
Abstract
Background
Heterozygous loss-of-function mutations in the chromodomain helicase DNA-binding protein 7 (CHD7) gene cause CHARGE syndrome characterized by various congenital anomalies. A majority of patients with CHARGE syndrome present with congenital hypogonadotropic hypogonadism (HH), and combined pituitary hormone deficiency (CPHD) can also be present. Whereas CHD7 mutations have been identified in some patients with isolated HH without a diagnosis of CHARGE syndrome, it remains unclear whether CHD7 mutations can be identified in patients with CPHD who do not fulfill the criteria for CHARGE syndrome.
Case presentation
A 33-year-old woman was admitted to our hospital. She had primary amenorrhea and was at Tanner stage 2 for both pubic hair and breast development. She was diagnosed with CPHD (HH, growth hormone deficiency, and central hypothyroidism), and a heterozygous rare missense mutation (c.6745G > A, p.Asp2249Asn) in the CHD7 gene was identified. Our conservation analysis and numerous in silico analyses suggested that this mutation had pathogenic potential. She had mild intellectual disability, a minor feature of CHARGE syndrome, but did not fulfill the criteria for CHARGE syndrome.
Conclusions
We report a rare case of CPHD harboring CHD7 mutation without CHARGE syndrome. This case provides valuable insights into phenotypes caused by CHD7 mutations. CHD7 mutations can have a continuous phenotypic spectrum depending on the severity of hypopituitarism and CHARGE features. Therefore, we would like to propose a novel concept of CHD7-associated syndrome.
Similar content being viewed by others

Background
Heterozygous loss-of-function mutations in the chromodomain helicase DNA-binding protein 7 (CHD7) gene constitute the major pathogenic cause of CHARGE syndrome [1, 2]. CHARGE syndrome is a rare disorder characterized by various congenital anomalies including coloboma of the eye, heart defects, choanal atresia, retardation of growth and development, genital hypoplasia, and ear abnormalities [3, 4]. Hypothalamic-pituitary dysfunction is common in CHARGE syndrome and approximately 60–80% of individuals with CHARGE syndrome present with congenital hypogonadotropic hypogonadism (HH) [5,6,7,8,9,10,11]. Combined pituitary hormone deficiency (CPHD) can also be present. Growth hormone (GH) and thyroid-stimulating hormone (TSH) deficiencies occur at rates of 9–34% [5,6,7,8] and 8–18% [6,7,8,9], respectively, and sporadic cases of adrenocorticotropic hormone (ACTH) deficiency have been reported [12, 13]. Rarely do structural pituitary abnormalities such as anterior pituitary hypoplasia occur in combination [13]. CHD7 mutations have also been identified in 5–19% of patients with isolated HH who were not diagnosed with CHARGE syndrome [11, 14,15,16,17,18,19,20]. However, it remains unclear whether CHD7 mutations can be detected in patients with CPHD who do not fulfill the criteria for CHARGE syndrome. Herein, we present a rare case of patient with CPHD, who despite not fulfilling the diagnostic criteria for CHARGE syndrome, presented with minor characteristics and a CHD7 missense mutation with pathogenic potential.
Case presentation
The patient provided written informed consent for the publication of this case report. A 33-year-old woman was admitted to our hospital for treatment of diabetes mellitus. She had been born full-term, with a weight of approximately 2800 g, with no perinatal or infantile developmental abnormalities although detailed information was unavailable. Around the age of 7 years, she had begun to experience a decline in academic performance and weight gain. She had been the first or second shortest in her class until approximately 15 years, after which her height begun to increase. At the age of 15 years, she visited a hospital with a chief complaint of amenorrhea. Further examination was not performed because her symptom had been attributed to obesity; however, she did not menstruate thereafter. At the age of 16 years, she got a job, but had to change employment frequently because she could not work for long periods at one job. At the age 33 years, she was incidentally diagnosed with diabetes at another clinic and referred to our hospital.
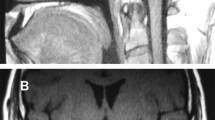
On admission, the patient’s height, weight, body mass index, waist circumference, and arm span were 161.1 cm, 97.1 kg, 37.4 kg/m2, 111.2 cm, and 160.0 cm, respectively. Her visceral fat area estimated via bioelectrical impedance analysis using EW-FA90 (Panasonic Corporation, Osaka, Japan) was 166 cm2. She had no history of head trauma or head surgery, and neither drank alcohol nor took any medications. Although her maternal grandmother had obesity and diabetes, she had no other family members with obesity, diabetes, or endocrinological disorders, and there were no consanguineous marriages in her family. She had primary amenorrhea and was at Tanner stage 2 for both pubic hair and breast development. Her intelligence quotient, as determined by the Wechsler Adult Intelligence Scale, fourth edition, was 60, indicating mild intellectual disability. Her laboratory findings are presented in Table 1. Her hemoglobin A1c level was 9.7%. She had thrombocytopenia, prolonged prothrombin time, and elevated liver fibrosis marker levels, suggestive of liver cirrhosis. Viral hepatitis, autoimmune hepatitis, primary biliary cholangitis, Wilson’s disease, and alcoholic hepatitis were unlikely differential diagnoses, suggesting that her liver dysfunction was caused by nonalcoholic steatohepatitis (NASH). Her endocrinological findings are presented in Tables 1 and 2. HH and growth hormone deficiency (GHD) were confirmed by comparing baseline hormonal levels and the results of stimulation tests. Central hypothyroidism (CH) was diagnosed due to low free thyroxine level and normal or mildly elevated TSH levels at the baseline, although the TSH level responded normally to thyrotropin-releasing hormone (TRH) stimulation. Plasma ACTH levels showed normal responses in both the insulin tolerance test (ITT) and corticotropin-releasing hormone (CRH) stimulation test. Cortisol response was almost normal in the rapid ACTH stimulation test but blunted in the ITT and CRH stimulation test. Based on these results and the absence of clinical signs of adrenal insufficiency, central adrenal insufficiency was not apparent. Her prolactin level responded normally to TRH stimulation. Thus, she was diagnosed with CPHD (combined HH, GHD, and CH). Brain magnetic resonance imaging (MRI) revealed anterior pituitary hypoplasia (Fig. 1). There were no other abnormal intracranial findings, including those of the posterior pituitary and pituitary stalk. Abdominal MRI showed morphological features of cirrhosis (Fig. 2a) and esophagogastroduodenoscopy (EGD) showed esophageal varices (Fig. 2b). Liver biopsy revealed severe fibrosis (Fig. 2c). Although there was no histological evidence of NASH, such as severe steatosis or ballooning, the clinical course and laboratory findings shown in Table 1 suggested that the liver cirrhosis was caused by burnt-out NASH. Based on these results, we diagnosed her with CPHD (combined HH, GHD, and CH) with comorbid diabetes, severe obesity, and liver cirrhosis probably due to NASH. She started treatment with recombinant GH, levothyroxine sodium, and estrogen/progesterone therapy. Her glycemic control was remarkably improved by treatment with linagliptin and empagliflozin.

Gadolinium-enhanced brain magnetic resonance imaging showing anterior pituitary hypoplasia (circle)

a: Abdominal magnetic resonance imaging showing morphological features characteristic of cirrhosis such as irregularity of the liver surface, enlargement of the left lobe, and splenomegaly (arrowhead). b: Esophagogastroduodenoscopy showing esophagus varices (arrowhead). c: Histological analysis of the liver with Masson’s trichrome staining showing severe fibrosis (arrow) and mild steatosis (arrowhead)
Her lack of secondary sexual characteristics made us suspect that genetic abnormalities may have contributed to her pituitary hormone deficiency. We analyzed the coding and splicing regions of 17 major genes associated with congenital hypopituitarism (HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, SOX2, SOX3, CHD7, FGF8, FGFR1, GLI2, IGSF1, KISS1R, SOX10, and WDR11) using the NextSeq Sequencing System (Illumina, San Diego, CA, USA) at the Kazusa DNA Research Institute (Kisarazu, Japan) [21]. As a result, a heterozygous missense mutation (c.6745G > A, p.Asp2249Asn) in the CHD7 gene was identified. This mutation had not been reported in ClinVar and the CHD7 database (www.chd7.org), and the minor allele frequency was < 0.1% in several population databases (0.0011% in Genome Aggregation Database v2.1.1, 0.0015% in Trans-Omics for Precision Medicine, 0% in the Human Genetic Variation Database, and 0.099% in the Japanese Multi Omics Reference Panel [14KJPN]). The nucleotide position showed a phyloP conservation score [22] of 7.526 for 100 vertebrates, a PhastCons score [22] of 1.00 for 100 vertebrates, and a Genomic Evolutionary Rate Profiling score [23] of 5.3 for mammalian alignment, indicating that this mutation occurred in a highly conserved nucleotide. Numerous in silico prediction tools such as Polymorphism Phenotyping v2 [24], MutationTaster [25], Functional Analysis through Hidden Markov Models [26], Mendelian Clinically Applicable Pathogenicity [27], and Combined Annotation-Dependent Depletion [28] (CADD score, 25.4) suggested that this mutation causes changes in protein function, although the mutation was considered to have uncertain significance according to the American College of Medical Genetics and Genomics (ACMG) guidelines [29]. Taken together, the CHD7 missense mutation in this patient was suggested to be pathogenic and contributory to the CPHD.
The patient underwent ophthalmologic and otorhinolaryngologic examinations, brain MRI, echocardiography, abdominal MRI, and EGD for the evaluation of CHARGE features. Apart from CPHD and intellectual disability, there was no evidence of CHARGE or CHARGE-like features such as coloboma of the eyes, atresia of choanae, anomalies of the semicircular canals, external or middle ear anomalies, hearing loss, hyposmia, olfactory bulb hypoplasia, cranial nerve dysfunctions, cleft lip/palate, anomalies of the mediastinal viscera, and renal anomalies.
Discussion and conclusions
Herein, we present the clinical history of a 33-year-old woman with CPHD (HH, GHD, and CH) alongside comorbid diabetes, severe obesity, and liver cirrhosis probably due to NASH. Despite not fulfilling the criteria for CHARGE syndrome, the patient had intellectual disability, one of its minor features, and a CHD7 missense mutation with pathogenic potential.
Whether the CHD7 mutations can be detected in patients with CPHD who do not satisfy the criteria for CHARGE syndrome remains to be elucidated. Recently, a synonymous CHD7 mutation predicted to affect splicing had been identified in one of 80 patients with CPHD who underwent a comprehensive genetic examination [30]. However, this report does not include a detailed description of CHARGE features. Several studies have highlighted the importance of careful evaluation of CHARGE features in patients with CHD7 mutations, as a subset of patients with apparently isolated HH harboring CHD7 mutations were subsequently found to have multiple CHARGE features and were reclassified as having CHARGE syndrome [31,32,33]. Our patient is a rare case of CPHD harboring a CHD7 mutation that, after detailed examinations, does not certainly fulfill the criteria for CHARGE syndrome.
In patients with isolated HH harboring CHD7 mutations without CHARGE syndrome, some patients have minor CHARGE features, such as hearing loss or intellectual disability [11, 15,16,17,18,19,20]. In addition, CHD7 mutations in CHARGE syndrome are typically truncating and can lead to serious genetic dysfunction, whereas mutations in isolated HH with or without minor CHARGE features are predominantly of the missense type that has less impact on gene function compared with truncating mutation [15, 16]. Furthermore, following the classification of missense mutations in patients with isolated HH based on ACMG guidelines, minor CHARGE features have been reported to be more commonly present in patients with pathogenic or likely pathogenic mutations than in those with uncertain significance mutations [20]. These pieces of evidence suggest that isolated HH harboring CHD7 mutations is a mild form of CHARGE syndrome. Given that our patient had intellectual disability, which is a minor feature of CHARGE syndrome, and that the CHD7 mutation was of the missense type, our patient with CPHD may also be considered as having a mild form of CHARGE syndrome. Moreover, based on the above findings, including those of our patient, we speculate that CHD7 mutations may have a continuous phenotypic spectrum depending on the severity of hypopituitarism and CHARGE features, and propose a novel concept of CHD7-associated syndrome, including CHARGE syndrome (Fig. 3).

The concept of CHD7-associated syndrome. CHD7 mutations can have a continuous phenotypic spectrum depending on the severity of hypopituitarism and CHARGE features. HH, congenital hypogonadotropic hypogonadism; CHD7, chromodomain-helicase-DNA-binding protein 7; CPHD, combined pituitary hormone deficiency
Previous studies have indicated that HH caused by CHD7 mutations is due to hypothalamic gonadotropin-releasing hormone (GnRH) deficiency resulting from the impaired migration of GnRH-synthesizing neurons that migrate alongside the olfactory fibers during embryonic development [11, 34]. This postulated etiology is consistent with the observations that most individuals with CHARGE syndrome have olfactory dysfunction and do not have abnormalities in other anterior pituitary hormone levels or pituitary morphology. However, a subset of individuals with CHARGE syndrome can have CPHD [5,6,7,8,9, 12, 13], and some of these patients can demonstrate structural pituitary abnormalities such as anterior pituitary hypoplasia [13], similar to that observed in our patient. CHD7 is expressed in the olfactory epithelium associated with the migration of GnRH neurons as well as the developing anterior pituitary and hypothalamus in both mice and humans [35, 36]. CHD7 has also been identified as a SOX2 transcriptional cofactor [37]. In addition, recent studies have shown that CHD7-deficient mice have hypoplastic Rathke’s pouches [38] and reduced OTX2 mRNA expression [39]. Both SOX2 and OTX2 are associated with pituitary development and differentiation [40, 41]. These pieces of evidence suggest that CHD7 potentially plays a role in the development and function of the hypothalamic-pituitary axis and that CHD7 mutations can result in variable degrees of hypopituitarism, as well as HH. Further studies are needed to elucidate more detailed mechanisms underlying the relationship between CHD7 and the hypothalamic-pituitary axis.
Our patient had experienced short stature until approximately 15 years of age, after which she had grown taller and had attained normal height despite GHD. Growth without GH has been reported in rare cases of hypopituitarism untreated until adulthood [42,43,44]. Hyperinsulinemia, hyperleptinemia, or delayed epiphyseal maturation caused by concomitant HH have been postulated as possible underlying mechanisms [44, 45]. Several factors such as endocrine, paracrine, intracellular, or extracellular matrix factors may also contribute to normal growth in GHD [46]. In fact, previous evidence has shown that the GH–insulin-like growth factor-1 (IGF-1) axis is just one of many regulatory systems that control height [46].
In our patient, long-term untreated GHD and CH may have contributed to the development of diabetes, severe obesity, and liver cirrhosis probably due to NASH [47]. In particular, GHD can induce NASH, and liver fibrosis is associated with a lower IGF-1 level even in adolescents and young adults with GHD [48]. GH replacement therapy improves liver dysfunction and histological hepatic characteristics, including steatosis and fibrosis, as well as metabolic abnormalities [49, 50]. However, it is unclear to what extent GH replacement therapy would improve the liver function of our patient because of the severe advanced cirrhosis. Therefore, we will carefully monitor the effect of the treatment on her liver function.
The present case report had several limitations. First, we could not perform genetic analyses on the proband’s parents. Most CHD7 truncating mutations in typical CHARGE syndrome are heterozygous de novo, whereas CHD7 missense mutations in isolated HH are often inherited from unaffected parents, suggesting incomplete penetrance of the phenotype of pituitary hormone deficiencies in individuals harboring CHD7 missense mutations [15,16,17,18]. Thus, the CHD7 mutation observed in our patient may be inherited from either unaffected parent. Second, a limited number of genes associated with pituitary hormone deficiencies were analyzed. Although various genes associated with congenital pituitary hormone deficiencies have been identified, a majority of the related genes remain unknown [51]. In addition, oligogenic inheritance has been observed in a number of cases of isolated HH [16,17,18, 52]. Therefore, some unknown gene mutations in addition to the observed CHD7 mutation may contribute to the phenotype of our patient in an additive or synergistic manner, and the oligogenicity may explain the incomplete penetrance or phenotypic variations in patients harboring CHD7 mutations. Third, the observed CHD7 mutation was not confirmed by Sanger sequencing. However, the next-generation sequencing workflow we employed has been shown to have high specificity and can omit the need for confirmatory assessment by Sanger sequencing for variants with high-quality scores [21]. Finally, as is the case with most previously reported cases of CHD7 missense mutations, we were unable to confirm the functional abnormality of the observed CHD7 mutation via in vitro or in vivo experiments. Despite these limitations, the findings that our patient’s CHD7 mutation was an ultra-rare variant (< 0.1%), occurred at a highly conserved nucleotide, and was predicted to be damaging by numerous major bioinformatic tools, strongly suggest that this missense mutation was associated with her phenotype.
In conclusion, we report a rare case of CPHD with a minor feature harboring a CHD7 mutation. This case does not only imply that CHD7 plays a potential role in the development and function of the hypothalamic-pituitary axis but provides valuable insights into phenotypes caused by CHD7 mutations. CHD7 mutations can have a continuous phenotypic spectrum depending on the severity of hypopituitarism and CHARGE features. Therefore, we would like to propose a novel concept of CHD7-associated syndrome.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the DDBJ repository, accession number DRA014912.
Abbreviations
- CHD7:
-
Chromodomain helicase DNA-binding protein 7
- HH:
-
Hypogonadotropic hypogonadism
- CPHD:
-
Combined pituitary hormone deficiency
- GH:
-
Growth hormone
- TSH:
-
Thyroid-stimulating hormone
- ACTH:
-
Adrenocorticotropic hormone
- NASH:
-
Nonalcoholic steatohepatitis
- GHD:
-
Growth hormone deficiency
- CH:
-
Central hypothyroidism
- TRH:
-
Thyrotropin-releasing hormone
- ITT:
-
Insulin tolerance test
- CRH:
-
Corticotropin-releasing hormone
- MRI:
-
Magnetic resonance imaging
- EGD:
-
Esophagogastroduodenoscopy
- ACMG:
-
American college of medical genetics and genomics
- GnRH:
-
Gonadotropin-releasing hormone
- IGF-1:
-
Insulin-like growth factor-1
References
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–7.
Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33:1149–60.
Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A:306–8.
Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170A:344–54.
Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, et al. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005;90:5621–6.
Asakura Y, Toyota Y, Muroya K, Kurosawa K, Fujita K, Aida N, et al. Endocrine and radiological studies in patients with molecularly confirmed CHARGE syndrome. J Clin Endocrinol Metab. 2008;93:920–4.
Shoji Y, Ida S, Etani Y, Yamada H, Kayatani F, Suzuki Y, et al. Endocrinological Characteristics of 25 Japanese Patients with CHARGE Syndrome. Clin Pediatr Endocrinol. 2014;23:45–51.
Legendre M, Abadie V, Attié-Bitach T, Philip N, Busa T, Bonneau D, et al. Phenotype and genotype analysis of a French cohort of 119 patients with CHARGE syndrome. Am J Med Genet C Semin Med Genet. 2017;175:417–30.
Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148:410–4.
Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011;48:334–42.
Balasubramanian R, Crowley WF Jr. Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am J Med Genet C Semin Med Genet. 2017;175:507–15.
James PA, Aftimos S, Hofman P. CHARGE association and secondary hypoadrenalism. Am J Med Genet A. 2003;117A:177–80.
Gregory LC, Gevers EF, Baker J, Kasia T, Chong K, Josifova DJ, et al. Structural pituitary abnormalities associated with CHARGE syndrome. J Clin Endocrinol Metab. 2013;98:E737–43.
Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–9.
Marcos S, Sarfati J, Leroy C, Fouveaut C, Parent P, Metz C, et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab. 2014;99:E2138–43.
Balasubramanian R, Choi JH, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, et al. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc Natl Acad Sci U S A. 2014;111:17953–8.
Stamou MI, Cox KH, Crowley WF Jr. Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the “-omics” era. Endocr Rev. 2015;36:603–21.
Gonçalves CI, Patriarca FM, Aragüés JM, Carvalho D, Fonseca F, Martins S, et al. High frequency of CHD7 mutations in congenital hypogonadotropic hypogonadism. Sci Rep. 2019;9:1597.
Li JD, Wu J, Zhao Y, Wang X, Jiang F, Hou Q, et al. Phenotypic Spectrum of Idiopathic Hypogonadotropic Hypogonadism Patients With CHD7 Variants From a Large Chinese Cohort. J Clin Endocrinol Metab. 2020;105:dgz182.
Sun B, Wang X, Mao J, Zhao Z, Zhang W, Nie M, et al. Classification of CHD7 Rare Variants in Chinese Congenital Hypogonadotropic Hypogonadism Patients and Analysis of Their Clinical Characteristics. Front Genet. 2021;12: 770680.
Fujiki R, Ikeda M, Yoshida A, Akiko M, Yao Y, Nishimura M, et al. Assessing the Accuracy of Variant Detection in Cost-Effective Gene Panel Testing by Next-Generation Sequencing. J Mol Diagn. 2018;20:572–82.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–21.
Cooper GM, Stone EA, Asimenos G; NISC Comparative Sequencing Program, Green ED, Batzoglou S, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Budny B, Zemojtel T, Kaluzna M, Gut P, Niedziela M, Obara-Moszynska M, et al. SEMA3A and IGSF10 are novel contributors to combined pituitary hormone deficiency (CPHD). Front Endocrinol (Lausanne). 2020;11:368.
Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71.
Bergman JE, de Ronde W, Jongmans MC, Wolffenbuttel BH, Drop SL, Hermus A, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. 2012;97:E858–62.
Xu C, Cassatella D, van der Sloot AM, Quinton R, Hauschild M, De Geyter C, et al. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet Med. 2018;20:872–81.
Teixeira L, Guimiot F, Dodé C, Fallet-Bianco C, Millar RP, Delezoide AL, et al. Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J Clin Invest. 2010;120:3668–72.
Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide AL, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43:211–7.
Bergman JE, Bosman EA, van Ravenswaaij-Arts CM, Steel KP. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur J Hum Genet. 2010;18:171–7.
Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet. 2011;43:607–11.
Hurd EA, Capers PL, Blauwkamp MN, Adams ME, Raphael Y, Poucher HK, et al. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18:94–104.
Layman WS, Hurd EA, Martin DM. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum Mol Genet. 2011;20:3138–50.
Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, et al. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab. 2010;95:756–64.
Jayakody SA, Andoniadou CL, Gaston-Massuet C, Signore M, Cariboni A, Bouloux PM, et al. SOX2 regulates the hypothalamic-pituitary axis at multiple levels. J Clin Invest. 2012;122:3635–46.
Papastathopoulou L, Tzanela M, Vlassopoulou V, Vassiliadi D, Thalassinos N. Untreated hypopituitarism due to absence of the pituitary stalk with normal adult height: report of two cases. Endocrine. 2006;29:175–9.
Ioachimescu AG, Hamrahian AH, Stevens M, Zimmerman RS. The pituitary stalk transection syndrome: multifaceted presentation in adulthood. Pituitary. 2012;15:405–11.
Wada S, Minagawa A, Imamaki K, Suda S, Yamanaka K, Iitaka M, et al. A patient of hypogonadotropic hypogonadism accompanied by growth hormone deficiency and decreased bone mineral density who attained normal growth. Intern Med. 2000;39:641–5.
Phillip M, Moran O, Lazar L. Growth without growth hormone. J Pediatr Endocrinol Metab. 2002;15(Suppl 5):1267–72.
Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015;11:735–46.
Gariani K, Jornayvaz FR. Pathophysiology of NASH in endocrine diseases. Endocr Connect. 2021;10:R52-65.
Kang SJ, Kwon A, Jung MK, Chae HW, Kim S, Koh H, et al. High Prevalence of Nonalcoholic Fatty Liver Disease Among Adolescents and Young Adults With Hypopituitarism due to Growth Hormone Deficiency. Endocr Pract. 2021;27:1149–55.
Nishizawa H, Iguchi G, Murawaki A, Fukuoka H, Hayashi Y, Kaji H, et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur J Endocrinol. 2012;167:67–74.
Matsumoto R, Fukuoka H, Iguchi G, Nishizawa H, Bando H, Suda K, et al. Long-term effects of growth hormone replacement therapy on liver function in adult patients with growth hormone deficiency. Growth Horm IGF Res. 2014;24:174–9.
Bosch I, Ara L, Katugampola H, Dattani MT. Congenital Hypopituitarism During the Neonatal Period: Epidemiology, Pathogenesis, Therapeutic Options, and Outcome. Front Pediatr. 2021;8: 600962.
Cangiano B, Swee DS, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. 2021;140:77–111.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YO made substantial contributions to the conception or design of the work, the acquisition, analysis and interpretation of data for the work, and the drafting of the work. KT HN, AT, IM, YU, and YM made substantial contributions to the acquisition and analysis of data for the work, and the revising of the work. HS, MK, and TY made substantial contributions to analysis and interpretation of data for the work, and the revising of the work. All authors approved the manuscript to be published, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient provided written informed consent for the publication of this case report.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Obata, Y., Takayama, K., Nishikubo, H. et al. Combined pituitary hormone deficiency harboring CHD7 gene missense mutation without CHARGE syndrome: a case report. BMC Endocr Disord 23, 118 (2023). https://doi.org/10.1186/s12902-023-01373-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-023-01373-8