
Abstract
The reaction of pyrrolo[3,2,1-ij]quinoline-1,2-diones with methyl (het)aryl ketones gave the corresponding 1-(het)arylmethylidenepyrroloquinolin-2-ones, and 1,3-dipolar cycloaddition of the latter to azomethine ylide generated from sarcosine and paraformaldehyde afforded 4-acyl-1,4′,4′,6′-tetramethylspiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′(4′H)-ones. The synthesized compounds were evaluated for inhibitory activity against blood coagulation factors Xa and XIa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Analysis of published data showed that search for inhibitors of factor Xa which is common for extrinsic and intrinsic blood coagulation pathways is one of the main lines in the design of efficient anticoagulants [1–6]. According to recent data, tetrahydroquinoline derivatives were found among these compounds [7]. Computer design of factor Xa inhibitors [8] revealed fairly active compounds in the series of 2,2,4-trimethyl-1,2-dihydroquinoline [9] and 4H-pyrrolo[3,2,1-ij]quinoline-1,2-dione derivatives [10–11]. The latter are inhibitors of protein kinases [12]. The strategy of search for new factor Xa inhibitors implies, among other methods, molecular hybridization [13], i.e., combination of two and more pharmacophores in a single molecule, which is achieved mainly by using selective coupling reactions. As applied to 4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones, such approach is possible, e.g., when a carbonyl group is present in the 1-position [14, 15]. Later on, this was also demonstrated for 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones [16, 17]. With the goal of searching for new inhibitors of factors Xa and XIa via molecular hybridization, it seemed promising to combine 4H-pyrrolo[3,2,1-ij]quinoline scaffold with pharmacophoric aroylmethylidene [18] and spirooxindole systems [19, 20]. In particular, it is known that 2-oxindoles are privileged substructures in the design of new drugs since many 2-oxindole derivatives have passed clinical trials for cancer therapy [21–23].
The present work was aimed at synthesizing new ethylidene and spiro-pyrrolidine derivatives of pyrrolo[3,2,1-ij]quinolin-2-ones and studying their inhibitory activity against factors Xa and XIa.
An activated double bond can be built up by crotonization of the carbonyl group in position 1 of 4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones with acetophenone and its heterocyclic analogs. In the case of isatin, which is a structural analog of 4H-pyrrolo[3,2,1-ij]quinoline-1,2-dione, this can be achieved via a two-step synthesis [24–29]. In the first step, which is catalyzed by di- or trialkylamine, tertiary alcohol is formed. In the second step, the alcohol undergoes dehydration in the presence of acetic and hydrochloric acids with the formation of the corresponding cyclic chalcone. Acetophenones reacted with dihydropyrrolo[3,2,1-ij]quinoline-1,2-diones in a similar way [17].
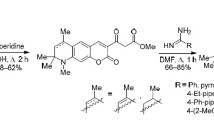
This synthetic approach proved to be applicable to 4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones 1. Their reaction with substituted acetophenones, 2-acetylpyridine, and 2-acetylthiophene gave 1-(het)arylmethylidenepyrroloquinolin-2-ones 3a–3i in good yields (Scheme 1). The product separated from the reaction mixture even in the first step, when the initial compounds were heated in boiling ethanol in the presence of diethylamine. The subsequent treatment of the filtrate with a mixture of aqueous HCl and acetic acid completed dehydration of intermediate tertiary alcohol and allowed good yields to be achieved without isolation of the latter.

1.
Compounds 3a–3i can be formed as E and Z isomers. According to the 1H NMR, IR, and HPLC/MS data, only one of the possible isomers was formed in our case. Stereochemical studies of structurally related 3-acylmethylidene-2-oxindoles showed that the E isomer is preferred and that Z isomer is formed only when a substituent is present in the 4-position of isatin [27]. The corresponding position in pyrroloquinoline-1,2-dione molecules 1 is C9, and our attempts to obtain compound like 3 from a 9-substituted 4H-pyrrolo[3,2,1-ij]quinoline-1,2-dione were unsuccessful. The E configuration of the exocyclic double bond was also confirmed by the NOESY spectrum of 3h, which displayed no coupling between the CH= and 9-H protons (δ 7.44/7.73 ppm) possible for the Z isomer. Presumably, preferential formation of the E isomer with formally trans-oriented carbonyl groups is related to the energy factor: in the case of the Z isomer, larger carbonyl oxygen atoms would appear close to each other.
The reaction of azomethine ylilides with alkenes underlies one of the main synthetic approaches to pyrrolidines [30]. In order to construct a pyrrolidine fragment spiro-fused to the pyrrolo[3,2,1-ij]quinoline system, compounds 3a–3d and 3h were used as dipolarophiles in 1,3-dipolar cycloaddition with azomethine ylides generated in situ from sarcosine and paraformaldehyde [31–33] (Scheme 2).

2.
Fast depolymerization of paraformaldehyde and decarboxylation of sarcosine in boiling toluene leads to generation of azomethine ylide which adds to the activated exocyclic C=C double bond of 3, yielding 4-aroyl-8′-R-1,4′,4′,6′-tetramethyl-4′H-spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-ones 4a–4e (Scheme 3). Taking into account the assumed E configuration of the exocyclic double bond, only one diastereoisomer is formed in which the acyl fragment R2 and benzene ring of the pyrroloquinoline moiety are arranged cis with respect to each other. The formation of only one stereoisomer was confirmed by the 1H and 13C NMR spectra which contained only one set of signals.

3.
The 1H NMR spectra of 4a–4e characteristically showed signals from methylene protons in the pyrrolidine ring as doublets at δ 2.6 and 2.8 ppm and doublets of doublets at δ 3.6 and 4.5 ppm. In the 13C NMR spectra of 4a–4e, 4′-methyl carbon signals were observed at δC 25.61 and 27.01 ppm, methylene carbons resonated at δC 53.25 and 67.71 ppm, and carbonyl carbon signals were located at δC 177.00 and 197.82 ppm. The IR spectra of 4a–4e displayed absorption bands at 2769–2798 (NCH3), 1652–1687 (NC=O), 1698–1703 (C=O), and 1614–1615 cm–1 (C=C). Compounds 3 and 4 were characterized by HPLC retention times of 5.3–5.9 and 1.4–1.5 min, respectively.
Compounds 3f–3i and 4a–4e were evaluated for their in vitro inhibitory activity against factors Xa and XIa. Compounds 4 showed no inhibitory activity, whereas compounds 3 moderately inhibited factors Xa and XIa (Table 1). These findings prompted us to continue studies aimed at rational molecular design of efficient anticoagulants based on pyrrolo[3,2,1-ij]quinolin-2-ones.
In summary, we have synthesized new pyrrolo[3,2,1-ij]quinolin-2-ones containing acylmethylidene and spiro-pyrrolidine moieties and tested them for their inhibitory activity against blood coagulation factors Xa and XIa.
EXPERIMENTAL
The 1H and 13C NMR spectra were recorded on Bruker DRX-500 (500 and 125.76 MHz, respectively) and Bruker AM-300 spectrometers using DMSO-d6 as solvent and tetramethylsilane as internal standard. HPLC/MS analyses were carried out with an Agilent Infinity 1260 liquid chromatograph coupled with an Agilent 6230 TOF mass-selective detector; Poroshell 120 EC-C18 column, 4.6×50 mm, particle size 2.7 μm; column temperature 28°C; mobile phase: 0.1% formic acid in MeCN (A)/0.1% formic acid in water (B), gradient 0–100%: A, 3.5 min, 50%; A, 1.5 min, 50–100%; B, 3.5 min, 50%; B, 1.5 min, 50–0%; flow rate 0.4 mL/min; electrospray ionization, capillary voltage –3.5 kV; fragmentor voltage +191 V; OctRF +66 V, positive polarity. The IR spectra were recorded on a Bruker Vertex-70 spectrometer with Fourier transform. The melting points were measured with a Stuart SMP30 melting point apparatus. The purity of the initial compounds and products was checked, and the progress of reactions was monitored, by TLC on Merck TLC Silica gel 60 F254 plates using CHCl3–MeOH (10:1) as eluent; spots were visualized under UV light. Initial compounds 1a–1c were synthesized according to the procedure described in [34].
The inhibitory activity of the synthesized compounds against blood coagulation factors Xa and XIa was evaluated by measuring the kinetics of hydrolysis of the corresponding enzyme-specific substrates in the presence of tested compounds. These specific substrates were S2765 (Z-D-Arg-Gly-Arg-pNA, 2HCl) for factor Xa and S2366 (pyroGlu-Pro-Arg-pNA·HCl) for factor XIa. The substrates were obtained from Chromogenix (Instrumentation Laboratory Company, Lexington, MA 02421, USA).
Each well of a 96-well microplate was charged with a buffer containing 140 mM NaCl, 20 mM HEPES, 0.1% PEG (6000) (pH 8.0), and factor Xa (final concentration 2.5 nM) or XIa (final concentration 0.8 nM), substrate S2765 (final concentration 200 μM) or S2366 (200 μM), and inhibitor (30 μM), and DMSO (≤2%) were added. The kinetics of formation of p-nitroaniline were measured with a THERMOmax Microplate Reader (Molecular Devices Corporation, Sunnyvale, California) at λ 405 nm. The initial hydrolysis rate was determined from the initial slope of the kinetic curve. The rate of substrate hydrolysis by the enzyme in the presence of an inhibitor was normalized to the rate of hydrolysis in the absence of inhibitor. The results were processed using GraphPad Prism (GraphPad, 2365 Northside Dr, San Diego, CA 92108, USA) and OriginPro 8 (OriginLab Corporation, One Roundhouse Plaza, Suite 303 Northampton, MA 01060 USA) (Table 1).
8-Substituted 4,4,6-trimethyl-1-[2-oxo-2-(het)arylethylidene]-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones 3a–3i (general procedure). A mixture of 2 mmol of pyrrolo[3,2,1-ij]quinoline-1,2-dione 1a–1c and 3 mmol of (het)aryl methyl ketone 2a–2e in 15 mL of ethanol and 1 mL of diethylamine was refluxed for 4– 6 h. After cooling, the precipitate was filtered off, 1 mL of concentrated aqueous HCl and 1 mL of acetic acid were added to the filtrate, and the mixture was refluxed for 2–3 h. After cooling, the precipitate was filtered off, washed with ethanol, dried, and combined with the precipitate isolated previously. The product was purified by recrystallization from propan-2-ol containing a few drops of acetic acid.
(E)-4,4,6-Trimethyl-1-(2-oxo-2-phenylethylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3a). Yield 0.45 g (68%), red–brown crystals, mp 162–164°C. IR spectrum, ν, cm–1: 1705 s (C=O), 1655 s (NC=O), 1614 (C=C), 1598 m (COC=C). 1H NMR spectrum, δ, ppm: 1.65 s (6H, 4-CH3), 1.99 d (3H, 6-CH3, J = 1.4 Hz), 5.46 d (1H, 5-CH, J = 1.4 Hz), 6.93 t (1H, 8-H, J = 7.8 Hz), 7.21 d (1H, 7-H, J = 7.7 Hz), 7.61 t (2H, Harom, J = 7.6 Hz), 7.73 t (1H, Harom, J = 7.6 Hz), 7.75 s [1H, C(O)CH], 7.80 d (1H, 9-H, J = 7.7 Hz), 8.08 d (2H, Harom, J = 7.3 Hz). Mass spectrum: m/z 330.1494 [M + H]+. C22H19NO2. Calculated: M 330.1490.
(E)-4,4,6-Trimethyl-1-[2-oxo-2-(pyridin-2-yl)ethylidene]-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3b). Yield 0.41 g (62%), red crystals, mp 198–200°C. IR spectrum, ν, cm–1: 1704 s (C=O), 1655 s (NC=O), 1612 m (C=C), 1597 m (COC=C). 1H NMR spectrum, δ, ppm: 1.65 s (6H, 4-CH3), 2.00 d (3H, 6-CH3, J = 1.4 Hz), 5.46 d (1H, 5-H, J = 1.4 Hz), 7.00 t (1H, 8-H, J = 7.8 Hz), 7.25 d (1H, 7-H, J = 7.6 Hz), 7.74 d.d.d (1H, 4′-H, J = 1.3, 4.7, 7.5 Hz), 8.11 t.d (1H, 5′-H, J = 1.7, 7.5 Hz), 8.17 d.t (1H, 3′-H, J = 1.2, 7.8 Hz), 8.25 d (1H, 9-H, J = 7.7 Hz), 8.40 s [1H, C(O)CH], 8.82 d.t (1H, 6′-H, J = 1.2, 4.6 Hz). Mass spectrum: m/z: 331.1440 [M + H]+. C21H18N2O2. Calculated: M 331.1442.
(E)-4,4,6-Trimethyl-1-[2-oxo-2-(thiophen-2-yl)ethylidene]-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3c). Yield 0.48 g (72%), brown crystals, mp 165–167°C. IR spectrum, ν, cm–1: 1709 s (C=O), 1649 s (NC=O), 1612 m (C=C), 1600 m (COC=C). 1H NMR spectrum (300 MHz), δ, ppm: 1.64 s (6H, 4-CH3), 1.99 s (3H, 6-CH3), 5.46 s (1H, 5-H), 6.96 t (1H, 8-H, J = 7.8 Hz), 7.22 d (1H, 7-H, J = 7.6 Hz), 7.32 t (1H, HTh, J = 4.3 Hz), 7.67 s [1H, C(O)CH], 8.08 d (1H, 9-H, J = 7.8 Hz), 8.13–8.18 m (2H, HTh). Mass spectrum: m/z: 336.1049 [M + H]+. C20H17NO2S. Calculated: M 336.1053.
(E)-4,4,6,8-Tetramethyl-1-(2-oxo-2-phenylethylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3d). Yield 0.51 g (75%), brown crystals, mp 212–214°C. IR spectrum, ν, cm–1: 1701 s (C=O), 1653 s (NC=O), 1618 m (C=C), 1602 m (COC=C). 1H NMR spectrum, δ, ppm: 1.63 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.0 Hz), 2.24 s (3H, 8-CH3), 5.45 s (1H, 5-H), 7.05 s (1H, 7-H), 7.61 t (2H, Harom, J = 7.7 Hz), 7.66 s [1H, C(O)CH], 7.71–7.75 m (2H, Harom, 9-H), 8.08 d (2H, Harom, J = 7.3 Hz). Mass spectrum: m/z: 344.1643 [M + H]+. C23H21NO2. Calculated: M 344.1646.
(E)-1-[2-(4-Bromophenyl)-2-oxoethylidene]-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3e). Yield 0.56 g (67%), brown crystals, mp 184–186°C. IR spectrum, ν, cm–1: 1702 s (C=O), 1654 s (NC=O), 1617 m (C=C), 1602 m (COC=C). 1H NMR spectrum, δ, ppm: 1.62 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.3 Hz), 2.23 s (3H, 8-CH3), 5.44 d (1H, 5-H, J = 1.4 Hz), 7.05 s (1H, 7-H), 7.69 s [1H, C(O)CH], 7.70 s (1H, 9-H), 7.81 d (2H, Harom, J = 8.6 Hz), 8.01 d (2H, Harom, J = 8.6 Hz). Mass spectrum: m/z 422.0755 [M + H]+. C23H20BrNO2. Calculated: M 422.0751.
(E)-1-[2-(4-Methoxyphenyl)-2-oxoethylidene]-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3f). Yield 0.49 g (66%), red–brown crystals, mp 181–183°C. IR spectrum, ν, cm–1: 1706 s (C=O), 1654 s (NC=O), 1615 m (C=C), 1594 s (COC=C). 1H NMR spectrum, δ, ppm: 1.63 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.3 Hz), 2.23 s (3H, 8-CH3), 3.88 s (3H, OCH3), 5.44 d (1H, 5-H, J = 1.4 Hz), 7.03 s (1H, 7-H), 7.12 d (2H, Harom, J = 8.9 Hz), 7.63 s [1H, C(O)CH], 7.70 s (1H, 9-H), 8.07 d (2H, Harom, J = 8.9 Hz). Mass spectrum: m/z 347.1755 [M + H]+. C24H23NO3. Calculated: M 347.1752.
(E)-4,4,6,8-Tetramethyl-1-[2-oxo-2-(thiophen-2-yl)ethylidene]-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3g). Yield 0.47 g (67%), brown crystals, mp 190–192°C. IR spectrum, ν, cm–1: 1703 s (C=O), 1648 s (NC=O), 1614 m (C=C), 1600 m (COC=C). 1H NMR spectrum, δ, ppm: 1.63 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.3 Hz), 2.27 s (3H, 8-CH3), 5.44 d (1H, 5-H, J = 1.4 Hz), 7.06 s (1H, 7-H), 7.32 d.d (1H, HTh, J = 1.0, 4.9 Hz), 7.64 s [1H, C(O)CH], 7.95 s (1H, 9-H), 8.15 d.d (1H, HTh, J = 1.0, 4.9 Hz), 8.16 d.d (1H, HTh, J = 1.0, 4.9 Hz). Mass spectrum: m/z 350.1208 [M + H]+. C21H19NO2S. Calculated: M 350.1210.
(E)-8-Methoxy-4,4,6-trimethyl-1-(2-oxo-2-phenylethylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3h). Yield 0.56 g (78%), violet crystals, mp 180–182°C. IR spectrum, ν, cm–1: 1702 s (C=O), 1652 s (NC=O), 1604 m (COC=C). 1H NMR spectrum, δ, ppm: 1.63 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.3 Hz), 3.70 s (3H, OCH3), 5.49 d (1H, 5-H, J = 1.4 Hz), 6.81 d (1H, 7-H, J = 2.4 Hz), 7.44 d (1H, 9-H, J = 2.4 Hz), 7.61 t (2H, Harom, J = 7.9 Hz), 7.71–7.75 m [2H, Harom, C(O)CH], 8.08 d (2H, Harom, J = 7.2 Hz). Mass spectrum: m/z 360.1597 [M + H]+. C23H21NO3. Calculated: M 360.1595.
(E)-8-Methoxy-4,4,6-trimethyl-1-[2-oxo-2-(pyridin-2-yl)ethylidene]-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (3i). Yield 0.45 g (62%), violet crystals, mp 209–211°C. IR spectrum, ν, cm–1: 1699 s (C=O), 1658 s (NC=O), 1604 m (COC=C). 1H NMR spectrum, δ, ppm: 1.63 s (6H, 4-CH3), 1.99 d (3H, 6-CH3, J = 1.3 Hz), 3.79 s (3H, OCH3), 5.49 d (1H, 5-H, J = 1.4 Hz), 6.85 d (1H, 7-H, J = 2.4 Hz), 7.74 d.d.d (1H, 4′-H, J = 1.2, 4.5, 7.5 Hz), 7.92 d (1H, 9-H, J = 2.4 Hz), 8.10 t.d (1H, 5′-H, J = 1.7, 7.5 Hz), 8.18 d.t (1H, 3′-H, J = 1.2, 7.8 Hz), 8.39 s [1H, C(O)CH], 8.82 d.t (1H, 6′-H, J = 1.2, 4.6 Hz). Mass spectrum: m/z: 361.1544 [M + H]+. C22H20N2O3. Calculated: M 361.1548.
Spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-ones 4a–4e (general procedure). A mixture of 1.5 mmol of 2-oxo-2-(het)arylethylidene-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one 3a–3d or 3h, 4.5 mmol of sarcosine, and 7.5 mmol of paraformaldehyde in 15 mL of anhydrous toluene was refluxed for 1–1.5 h in a flask equipped with a Dean–Stark trap. The solution became colorless during the reaction. After completion of the reaction, the solvent was removed under reduced pressure, the residue was dispersed in water, and the precipitate was filtered off, dried, and recrystallized from propan-2-ol.
4-Benzoyl-1,4′,4′,6′-tetramethyl-4′H-spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-one (4a). Yield 0.37 g (64%), white powder, mp 173–175°C. IR spectrum, ν, cm–1: 2769 m (NCH3), 1703 s (C=O), 1687 s (NC=O), 1614 m (C=C). 1H NMR spectrum, δ, ppm: 1.31 s (3H, 4′-CH3), 1.51 s (3H, 4′-CH3), 1.81 d (3H, 6′-CH3, J = 1.3 Hz), 2.36 s (3H, NCH3), 2.63 d (1H, CH2, J = 8.7 Hz), 2.71 t (1H, CH2, J = 8.8 Hz), 2.78 d (1H, CH2, J = 8.7 Hz), 3.60 d.d (1H, CH2, J = 5.5, 9.1 Hz), 4.50 d.d (1H, CH, J = 5.5, 8.5 Hz), 5.18 d (1H, 5′-H, J = 1.4 Hz), 6.81 t (1H, 8′-H, J = 7.6 Hz), 6.85 d.d (1H, 7′-H, J = 1.2, 7.7 Hz), 6.93 d.d (1H, 9′-H, J = 1.2, 7.3 Hz), 7.24–7.31 m (4H, Harom), 7.41–7.44 m (1H, Harom). Mass spectrum: m/z 387.2066 [M + H]+. C25H26N2O2. Calculated: M 387.2068.
1,4′,4′,6′-Tetramethyl-4-(pyridine-2-carbonyl)-4′H-spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-one (4b). Yield 0.36 g (62%), white powder, mp 150–152°C. IR spectrum, ν, cm–1: 2798 m (NCH3), 1699 s (C=O), 1652 m (NC=O), 1615 m (C=C). 1H NMR spectrum (300 MHz), δ, ppm: 1.52 s (3H, 4′-CH3), 1.62 s (3H, 4′-CH3), 1.77 s (3H, 6′-CH3), 2.36 s (3H, NCH3), 2.52–2.73 m (3H, CH2), 3.59 br.s (1H, CH2), 4.70 br.s (1H, CH), 5.21 s (1H, 5′-H), 6.62–6.82 m (3H, 7′-H, 8′-H, 9′-H), 7.35–7.46 m (2H, HPy), 7.70 t (1H, HPy, J = 7.2 Hz), 8.45 br.s (1H, HPy). Mass spectrum: m/z 388.2025 [M + H]+. C24H25N3O2. Calculated: M 388.2021.
1,4′,4′,6′-Tetramethyl-4-(thiophene-2-carbonyl)-4′H-spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-one (4c). Yield 0.39 g (66%), white powder, mp 159–161°C. IR spectrum, ν, cm–1: 2769 m (NCH3), 1703 s (C=O), 1665 s (NC=O), 1614 w (C=C). 1H NMR spectrum, δ, ppm: 1.46 s (3H, 4′-CH3), 1.59 s (3H, 4′-CH3), 1.84 d (3H, 6′-CH3, J = 1.3 Hz), 2.35 s (3H, NCH3), 2.64 d (1H, CH2, J = 8.7 Hz), 2.69 t (1H, CH2, J = 8.9 Hz), 2.78 d (1H, CH2, J = 8.7 Hz), 3.55 d.d (1H, CH2, J = 5.7, 9.1 Hz), 4.36 d.d (1H, CH, J = 5.5, 8.5 Hz), 5.25 d (1H, 5′-H, J = 1.4 Hz), 6.82 t (1H, 8′-H, J = 7.5 Hz), 6.88 d (1H, 7′-H, J = 7.6 Hz), 6.96 d (1H, 9′-H, J = 7.4 Hz), 7.05 t (1H, HTh, J = 4.1 Hz), 7.42 d.d (1H, HTh, J = 1.1,3.9 Hz), 7.80 d (1H, HTh, J = 5.0 Hz). Mass spectrum: m/z 393.1629 [M + H]+. C23H24N2O2S. Calculated: M 393.1633.
4-Benzoyl-1,4′,4′,6′,8′-pentamethyl-4′H-spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-one (4d). Yield 0.38 g (63%), white powder, mp 182–184°C. IR spectrum, ν, cm–1: 2788 m (NCH3), 1702 s (C=O), 1686 s (NC=O), 1614 w (C=C). 1H NMR spectrum, δ, ppm: 1.30 s (3H, 4′-CH3), 1.50 s (3H, 4′-CH3), 1.79 d (3H, 6′-CH3, J = 1.3 Hz), 2.19 s (3H, 8′-CH3), 2.36 s (3H, NCH3), 2.62 d (1H, CH2, J = 8.7 Hz), 2.68 t (1H, CH2, J = 8.8 Hz), 2.75 d (1H, CH2, J = 8.7 Hz), 3.60 d.d (1H, CH2, J = 5.4, 9.1 Hz), 4.50 d.d (1H, CH, J = 5.4, 8.4 Hz), 5.15 d (1H, 5′-H, J = 1.4 Hz), 6.68 s (1H, 7′-H), 6.76 s (1H, 9′-H), 7.24–7.32 m (4H, Harom), 7.40–7.44 m (1H, Harom). 13C NMR spectrum, δC, ppm: 16.87, 21.04, 26.61, 27.07, 41.58, 53.25, 56.06, 56.43, 57.12, 67.71, 117.38, 121.71, 124.48, 124.58, 127.33, 128.36, 129.63, 130.64, 132.47, 135.37, 137.24, 177.00, 197.82. Mass spectrum: m/z 401.2228 [M + H]+. C26H28N2O2. Calculated: M 401.2225.
4-Benzoyl-8′-methoxy-1,4′,4′,6′-tetramethyl-4′H-spiro[pyrrolidine-3,1′-pyrrolo[3,2,1-ij]quinolin]-2′-one (4e). Yield 0.42 g (67%), white powder, mp 134–136°C. IR spectrum, ν, cm–1: 2780 m (NCH3), 1698 s (C=O), 1680 s (NC=O), 1614 m (C=C). 1H NMR spectrum (300 MHz), δ, ppm: 1.30 s (3H, 4′-CH3), 1.50 s (3H, 4′-CH3), 1.80 s (3H, 6′-CH3), 2.37 s (3H, NCH3), 2.62–2.77 m (3H, CH2), 3.61 d.d (1H, CH2, J = 5.3, 9.1 Hz), 3.68 s (3H, OCH3), 4.50 d.d (1H, CH, J = 5.3, 8.3 Hz), 5.20 s (1H, 5′-H), 6.44 d (1H, 7′-H, J = 2.0 Hz), 6.53 d (1H, 9′-H, J = 2.0 Hz), 7.24–7.36 m (4H, Harom), 7.43 t (1H, Harom, J = 7.0 Hz). Mass spectrum: m/z: 417.2177 [M + H]+. C26H28N2O3. Calculated: M 417.2174.
REFERENCES
Zbinden, K.G., Anselm, L., Banner, D.W., Benz, J.,Blasco, F, Décoret, J., Himber, J., Kuhn, B., Panday, N., Ricklin, F., Risch, Ph., Schlatter, D., Stahl, M., Thomi, S., Unger, R., and Haap, W., Eur. J. Med. Chem., 2009, vol. 44, p. 2787. https://doi.org/10.1016/j.ejmech.2008.12.025
Anselm, L., Banner, D.W., Benz, J., Zbinden, K.G., Himber, J., Hilpert, H., Huber, W., Kuhn, B., Mary, J.-L., Otteneder, M.B., Panday, N., Ricklin, F., Stahl, M., Thomi, S., and Haap, W., Bioorg. Med. Chem. Lett., 2010, vol. 20, p. 5313. https://doi.org/10.1016/j.bmcl.2010.06.126
Pinto, D.J.P., Smallheer, J.M., Cheney, D.L., Knabb, R.M., and Wexler, R.R., J. Med. Chem., 2010, vol. 53, p. 6243. https://doi.org/10.1021/jm100146h
Trstenjak, U., Ilaš, J., and Kikelj, D., Med. Chem. Commun., 2014, vol. 5, p. 197. https://doi.org/10.1039/c3md00250k
Vavilova, T.V., Kardiologiya, 2019, vol. 59, p. 28. https://doi.org/10.18087/cardio.n951
Podoplelova, N.A., Sulimov, V.B., Ilin, I.S., Tashilova, A.S., Panteleev, M.A., Ledeneva, I.V., and Shikhaliev, Kh.S., Pediatr. Hematol. Oncol. Immunopathol., 2020, vol. 19, p. 139. https://doi.org/10.24287/1726-1708-2020-19-139-157
Santana-Romo, F., Lagos, C.F., Duarte, Y., Castillo, F., Moglie, Y., Maestro, M.A., Charbe, N., and Zacconi, F.C., Molecules, 2020, vol. 25, p. 491. https://doi.org/10.3390/molecules25030491
Kabankin, A.S., Sinauridze, E.I., Lipets, E.N., and Ataullakhanov, F.I., Biochemistry (Moscow), 2019, vol. 84, p. 119. https://doi.org/10.1134/S0006297919020032
Ilin, I., Lipets, E., Sulimov, A., Kutov, D., Shikhaliev, Kh., Potapov, A., Krysin, M., Zubkov, F., Sapronova, L., Ataullakhanov, F., and Sulimov, V., J. Mol. Graphics Modell., 2019, vol. 89, p. 215. https://doi.org/10.1016/j.jmgm.2019.03.017
Sulimov, V.B., Gribkova, I.V., Kochugaeva, M.P., Katkova, E.V., Sulimov, A.V., Kutov, D.C., Shikhaliev, Kh.S., Medvedeva, S.M., Krysin, M.Yu., Sinauridze, E.I., and Ataullakhanov, F.I., BioMed Res. Int., 2015, vol. 2015, article ID 120802. https://doi.org/10.1155/2015/120802
Medvedeva, S.M., Potapov, A.Yu., Gribkova, I.V., Katkova, E.V., Sulimov, V.B., and Shikhaliev, Kh.S., Pharm. Chem. J., 2018, vol. 51, p. 975. https://doi.org/10.1007/s11094-018-1726-4
Novichikhina, N.P., Shestakov, A.S., Potapov, A.Yu., Kosheleva, E.A., Shatalov, G.V., Verezhnikov, V.N., Vandyshev, D.Yu., Ledeneva, I.V., and Shikhaliev, Kh.S., Russ. Chem. Bull., Int. Ed., 2020, vol. 69, p. 787. https://doi.org/10.1007/s11172-020-2834-3
Gao, F., Ye, L., Wang, Y., Kong, F., Zhao, Sh., Xiao, J., and Huang, G., Eur. J. Med. Chem., 2019, vol. 183, article ID 111678. https://doi.org/10.1016/j.ejmech.2019.111678
Leshcheva, E.V., Shikhaliev, Kh.S., Shatalov, G.V., and Ermolova, G.I., Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol., 2003, vol. 46, p. 105.
Medvedeva, S.M., Sabynin, A.L., and Shikhaliev, Kh.S., Russ. Chem. Bull., Int. Ed., 2014, vol. 63, p. 2693. https://doi.org/10.1007/s11172-014-0801-6
Vahedi, H., Baradarani, M.M., Rashidi, A., and Joule, J.A., J. Heterocycl. Chem., 2015, vol. 52, p. 1208. https://doi.org/10.1002/jhet.2237
Mazaheri, F., Saatluo, B.E., Baradarani, M.M., and Joule, J.A., J. Heterocycl. Chem., 2017, vol. 54, p. 147. https://doi.org/10.1002/jhet.2555
Gupta, A.K., Bharadwaj, M., and Mehrotra, R., J. Heterocycl. Chem., 2019, vol. 56, p. 703. https://doi.org/10.1002/jhet.3424
Santos, M.M.M., Tetrahedron, 2014, vol. 70, p. 9735. https://doi.org/10.1016/j.tet.2014.08.005
Xia, M. and Ma, R.-Zh., J. Heterocycl. Chem., 2014, vol. 51, p. 539. https://doi.org/10.1002/jhet.1114
Gupta, A.K., Bharadwaj, M., Kumar, A., and Mehrotra, R., Top. Curr. Chem., 2017, vol. 375, p. 3. https://doi.org/10.1007/s41061-016-0089-0
Leoni, A., Locatelli, A., Morigi, R., and Rambaldi, M., Expert. Opin. Ther. Pat., 2016, vol. 26, p. 149. https://doi.org/10.1517/13543776.2016.1118059
Prakash, C.R. and Raja, S., Mini-Rev. Med. Chem., 2012, vol. 12, p. 98. https://doi.org/10.2174/138955712798995039
Dandia, A., Jain, A.K., Laxkar, A.K., and Bhati, D.S., Tetrahedron, 2013, vol. 69, p. 2062. https://doi.org/10.1016/j.tet.2012.12.021
Shaabanzadeha, M. and Khabarib, F., J. Heterocycl. Chem., 2010, vol. 47, p. 949. https://doi.org/10.1002/jhet.394
Singh, D., Fatma, Sh., Ankit, P., Mishra, P., Singh, S., Singh, Sh.B., and Singh, J., Synth. Commun., 2013, vol. 43, p. 3072. https://doi.org/10.1080/00397911.2013.769604
Edeson, S.J., Jiang, J., Swanson, S., Procopiou, P.A., Adams, H., Meijer, A.J.H.M., and Harrity, J.P.A., Org. Biomol. Chem., 2014, vol. 12, p. 3201. https://doi.org/10.1039/c4ob00496e
Gangarapu, K., Thumma, G., Manda, S., Jallapally, A., Jarapula, R., and Rekulapally, S., Med. Chem. Res., 2017, vol. 26, p. 819. https://doi.org/10.1007/s00044-017-1781-5
Nikoofar, K. and Peyrovebaghi, S.S., J. Chin. Chem. Soc., 2020, vol. 67, p. 1303. https://doi.org/10.1002/jccs.201900365
Pandey, G., Banerjee, P., and Gadre, S.R., Chem. Rev., 2006, vol. 106, p. 4484. https://doi.org/10.1021/cr050011g
Tsuge, O., Kanemasa, Sh., Ohe, M., and Takenaka, Sh., Bull. Chem. Soc. Jpn., 1987, vol. 60, p. 4079. https://doi.org/10.1246/bcsj.60.4079
Shvets, A.A. and Kurbatov, S.V., Chem. Heterocycl. Compd., 2009, vol. 45, p. 866. https://doi.org/10.1007/s10593-009-0344-1
Huang, Y., Min, W., Wu, Q.-W., Sun, J., Shi, D.-H., and Yan, Ch.-G., New J. Chem., 2018, vol. 42, p. 16211. https://doi.org/10.1039/c8nj03813a
Lescheva, E.V., Medvedeva, S.M., and Shikhaliev, Kh.S., J. Org. Pharm. Chem., 2014, vol. 12, p. 15. https://doi.org/10.24959/ophcj.14.798
Funding
This study was performed under financial support by the Russian Science Foundation (project no. 18-74-10097).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare the absence of conflict of interest.
Rights and permissions
About this article

Cite this article
Novichikhina, N.P., Skoptsova, A.A., Shestakov, A.S. et al. Synthesis and Anticoagulant Activity of New Ethylidene and Spiro Derivatives of Pyrrolo[3,2,1-ij]quinolin-2-ones. Russ J Org Chem 56, 1550–1556 (2020). https://doi.org/10.1134/S1070428020090080
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070428020090080