
Abstract
The pituitary stalk transection syndrome was characterized after introducing the MRI scan in the evaluation of children with hypopituitarism. Its prevalence and natural history into adulthood have not been clearly established. We present 4 cases of stalk transection syndrome diagnosed by the adult endocrinologist that reflect its pleiotropic manifestations. In all cases, MRI showed pathognomonic findings with small anterior pituitary, diminutive or absent infundibulum and ectopic posterior pituitary at the median eminence. Clinical presentation occurred in childhood or the second decade of life. The hormonal deficits were variable in severity and onset, with adrenal insufficiency diagnosed in the second and forth decade in three patients, and absent in another. Growth hormone deficiency was diagnosed before age 10 in three cases and at age 20 in one case with normal spontaneous linear growth. Hypothyroidism had onset in the first or second decade of life and hypogonadism was diagnosed during work-up for lack of pubertal development in all cases. The pituitary stalk transection syndrome should be considered in patients who were previously thought to have idiopathic GH deficiency or multiple pituitary hormone deficiencies. Presence of MRI characteristics compatible with the pituitary stalk transection syndrome should prompt a full pituitary hormonal evaluation. Long-term follow-up by the adult endocrinologist is warranted as new hormone deficiencies might appear later in life.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pituitary gland integrates stimulatory and inhibitory central and peripheral signals to secrete hormones by five highly differentiated cell types: somatotroph, gonadotrophs, thyrotrophs, lactotrophs and cortricophtrophs. Essential for pituitary function are the hypothalamic releasing and inhibiting hormones transported via the hypophyseal portal system. The pituitary gland cells harbor specific G-protein-coupled receptors for the hypothalamic regulating factors [1]. Impaired transport or activity of these peptides can cause pituitary gland hypoplasia which can affect the entire pituitary gland or can be selective [2]. The stalk transection (or dysgenesis) syndrome is an example of functional disruption of the hypothalamic-anterior pituitary unit with significant endocrine consequences usually observed early in life.
The pituitary stalk transection was described after introduction of MRI with the following characteristics: small anterior pituitary gland, thin or absent infundibulum after gadolinium administration and ectopic posterior pituitary location [3]. Exact prevalence is unknown, but a recent study in 231 adult patients with hypopituitarism showed that 26 (11.2%) had such MRI characteristics [4]. Another study in 35 patients with previously diagnosed “idiopathic” hypopituitarism, showed MRI diagnosis of stalk transection syndrome in 15 (43%) [5]. Such prevalence seems high based on authors’ experience in treating adults with hypopituitarism.
Patients with pituitary stalk transection usually present in the first decade of life with growth retardation due to GH deficiency; this can be isolated or associated with other pituitary hormone deficiencies with onset in childhood [6]. Possible causative factors include defective migration of the pituitary gland during intrauterine life or trauma-related ischemia with subsequent reorganization of infundibular axons and development of an ectopic posterior pituitary. The natural history of the disease into adulthood has not been established as most previous studies were cross-sectional [3, 7, 8].
Here we present 4 cases of stalk transection syndrome diagnosed by the adult endocrinologist that illustrate an evolving spectrum of manifestations into adulthood.
Case reports
Case 1
A 20 year old woman was referred for evaluation of possible growth hormone (GH) deficiency.
The patient was the result of a full term pregnancy with breech delivery. She was a single child of healthy parents with heights of 162.6 cm (mother) and 182.9 cm (father; mid-parental target height: 162–180 cm). At birth, the patient’s weight was 4,280 grams and length was 53.3 cm. At 2 months of age, she suffered a severe head trauma during a motor vehicle accident with two skull fractures. Due to cognitive delays, she attended special classes in school. Her growth velocity in childhood was appropriate. At age 14, she was seen by a Pediatric Endocrinologist for primary amenorrhea and absence of secondary sex characteristics. She denied polyuria, polydipsia or anosmia. At that time, her height was 158.8 cm (35th percentile), weight 60.9 kg (75th percentile), and the sexual secondary characteristics were absent (Tanner stage I). Bone age was 11.0 years, i.e., less than 2 standard deviation below chronological age. Laboratory work showed: prolactin of 19.27 ng/ml (normal: 1.4–24), LH < 0.1 UI/l (normal: 1–12), FSH 0.7 IU/l (normal: 2–11), TSH of 9.86 mUI/L (normal: 0.5–5.5) and low free T4 of 0.4 ng/dl (normal: 0.7–1.8). Pelvic ultrasound showed an infantile uterus. She was started on levothyroxine and estrogen replacement. At age 16, while taking antibiotics for pharyngitis, she developed emesis and diarrhea, requiring hospital admission. A morning cortisol level was 3 μg/dl (normal: 5–25 μg/dl) and she was started on hydrocortisone replacement. She started to have menstrual cycles at age 19 while taking oral contraceptives. She grew 3.8 cm after age 14.
Upon presentation to the adult Endocrinologist at age 20, her complaints were fatigue and inability to concentrate. She was taking levothyroxine 200 mcg/day, hydrocortisone 5 mg daily and ethinyl estradiol/ethynodiol diacetate 1 mg/35 μg. Her height was 162.6 cm, weight 75.0 kg and she appeared young for her age. Otherwise, exam was unremarkable. Laboratory studies showed slightly elevated prolactin of 26.7 ng/ml (normal: 2–17.4), low IGF-1 of 15 ng/ml (normal: 182–780), TSH < 0.005 mIU/l, free T4 of 1.7 ng/dl (normal: 0.7–1.8), and free T3 of 3.9 pg/ml (normal: 1.5–3.5). The MRI of the pituitary gland showed absence of the infundibulum before and after contrast administration, small anterior pituitary, and ectopic posterior pituitary located at the infundibular recess of the third ventricle.
The dose of hydrocortisone was increased to 10 mg in the morning and 2.5 mg in the afternoon (approximately 7 mg/m2/day), and the dose of levothyroxine was decreased to 175 μg/day. The patient also started on growth hormone replacement, which she subsequently stopped due to the inconvenience of daily injections.
Case 2
A 19 year-old male college student presented for transition from Pediatric to the Adult Endocrinologist.
The patient was a result of an uncomplicated full-term pregnancy with normal vaginal delivery. He was the only child of healthy parents with heights of 165.5 cm (mother) and 183 cm (father). The perinatal course was unremarkable. At birth, weight was 4,507 g and length 54.6 cm. He was diagnosed with short stature secondary to GHD at age 4 and took growth hormone replacement until age 18 with good response of linear growth. At age 7, he was diagnosed with hypothyroidism and started on thyroid hormone replacement. He did not undergo pubertal changes until testosterone initiated at age 15 by the Pediatric Endocrinologist. He denied polydipsia, polyuria, galactorrhea, gynecomastia or anosmia. Past surgical history was remarkable for upper respiratory tract surgery at age 2 and orchidopexy at 11, both uncomplicated.
When he presented at the adult Endocrinologist at age 19, he had no specific complaints. He was taking levothyroxine 150 μg daily and testosterone cypionate 100 mg intramuscularly monthly. Height was 188 cm, and weight 88.2 kg. Physical exam was remarkable for small testes (3 cm3) and lack of facial hair. The pubic hair distribution and penile size were normal. The hormonal evaluation showed: low IGF-1 of 42 ng/ml (normal: 182–780), slightly elevated prolactin 22.7 ng/ml (normal: 2.0–14.0), TSH 0.032 mU/l (normal: 0.5–5.5), total T4 of 7.9 μg/dl (5.0–11.0) and free thyroxine index of 7.1 μg/dl (normal: 6.0–11.0). ACTH (Cosyntropin) stimulation test (250 μg) showed low baseline serum cortisol of 1.6 μg/dl that increased only to 4.4 and 5.3 μg/dl at 30 and 60 min. The MRI of the pituitary gland showed absence of the infundibulum before and after contrast administration, a small amount of enhancing soft tissue in the pituitary fossa, and ectopic posterior pituitary situated posterior to the optic chiasm.
The patient was started on hydrocortisone 10 mg in am and 5 mg in pm. The thyroid and testosterone supplementation were continued. He was not interested to resume GH replacement due to lack of symptoms.
Case 3
A 36 year old woman presented for fatigue and weight gain with abdominal fat deposition despite regular exercise.
The patient was born at 39 weeks of gestation after a normal pregnancy. Both parents had heights of 183 cm and patient’s sisters had heights of 170 and 180 cm, respectively. She was delivered with difficulties due to breech presentation, and needed resuscitation and neonatal ICU care for few days. She stopped growing at age 5. She had an abnormal exercise GH stimulation test at age 6 and abnormal arginine and insulin tolerance tests at age 8. Peak GH during insulin tolerance test was <1 ng/ml with normal cortisol of 18 μg/dl and blood glucose of 28 mg/dl. At age 8, thyroid tests showed normal TSH of 3.8 mIU/l and total T4 of 5.3 μg/dl (normal: 5.0–11.0). She took growth hormone replacement off and on between ages 8 and 18. She was diagnosed with hypothyroidism in her 20’s, and required progressively higher doses of levothyroxine over the years. She did not develop secondary sexual characteristics and did not have spontaneous menstrual cycles until starting oral contraceptives in her early 20’s. Her karyotype was 46,XX. She underwent in vitro fertilization at age 34, which did not result in pregnancy despite symptoms of overstimulation. At age 36, she was told that she had polyglandular autoimmune deficiency and was started on dexamethasone 0.5 mg at bedtime. The patient denied galactorrhea, anosmia and polyuria.
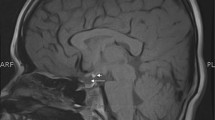
When she presented to our office, she complained of fatigue, weight gain and abdominal fat deposition despite regular exercise. She was taking dexamethasone 0.5 mg at bedtime, and levothyroxine 88 mcg daily. Height was 160 cm, and weight 67.5 kg. The exam was only remarkable for absence of axillary and pubic hair without features of hypercortisolism. Laboratory tests showed prolactin 15.6 ng/ml (normal: 2–17.4), IGF-1 < 15 ng/ml (normal: 114–492), ACTH of 8 pg/ml (normal: 5–50) and low a.m. cortisol of 1.5 μg/dl (after stopping the dexamethasone for 3 days). The thyroid panel while taking levothyroxine was normal, with TSH 1.46 mUI/l, free T4 of 1.1 μg/dl (0.7–1.8), free T3 of 2.4 ng/dl (1.8–4.6) and undetectable microsomal antibodies. The ACTH stimulation test (250 μg) showed baseline cortisol of 1.6, which only increased to 2.6 and 4.5 μg/dl at 30 and 60 min. MRI of the pituitary gland was performed and showed a small sella with a thin rim of anterior pituitary along the margins of the sella with uniform enhancement following gadolinium administration (Fig. 1b). The neurohypophysis was ectopic along the floor of the hypothalamus in the region of the median eminence. The infundibulum was very small in caliber with minimal enhancement following gadolinium administration.

a Coronal pituitary MRI before and after contrast administration. Arrows represent ectopic pituitary at the floor of the third ventricle. The figure also illustrates the small anterior pituitary gland and absent infundibulum (case 4). b Sagital pituitary MRI before and after contrast administration (case 3)
The dexamethasone was replaced with hydrocortisone 10 mg in the morning and 5 mg in the afternoon (approx. 9 mg/m2/day). She was continued on the thyroid and estrogen/progesterone supplementation. She was also started on GH 0.2 mg subcutaneously daily which was gradually increased to 0.8 mg/day until IGF-1 normalized. Her fatigue, weight and abdominal girth improved on this regimen.
Case 4
A 21 year old college student presented with fatigue, muscle pain and decreased exercise tolerance.
The patient is a single child of healthy parents with heights of 178 and 165 cm. She was delivered after 40 weeks of normal gestation. Weight and length were normal at 3,700 g and 53.3 cm, respectively. Parents noticed she was the shortest among her peers at age 4. She did not have polyuria, polydipsia or anosmia. At age 5, IGF-1 was very low of 6 ng/ml, along with low total T4 of 4.4 mcg/dl and normal TSH of 1.45 mIU/l, and prolactin of 16.9 ng/ml. Clonidine stimulation test showed a peak GH of only 0.6 ng/ml. Her karyotype was 46,XX. She was started on growth hormone replacement and levothyroxine at age 5. She was not compliant with injection administration and discontinued them at age 15. She stopped the levothyroxine at age 19 after leaving her Pediatric Endocrinologist. She did not develop secondary sexual characteristics or spontaneous menstrual cycles. She started oral contraceptives at age 16, but stopped them after few months due to poor compliance.
When she presented to the adult endocrinology office, she complained of fatigue, muscle pain, small joint pain and decreased exercise tolerance. She was not taking any medications. Height was 138.4 cm, and weight 54.6 kg. The exam was remarkable for short stature, small hands and feet, increased adiposity in the abdominal region, breast Tanner stage III breasts and absence of axillary and pubic hair without other endocrine stigmata. She did not have a goiter or bradycardia. Laboratory tests showed prolactin of 8 ng/ml (normal: 1–24), undetectable FSH, LH, estradiol, undetectable IGF-1 (normal: 128–488), and GH of 0.02 ng/ml (nl: 0.03–10), ACTH 20 pg/ml (normal: 5–27) and a.m. cortisol 9.8 μg/dl. The thyroid panel showed TSH of 2.01 mUI/l, undetectable free T4 (normal: 0.5–1.6 ng/dl), and low free T3 of 2.2 ng/dl (2.4–4.2). The ACTH stimulation test (250 μg) showed baseline cortisol of 7.1 μg/dl, which increased to 24.4 μg/dl at 60 min. MRI of the pituitary gland (Fig. 1a) showed a small sella with a thin rim of anterior pituitary of 3 mm sagital diameter. The neurohypophysis was ectopic at the floor of the third ventricle. The infundibulum was not identified after gadolinium administration.
The patient was started on levothyroxine and estrogen/progesterone replacement. She is considering resuming growth hormone replacement.
Discussion
We present 4 cases of multiple pituitary hormone deficiencies (MPHD) with pleiotropic presentations in the context of the stalk transection syndrome diagnosed by the adult endocrinologist. All patients had a small anterior pituitary, diminutive caliber or absent infundibulum after gadolinium administration and ectopic posterior pituitary located close to the median eminence. All patients had TSH, FSH and LH deficiency and 3 patients (cases 2, 3 and 4) had growth failure due to GH deficiency diagnosed before age 5 (Table 1). In case 1, the patient almost achieved her mid-parental height, but symptomatic GH deficiency was detected at age 20. In cases 2 and 3, adrenal insufficiency was diagnosed late, at age 19 and 36, respectively. In case 4, adrenal function was normal at age 21 despite childhood-onset of GH, TSH, FSH and LH deficiency. Our cases indicate that new endocrine deficiencies can be encountered in the adult life in patients with MRI characteristics of pituitary stalk transection syndrome.
Differentiation of pituitary cell lineage during organogenesis depends on a temporally regulated cascade of tissue-specific regulators. Structural abnormalities of the pituitary gland may be heritable or sporadic. Transcription factor defects cause dominantly or recessively inherited MPHD with onset in childhood. The most frequently reported mutations involve the PROP1 gene; in these cases, the pituitary gland is hypoplastic or enlarged, while the infundibulum and posterior pituitary usually appear normal [9, 10]. Other genetic defects involve: POU1F1 or Pit-1 (GH, PRL and TSH deficiency), Hesx1 (MPHD and septo-optic dysplasia), SOX3 (infundibular hypoplasia, MPHD and mental retardation), SOX2 (MPHD, learning difficulties, esophageal atresia and anophtalmia), LX3 (GH, TSH, LH and FSH deficiency and rigid cervical spine), and LX4 (GH, TSH and ACTH deficiency, undescended posterior pituitary and pointed cerebellar tonsils) [11, 12]. The vast majority of previously reported cases of the stalk transection syndrome occurred sporadically. Even more, a recent study in 26 Spanish patients with hypopituitarism and the stalk transection syndrome revealed that only 2 consanguineous patients (4.3%) had a PROP1 gene mutation, while POU1F1, HEXS1 and LX-4 mutations were not detected [4]. In the 4 cases we present here, family history of endocrine disorders or short stature was negative. Also, prolactin level was elevated in cases 1 and 2 and normal in cases 3 and 4, while many gene mutations are usually associated with low or low-normal prolactin. Although evidence for systematic association of genetic defects with the stalk transection syndrome is lacking, future research might reveal new genetic mutations encoding signaling molecules or transcription factors in these patients.
The mechanism responsible for the pituitary stalk transection syndrome is unknown. One hypothesis includes a defective neural migration during embryogenesis, which goes along with midline central nervous system malformations (optic nerve hypoplasia, absence of septum pellucidum, Chiari 1 malformation) encountered in 20% children with these MRI characteristics [13, 14]. None of our patients had clinical or MRI characteristics of midline central nervous system malformations. Another hypothesis is that ischemic injury of the pituitary stalk triggers axonal reorganization of the infundibular stump in an ectopic posterior pituitary. The adenohypophysis is susceptible to ischemia and loss of function after disruption of the pituitary portal blood flow, as its systemic arterial blood supply originates from the inferior hypophyseal arterial branches, which predominantly serve the posterior pituitary [15]. This mechanism has been advocated in patients with history of breech delivery, perinatal hypoxia or trauma in infancy [16, 17]. One study in 27 children with pituitary transection syndrome showed that breech delivery occurred in 68% [18]. Two of our cases (1 and 3) had breech presentation upon delivery, perinatal stress or head trauma at a very young age.
Patients with stalk transection syndrome present with childhood-onset GH deficiency or MPHD [4, 6]. The posterior pituitary function is usually normal as the ectopic posterior pituitary continues to release hormones in the systemic circulation. Prolactin level is normal or slightly elevated due to lack of the dopaminergic inhibitory control. Indeed, none of our cases had diabetes insipidus, while prolactin was slightly elevated in 2 cases. Most cases of stalk transection syndrome present with growth retardation in childhood in association with GH deficiency [4, 13]. Our cases 2, 3 and 4 also had GH deficiency as presenting feature before age 5. However, case 1 was diagnosed with GH deficiency at age 20 despite spontaneously achieving the low end of the target mid-parental height. In this case, central hypothyroidism and hypogonadism were diagnosed in the second decade of life despite a history of head trauma at 2 months after birth. Although lack of estrogen contributed to the patient’s spontaneous linear growth during adolescence, it is unlikely that significant GH deficiency was present throughout her childhood due to the observed normal growth velocity. This may illustrate a rare occurrence of late onset of GH deficiency despite MRI evidence of stalk transection syndrome and MPHD. This patient was not evaluated for GHD during childhood due to absence of suggestive clinical characteristics. Other rare similar cases were previously reported, all with multiple pituitary hormone deficiencies in the context of the stalk transection syndrome untreated until 3rd–5th decade of life [19–23]. Possible explanations of attaining normal adult height in these patients include: lack of estrogen, normal prolactin secretion, other growth factors and later development of GH deficiency.
Imaging predictors of hormonal phenotype in patients with stalk transection syndrome (MPHD versus isolated GH deficiency) have been proposed. In a study of 14 patients with hypopituitarism ages 10–25 years, a thin visible infundibulum was identified in 8 after gadolinium administration; among them, 6 had isolated GH deficiency and 2 MPHD. In the same study, 5 of the 6 children in whom the infundibulum was not identified had MPHD [8]. Similarly, in a study of 44 patients ages 0.3–33 years with GH deficiency (21 isolated and 23 MPHD), 96% of patients with MPHD had stalk absence on the MRI scan [24]. A third study in 25 children with hypopituitarism ages 0.9–17.4 years, 12 of 14 with a visible infundibulum had isolated GHD. In the same study, 10 of 11 children without a visible infundibulum had MPHD. This study also suggested that ectopic posterior pituitary gland location at the level of the median eminence was more likely to be associated with MPHD (11/11 patients), while its location at lower stalk levels was associated with isolated GHD (12/14 patients) [7]. The most recent study of 26 adult patients with hypopituitarism did not support an association between the degree of hypopituitarism and pituitary stalk visibility or location of the ectopic posterior gland [4]. These discordant findings have probably a two-fold explanation: different timing of hormonal evaluation across studies and insufficient statistical power due to small numbers of patients.
Data with regards to the natural history of patients with transection syndrome into adulthood is sparse. These patients are likely to have persistent GH deficiency as shown by a study of 18 patients with childhood-onset hypopituitarism and ectopic neurohypophysis; those with absent infundibulum, ectopic neurohypophysis located at the median eminence and MPHD failed GH stimulation tests as adults [25]. Most patients exhibit progressive hormone deficiencies in the first 2 decades of life, as illustrated by a study of 26 patients, that showed that 46% had panhypopituitarism as adults [4]. In our case 3, adrenal insufficiency was not diagnosed until age 36, despite thorough testing during childhood. Case 4 had normal adrenal function at age 21 despite early onset of GH, TSH, FSH and LH deficiency. The cortricotroph cells are located in the central part of the adenohypophysis that surrounds the pituitary stalk and are likely less susceptible to ischemic stress compared to the gonadotroph and somatotroph cells. Late-onset adrenal insufficiency in the 3rd–4th decade of life has been previously reported in a few patients with childhood-onset GH deficiency and MPHD. One case report of a patient with pituitary stalk transection syndrome illustrates symptomatic onset of adrenal insufficiency at age 38 despite childhood-onset of GH, TSH, FSH and LH deficiency [26]. Another case of a patient with untreated hypopituitarism likely since childhood was diagnosed with stalk transection syndrome upon presentation with acute adrenal insufficiency at age 48 [27]. A few patients with PROP1 gene defects were also described with onset of adrenal insufficiency in the 3rd–5th decades of life, despite GH, TSH, FSH and LH deficiency apparent at a young age [10, 28]. The occurrence of late onset of adrenal insufficiency in patients with congenital hypopituitarism suggests that mechanisms other than failure of embryonic development of cortricotroph cells are at least partially responsible. The mechanism for progression to more pituitary deficiencies in adulthood is not clearly understood; one hypothesis relates to secondary atrophy of the pituitary infundibulum and/or adenohypophysis.
The importance of understanding the extent of the endocrine problem in patients with stalk transection syndrome is revealed by case reports of adrenal crisis in patients with undiagnosed congenital hypopituitarism [26, 28]. In case 3 presented here, the patient possibly had unrecognized adrenal insufficiency at the time of the in vitro fertilization attempt, which was unsuccessful. Patient education with regards to the importance of taking hormonal replacement and cortisol supplementation during acute illness may decrease the burden of morbidity in these patients [29]. All-cause and vascular mortality has been shown to be increased in patients with hypopituitarism compared to age- and gender-matched controls, although published series include mostly patients with pituitary adenomas who underwent surgical and radiation therapy [30]. Mortality studies in patients with childhood-onset hypopituitarism in general and stalk transection syndrome in particular are lacking.
In summary, the pituitary stalk transeaction (dysgenesis) syndrome should be considered in adults with hypopituitarism. Almost 25 years after the first description of this syndrome, adult endocrinologists diagnose this entity in patients who were previously thought to have idiopathic GH deficiency or MPHD. The presence of MRI characteristics compatible with the pituitary stalk transection syndrome should prompt a full pituitary hormonal evaluation. Long-term follow-up is necessary since the onset of pituitary hormone deficiencies may occur over several decades. Large cohort studies are needed to further elucidate the natural course of the disease and predictive factors of progressive MPHD.
References
Melmed S (2003) Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 112(11):1603–1618
Struthers RS, Vale WW, Arias C, Sawchenko PE, Montminy MR (1991) Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREB mutant. Nature 350(6319):622–624
Fujisawa I, Kikuchi K, Nishimura K, Togashi K, Itoh K, Noma S, Minami S, Sagoh T, Hiraoka T, Momoi T et al (1987) Transection of the pituitary stalk: development of an ectopic posterior lobe assessed with MR imaging. Radiology 165(2):487–489
Fernandez-Rodriguez E, Quinteiro C, Barreiro J, Marazuela M, Pereiro I, Peino R, Cabezas-Agricola JM, Dominguez F, Casanueva FF, Bernabeu I (2011) Pituitary stalk dysgenesis-induced hypopituitarism in adult patients: prevalence, evolution of hormone dysfunction and genetic analysis. Neuroendocrinology 93(3):181–188
Abrahams JJ, Trefelner E, Boulware SD (1991) Idiopathic growth hormone deficiency: MR findings in 35 patients. AJNR Am J Neuroradiol 12(1):155–160
Patkar D, Patankar T, Krishnan A, Prasad S, Shah J, Limdi J (1999) MR imaging in children with ectopic pituitary gland and anterior hypopituitarism. J Postgrad Med 45(3):81–83
Chen S, Leger J, Garel C, Hassan M, Czernichow P (1999) Growth hormone deficiency with ectopic neurohypophysis: anatomical variations and relationship between the visibility of the pituitary stalk asserted by magnetic resonance imaging and anterior pituitary function. J Clin Endocrinol Metab 84(7):2408–2413
Genovese E, Maghnie M, Beluffi G, Villa A, Sammarchi L, Severi F, Campani R (1997) Hypothalamic-pituitary vascularization in pituitary stalk transection syndrome: is the pituitary stalk really transected? The role of gadolinium-DTPA with spin-echo T1 imaging and turbo-FLASH technique. Pediatr Radiol 27(1):48–53
Deladoey J, Fluck C, Buyukgebiz A, Kuhlmann BV, Eble A, Hindmarsh PC, Wu W, Mullis PE (1999) Hot spot in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 84(5):1645–1650
Vallette-Kasic S, Barlier A, Teinturier C, Diaz A, Manavela M, Berthezene F, Bouchard P, Chaussain JL, Brauner R, Pellegrini-Bouiller I et al (2001) PROP1 gene screening in patients with multiple pituitary hormone deficiency reveals two sites of hypermutability and a high incidence of corticotroph deficiency. J Clin Endocrinol Metab 86(9):4529–4535
Irie Y, Tatsumi K, Kusuda S, Kawawaki H, Boyages SC, Nose O, Ichiba Y, Katsumata N, Amino N (1995) Screening for PIT1 abnormality by PCR direct sequencing method. Thyroid 5(3):207–211
Kelberman D, Dattani MT (2007) Hypopituitarism oddities: congenital causes. Horm Res 68(Suppl 5):138–144
Arifa N, Leger J, Garel C, Czernichow P, Hassan M (1999) Cerebral anomalies associated with growth hormone insufficiency in children: major markers for diagnosis? Arch Pediatr 6(1):14–21
Hamilton J, Blaser S, Daneman D (1998) MR imaging in idiopathic growth hormone deficiency. AJNR Am J Neuroradiol 19(9):1609–1615
Melmed S, Kleinberg D (2002) In: Williams textbook of endocrinology, 10th edn. Elsevier, Amsterdam, Holland, p 178
Kikuchi K, Fujisawa I, Momoi T, Yamanaka C, Kaji M, Nakano Y, Konishi J, Mikawa H, Sudo M (1988) Hypothalamic-pituitary function in growth hormone-deficient patients with pituitary stalk transection. J Clin Endocrinol Metab 67(4):817–823
Barbeau C, Jouret B, Gallegos D, Sevely A, Manelfe C, Oliver I, Pienkowski C, Tauber MT, Rochiccioli P (1998) Pituitary stalk transection syndrome. Arch Pediatr 5(3):274–279
Maghnie M, Larizza D, Triulzi F, Sampaolo P, Scotti G, Severi F (1991) Hypopituitarism and stalk agenesis: a congenital syndrome worsened by breech delivery? Horm Res 35(3–4):104–108
Badawy SZ, Pisarska MD, Wasenko JJ, Buran JJ (1994) Congenital hypopituitarism as part of suprasellar dysplasia. A case report. J Reprod Med 39(8):643–648
Den Ouden DT, Kroon M, Hoogland PH, Geelhoed-Duijvestijn PH, Wit JM (2002) A 43-year-old male with untreated panhypopituitarism due to absence of the pituitary stalk: from dwarf to giant. J Clin Endocrinol Metab 87(12):5430–5434
Kageyama K, Watanobe H, Nasushita R, Nishie M, Horiba N, Suda T (1998) A hypopituitary patient who attained tall stature without growth hormone. Intern Med 37(5):472–475
Papastathopoulou L, Tzanela M, Vlassopoulou V, Vassiliadi D, Thalassinos N (2006) Untreated hypopituitarism due to absence of the pituitary stalk with normal adult height: report of two cases. Endocrine 29(1):175–179
Wada S, Minagawa A, Imamaki K, Suda S, Yamanaka K, Iitaka M, Katayama S (2000) A patient of hypogonadotropic hypogonadism accompanied by growth hormone deficiency and decreased bone mineral density who attained normal growth. Intern Med 39(8):641–645
Kornreich L, Horev G, Lazar L, Schwarz M, Sulkes J, Pertzelan A (1998) MR findings in growth hormone deficiency: correlation with severity of hypopituitarism. AJNR Am J Neuroradiol 19(8):1495–1499
Leger J, Danner S, Simon D, Garel C, Czernichow P (2005) Do all patients with childhood-onset growth hormone deficiency (GHD) and ectopic neurohypophysis have persistent GHD in adulthood? J Clin Endocrinol Metab 90(2):650–656
Gotyo N, Doi M, Izumiyama H, Hirata Y (2007) Secondary adrenal insufficiency caused by adult development of pituitary stalk transection. Intern Med 46(20):1711–1715
Pentimone F, Riccioni S, Del Corso L (1999) Congenital hypopituitarism in a 48-year old adult. Natural course, hormonal study and MRI evidence. Panminerva Med 41(4):351–354
Lamesch C, Neumann S, Pfaffle R, Kiess W, Paschke R (2002) Adrenocorticotrope deficiency with clinical evidence for late onset in combined pituitary hormone deficiency caused by a homozygous 301–302delAG mutation of the PROP1 gene. Pituitary 5(3):163–168
Hakkaart-van Roijen L, Beckers A, Stevenaert A, Rutten FF (1998) The burden of illness of hypopituitary adults with growth hormone deficiency. Pharmacoeconomics 14(4):395–403
Sherlock M, Ayuk J, Tomlinson JW, Toogood AA, Aragon-Alonso A, Sheppard MC, Bates AS, Stewart PM (2010) Mortality in patients with pituitary disease. Endocr Rev 31(3):301–342
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ioachimescu, A.G., Hamrahian, A.H., Stevens, M. et al. The pituitary stalk transection syndrome: multifaceted presentation in adulthood. Pituitary 15, 405–411 (2012). https://doi.org/10.1007/s11102-011-0337-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-011-0337-9