the MIA PACa-2 cells over 48 h exposure (>92% cell viability). Free gemcitabine in solution demonstrated no significant cytotoxicity (cell viability >96%) over the concentration range of 5 to 50 μM after 24 h exposure, but viability had fallen to approximately 50% after 48 h. The CL increased gemcitabine cytotoxicity at high concentration (50 mM, p < 0.05) compared to gemcitabine solution. In contrast, treatment with gemcitabine PSL (drug to lipid ratio 4.2%, w/w) caused significant cytotoxicity (p < 0.05) after 24 h exposure, with cell viability reduced to 45 ± 9% at 50 μM, compared to both CL and gemcitabine solution (Fig. 6).

MIA PaCa-2 cell viability after exposure to free gemcitabine in solution and different gemcitabine-loaded liposomes (a: 24 h and b: 48 h). Results are mean ± SD, n = 6 from two independent experiments.
Pharmacokinetics in Rats
The plasma gemcitabine concentration-time profiles after i.v. administration of gemcitabine formulations are shown in Fig. 7 and the pharmacokinetic parameters in Table II. Compared with the gemcitabine solution, both PSL formulations (pre- and post-insertion method) resulted a significant reduction in gemcitabine’s plasma Cl, leading to an approximate 2.5- to 4-fold increase in the AUC0-∞. The elimination T1/2 and MRT of both PSL were also increased by approximately 5-fold and 3-fold, respectively, compared to gemcitabine solution.

Gemcitabine pharmacokinetic profiles in plasma following i.v. injection of gemcitabine solution or gemcitabine-loaded PSL in rats. Data are mean ± SD, n = 4.
Comparison of two PSL formulations also indicated significant differences in gemcitabine’s pharmacokinetics (p < 0.05) with the post-insertion PSL giving a significantly lower Cl and a smaller Vd, resulting in a 1.5-fold higher plasma exposure to gemcitabine. Interestingly, although both PSL formulations showed a different distribution phase in the initial 2 h after injection, they exhibited a similar elimination pattern thereafter with an elimination T1/2 of approximately 6 h.
Discussion
Conventional intravenous chemotherapy with liposomes frequently suffers from fast clearance caused by the RES and the limitation of efficient intra-cytoplasmic delivery (9). The use of PEGylated liposomes as drug carriers offers a potential means of altering a drug’s pharmacokinetics and lessen healthy tissue distribution to reduce toxicity, as has been demonstrated for Doxil® (28). With an improvement in endosome-escape ability by the post-insertion technique, PSL may deliver their cargo more efficiently to the cytoplasm and thus increase a drug’s availability at the intracellular site of action, compared to conventional liposomes (29,30). Therefore, long-circulation and high pH-sensitivity of the liposomes are both critical properties for in vivo systemic application of PSL. In this study, two methods (pre-insertion and post-insertion) were explored for PEGylation. The post-insertion method resulted PSL with a high pH-sensitivity, while maintaining the in vivo long-circulation properties.
Due to the pH-sensitivity of the lipids, it is impossible to load a weak base, such as gemcitabine, into PSL using active loading, which requires a low pH solution to be entrapped in the liposome core to create a trans-membrane pH gradient. Furthermore, our previous study (3) suggested that gemcitabine cannot be actively loaded using an ammonium sulphate gradient due to its low pKa (3.6). A low DL in liposomes may limit its anti-cancer effect, especially for drug resistant cells (31–33). Our modified SVI method increased the DL for gemcitabine to 4.2% w/w (16.8% mol), compared to the 10% mol reported by Bersani et al. (34).
As has been previously reported (15), PEGylation with either DSPE-mPEG2000 or DOPE-mPEG2000 significantly reduced the pH-sensitivity of the liposome in a concentration dependent manner (Fig. 1). Compared to DOPE-mPEG2000, DSPE-mPEG2000 could significantly enhance the pH-sensitivity of PSL (increased by 1–2% calcein release at pH 5.0). Therefore, only DSPE-mPEG2000 was chosen for the post-insertion study. In addition, based on our SVI method for gemcitabine loading, it is difficult to load gemcitabine at 60°C using PSL with less than 3% of DSPE-mPEG because of stability issue (particle size increased significantly to unacceptable level) and the macrophage-like cell uptake study, as a prediction of long-circulation, showed no further improvement in the ‘stealth’ property as DSPE-mPEG2000 increased from 3 to 5%. Therefore, except for investigation of pH sensitivity and uptake study by macrophage-like cells, all the other studies were carried out using PSL with 3% (in molar) DSPE-mPEG2000.
Apart from the shielding effect of the PEG layer to the pH-sensitive membrane to the proton (H+), another possible factor could be the reduction of the liposome’s zeta potential from −37 to −8 mV as a result of PEGylation, which caused a reduction in the attraction force between DOPE and H+ (Fig. 8). However the post-insertion technique produced PSL with more acid sensitivity than the traditional pre-insertion method as demonstrated by both calcein- and gemcitabine release studies. A possible mechanism for this is that the PEG chain, when grafted on both sides of the bilayer by the pre-insertion method, caused an increased viscosity of the inner liposomal liquid, and thus inhibiting the sterical transformation of the DOPE in the membrane in response to the lower pH conditions. With PEG on the external layer, even though the density of PEG chain is low, the concentration of PEG close to the liposome surface is high which may cause increased viscosity at the close-to-surface area and inhibited ion exchange, as well as transformation of DOPE (Fig. 8). Three possible mechanisms of PSL ‘endosome escape’ have been proposed: pH-dependent release, followed by passive diffusion from the endosome; destabilization of endosomal membrane; and membrane fusion of PSL with endosomes (4,29). The first may be the major mechanism, based on the finding that the efficiency of cytoplasmic delivery decreased when the molecular weight of the entrapped compound increased (35). Therefore an increased pH-dependant release is a possible mechanism to enhance intracellular delivery.

Possible mechanism for the enhanced pH-sensitivity of PSL by post-insertion.
In addition, the in vitro release study showed a faster release of gemcitabine from PSL than CL. This could also be partially due to the low transition temperature (Tc) of the main component DOPE in PSL than DPPC in the CL (−15°C versus 41°C). At the temperature of the release media (37°C), the fluidity of the liposome bilayers is higher than that at the Tc which could increase the permeability of the membrane to gemcitabine. This may also contribute to the smaller T1/2 of gemcitabine PSL relative to Doxil (27 h) (28). However, it does not follow that this will lead to a lower antitumor effect as a slow drug release rate at the tumour site may reduce the anti-tumour efficacy (36,37). Only the PSL prepared by post-insertion method showed an enhanced release at pH 6.5 (tumour extra-cellular pH) and this property may facilitate the drug diffusion into deeper area of tissue such as tumours or inflamed and infected areas which exhibit an acidic environment.
To differentiate endosome escape of liposomes after internalization, a novel calcein self-quench assay with confocal laser scanning microscopy was designed without the disturbance of the unreleased calcein after endocytosis. At low concentrations (0–20 μM, data not shown) fluorescence intensity of calcein is proportional to the concentration. However, the fluorescence of calcein is self-quenched at 80 mM. Therefore, the internalized PSL containing 80 mM calcein can only be recognised after endosome escape and dilution with cytoplasm.
Confocal microscopy indicated that the cytoplasmic delivery of PSL decreased after PEGylation, possibly due to a reduced uptake of liposome due to the grafted PEG, or a reduced pH-sensitivity of the liposome. Furthermore, the post-insertion method appeared to have a greater calcein release to the cytoplasm (less bright dots were observed and the florescence was more uniform than pre-insertion formulations), indicating an increased endosome escape. The more effective intracellular drug delivery of PSL was further supported by the greater in vitro cytotoxicity with gemcitabine PSL. Cytotoxicity towards MIA PaCa-2 was ranked in the order, post-inserted PSL ≥ pre-inserted PSL > CL > drug solution, which was consistent with the confocal microscopic observation for endosome escape.
With regard to possible ‘stealth’ properties, both the post- and pre-insertion methods reduced PSL uptake by RAW 264.7 cells by 50% and more (Fig. 3). Although, the pre-inserted PSL showed a slight decrease of the average uptake as DSPE-mPEG2000 increased from 1 to 5%, there was no statistically significant difference among all the PEGylated formulations. Thus, the model did not differentiate the RAW 264.7 cell uptake by pre- and post-insertion method although a slightly lower uptake was observed with post-insertion when 5% DSPE-mPEG2000 was used. In vivo, both PSL formulations resulted in 2.5 (pre-inserted) to 4-fold (post-inserted) increases in gemcitabine AUC and 4.5-fold increase in the elimination T½, compared to gemcitabine solution. PEGylation by post-insertion which increased PEG chain density in the PSL external layers appeared to offer an advantage over pre-insertion as the resulting PSL exhibited a decreased Vd and plasma clearance rate, resulting in an approximate 50% increase in gemcitabine’s plasma AUC. Since the in vivo efficacy of PSL depends strongly on the clearance rate by RES (6), an effective delivery system of PSL is highly dependent on their prolonged circulation time (38,39). A half-life of 6 h is generally considered to be sufficient to exploit better efficacy in vivo (40).
Conclusion
In the present study both PSL formulations demonstrated better intracellular delivery, greater cytotoxicity towards resistant pancreatic cancer cells, and superior in vivo exposure with a longer elimination half-life using gemcitabine as a model drug. The research highlighted that the PSL produced by the post-insertion method had better pH sensitivity, greater endosome escape capacity, and more optimal pharmacokinetics which is the prerequisite for all the in vivo systemic application of PSL.
Abbreviations
- AE:
-
Attachment efficiency
- CL:
-
Classical non-pH-sensitive PEGylated liposomes
- DL:
-
Drug loading
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- EE:
-
Entrapment efficiency
- MTT:
-
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide
- PC:
-
Pancreatic cancer
- PEG:
-
Polyethylene glycol
- PSL:
-
pH-sensitive liposomes
- RES:
-
Reticulo-endothelial system (RES)
- SVI:
-
Small Volume Incubation
- TEM:
-
Cryo-transmission electron microscopy
- TFHE:
-
Thin film hydration extrusion
References
Laouini A, Jaafar-Maalej C, Limayem-Blouza I, Sfar S, Charcosset C, Fessi H. Preparation, characterization and applications of liposomes: state of the Art. J Colloid Sci Biotechnol. 2012;1:147–68.
Hilbig A, Oettle H. Gemcitabine in the treatment of metastatic pancreatic cancer. Expert Rev Anticancer Ther. 2008;8:511–23.
Xu H, Paxton J, Lim J, Li Y, Zhang W, Duxfield L, et al. Development of high-content gemcitabine PEGylated liposomes and their cytotoxicity on drug-resistant pancreatic tumour cells. Pharm Res. 2014;31:2583–92.
Karanth H, Murthy RSR. pH-Sensitive liposomes—principle and application in cancer therapy. J Pharm Pharmacol. 2007;59:469–83.
Fattal E, Couvreur P, Dubernet C. “Smart” delivery of antisense oligonucleotides by anionic pH-sensitive liposomes. Adv Drug Deliv Rev. 2004;56:931–46.
Slepushkin VA, Simões S, Dazini P, Newman MS, Guo LS, Lima MCP, et al. Sterically stabilized pH-sensitive liposomes. J Biol Chem. 1997;272(4):2382–8.
Geisow MJ, Evans WH. pH in the endosome measurements during pinocytosis and receptor-mediated endocytosis. Exp Cell Res. 1984;150:36–46.
Murphy RF, Powers S, Cantor CR. Endosome pH measured in single cells by dual fluorescence flow cytometry : rapid acidification of insulin to pH 6. J Cell Biol. 1984;98:1757–62.
Sr S, Moreira JN, Fonsecab C, Düzgünes N, Lima MCP. On the formulation of pH-sensitive liposomes with long circulation times. Adv Drug Deliv Rev. 2004;56:947–65.
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103:405–18.
Leite EA, Souza CM, Carvalho-Júnior ÁD, Coelho LG, Lana ÂM, Cassali GD, et al. Encapsulation of cisplatin in long-circulating and pH-sensitive liposomes improves its antitumor effect and reduces acute toxicity. Int J Nanomedicine. 2012;7:5259–69.
Torchilin VP, Zhou F, Huang L. pH-sensitive liposomes. J Liposome Res. 1993;3:201–5.
III DEO, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102.
Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–36.
Hong M-S, Lim S-J, Oh Y-K, Kim C-K. pH-sensitive, serum-stable and long-circulating liposomes as a new drug delivery system. J Pharm Pharmacol. 2002;54:51–8.
Remaut K, Lucas B, Braeckmans K, Demeester J, Smedt SCD. Pegylation of liposomes favours the endosomal degradation of the delivered phosphodiester oligonucleotides. J Control Release. 2007;117:256–66.
Iden DL, Allen TM. In vitro and in vivo comparison of immunoliposomes made by conventional coupling techniques with those made by a new post-insertion approach. Biochim Biophys Acta. 2001;1513:207–16.
Li CL, Cui JX, Wang CX, Zhang L, Li YH, Zhang L, et al. Development of pegylated liposomal vinorelbine formulation using“post-insertion” technology. Int J Pharm. 2010;391:230–6.
Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C, Matsumoto S, et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68:157–64.
Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70(9):3606–17.
Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, et al. A Phase I Clinical, Plasma, and Cellular Pharmacology Study of Gemcitabine. J ClinOncol. 1991;9:491–8.
Storniolo AM, Allerheiligen SR, Pearce HL. Preclinical, pharmacologic, and phase I studies of gemcitabine. Semin Oncol. 1997;24:S7-2-S7.
Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233:134–44.
K-i O, Una K, Minatoa K, Tanakab K-I, Higakia K, Kimura T. Determinants for in vivo anti-tumor effects of PEG liposomal doxorubicin: importance of vascular permeability within tumors. Int J Pharm. 2008;359:234–40.
Sudimack JJ, Guo W, Tjarks W, Lee RJ. A novel pH-sensitive liposome formulation containing oleyl alcohol. Biochim Biophys Acta. 2002;1564:31–7.
El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22.
Costa P. An alternative method to the evaluation of similarity factor in dissolution testing. Int J Pharm. 2001;220:77–83.
Working PK, Newmanl MS, Huang SK, Mayhew E, Vaage J, Lasicl DD. Pharmacokiketics, biodistribution and therapeutic efficacy of doxorubicin encapsulated in stealth liposomes (doxil). J Liposome Res. 1994;4(1):667–87.
Simoes S, Slepushkin V, Duzgunes N, Lima MCP. On the mechanisms of internalization and intracellular delivery mediated by pH-sensitive liposomes. Biochim Biophys Acta. 2001;1515:23–37.
Paliwal SR, Paliwal R, Vyas SP. A review of mechanistic insight and application of pH-sensitive liposomes in drug delivery. Drug Deliv. 2014. In press.
Kim I-Y, Kang Y-S, Lee DS, Park H-J, Choi E-K, Oh Y-K, et al. Antitumor activity of EGFR targeted pH-sensitive immunoliposomes encapsulating gemcitabine in A549 xenograft nude mice. J Control Release. 2009;140:55–60.
Graeser R, Bornmann C, Esser N, Ziroli V, Jantscheff P, Unger C, et al. Antimetastatic effects of liposomal gemcitabine and empty liposomes in an orthotopic mouse model of pancreatic cancer. Pancreas. 2009;38:330–7.
Cosco D, Bulotta A, Ventura M, Celia C, Calimeri T, Perri G, et al. In vivo activity of gemcitabine-loaded PEGylated small unilamellar liposomes against pancreatic cancer. Cancer Chemother Pharmacol. 2009;64:1009–20.
Bersani S, Vila-Caballer M, Brazzale C, Barattin M, Salmaso S. pH-sensitive stearoyl-PEG-poly(methacryloyl sulfadimethoxine) decorated liposomes for the delivery of gemcitabine to cancer cells. Eur J Pharm Biopharm. 2014.
Chu C, Dijkstra J, Lai M, Hong K, Szoka FC. Efficiency of cytoplasmic delivery by pH-sensitive liposomes to cells in culture. Pharm Res. 1990;7(8):824–34.
Li C, Cui J, Li Y, Wang C, Li Y, Zhang L, et al. Copper ion-mediated liposomal encapsulation of mitoxantrone: The role of anions in drug loading, retention and release. Eur J Pharm Sci. 2008;34:333–44.
Cui J, Li C, Guo W, Li Y, Wang C, Zhang L, et al. Direct comparison of two pegylated liposomal doxorubicin formulations: is AUC predictive for toxicity and efficacy? J Control Release. 2007;118:204–15.
Maruyama K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv Drug Deliv Rev. 2011;63:161–9.
Yin H, Liao L, Fang J. Enhanced Permeability and Retention (EPR) effect based tumor targeting: the concept, application and prospect. JSM Clin Oncol Res. 2014;2(1):1010.
Fenske DB, Cullis PR. Liposomal nanomedicines. Expert Opin Drug Deliv. 2008;5(1):25–44.
ACKNOWLEDGMENTS AND DISCLOSURE
The financial support for this study was provided by the Marsden Fund by the Royal Society of New Zealand (Grant number UOA1201). Mr Hongtao Xu also wishes to acknowledge the support of a Doctorial Scholarship provided by The University of Auckland, New Zealand. We also would like to thank Mr Alan Gall from VJU for his technical support with the animal studies. The authors declare that they have no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, H., Paxton, J.W. & Wu, Z. Enhanced pH-Responsiveness, Cellular Trafficking, Cytotoxicity and Long-circulation of PEGylated Liposomes with Post-insertion Technique Using Gemcitabine as a Model Drug. Pharm Res 32, 2428–2438 (2015). https://doi.org/10.1007/s11095-015-1635-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1635-0