
Abstract
As a result of the toxicity of currently available anticancer drugs and the inefficiency of chemotherapeutic treatments, the design and discovery of effective and selective antitumor agents continues to be a hot topic in organic medicinal chemistry. Targeted therapy is a newer type of cancer treatment that uses drugs designed to interfere with specific molecules necessary for tumor growth and progression. This review explains the mechanism of regulation of p53 (tumor suppressor protein) by MDM2 and illustrates the role of targeting p53–MDM2 protein–protein interaction using small molecules as a new cancer therapeutic strategy. Spirocyclic oxindoles or spiro-oxindoles, with a rigid heterocyclic ring fused at the 3-position of the oxindole core with varied substitution around it, are the most efficacious class of small molecules which inhibit cell proliferation and induce apoptosis in cancer cells, leading to complete tumor growth regression without affecting activities of normal cells. In this review, we present a comprehensive account of the systematic development of and recent progress in diverse spiro-oxindole derivatives active as potent selective inhibitors of p53–MDM2 interaction with special emphasis on spiro-pyrrolidinyl oxindoles (the MI series), their mechanism of action, and structure–activity relationship. This review will help in understanding the molecular mechanism of p53 reactivation by spiro-oxindoles in tumor tissues and also facilitates the design and exploration of more potent analogues with high efficacy and low side effects for the treatment of cancer.
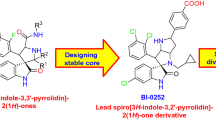
Graphical Abstract
Recent progress in spiro-oxindole derivatives as potent small molecule inhibitors of p53–MDM2 interaction, useful as anticancer agents, is described with reference to their mechanism of action and structure–activity relationship.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Cancer is one of the most devastating diseases and leading causes of death with a global estimate of 14.1 million new cases and 8.2 million cancer-related deaths in 2012 [1]. According to GLOBOCAN, developing countries have a relatively large population share, with around 57% of new cases and 65% of cancer-related deaths. The global burden of cancer is continuously increasing and the annual number of new cancer cases is expected to rise to 22 million within the next two decades. Therefore, research on the treatment of cancer is fundamental to improving outcomes for patients affected by the disease [2]. As the majority of chemotherapeutic agents work primarily through the inhibition of cell division, apart from cancer cells, other normal rapidly dividing cells like hair, gastrointestinal epithelium, and bone marrow are also affected by these drugs [3]. So, these conventional chemotherapeutics possess some serious side effects including damage of the immune system and other organs with rapidly proliferating cells as a result of nonspecific targeting, lack of solubility, and inability to enter the core of the tumors, resulting in impaired treatment with reduced dose and low survival rate [4]. Furthermore, it is widely accepted that drug-resistant cancer cells that remain alive after chemotherapy are responsible for the reappearance of tumors and the poor prognosis for patients [5]. The toxicity and inefficiency of currently available anticancer treatments, especially for advanced stages of the disease, have limited the optimization of clinical drug combinations and effective chemotherapeutic protocols [6]. So, there has been great concern about finding new types of drugs to combat cancer especially given the ever-increasing number of cases of resistance to existing anticancer drugs [7].
Advances in oncology have revolutionized cancer care and have significantly changed the treatment of cancer over the past 10 years [8, 9]. Breakthroughs in our understanding of the biology of the cancer cell in the last decade is the driving force for the development of a new generation of drugs that are targeted to the unique genetics of each tumor and each patient [10]. Targeted cancer therapies are designed to interfere with specific genes, proteins, or the tissue environment necessary for tumor growth and progression in the body [11, 12]. These factors are present in normal tissues, but are often mutated or overexpressed in tumor cells. The mechanisms of action and toxicities of targeted therapies differ from those of traditional cytotoxic chemotherapy. Targeted therapies are generally better tolerated than traditional chemotherapy [13]. These drugs attack cancer cells but leave healthy cells largely untouched, by blocking or switching off the molecular defects that cause cancer to grow and spread, resulting in better cancer control and better quality of life [8]. Many different types of targeted therapies are used to treat cancer, and many more are being developed [14]. Targeted drugs can be grouped, by how they work or what part of a cell they target, into two main categories: monoclonal antibodies [15] and small molecule inhibitors [13]. Small molecule inhibitors differ from monoclonal antibodies in several ways. They are usually administered orally rather than intravenously. They are chemically manufactured by a process that is often much less expensive than the bioengineering required for monoclonal antibodies. Small molecules can penetrate the cell membrane to interact with targets inside a cell and are designed to interfere with the enzymatic activity of the target protein [16]. Knowledge of the 3-D structure of large target protein macromolecules by X-ray crystallography permits the rational design of small molecules that mimic the stereochemical features of the macromolecule functional domains and generate virtual libraries of potential drug molecules to be used for in silico screening [17]. Therefore, small molecule inhibitors have emerged as a crucial component of therapy for many types of malignancies, including breast, colorectal, lung, and pancreatic cancers, as well as lymphoma, leukemia, and multiple myeloma [17,18,19]. Chemotherapy drugs are most effective when given in combination (combination chemotherapy). The rationale for combination chemotherapy is to use drugs that work by different mechanisms, thereby decreasing the likelihood of development of resistance by the cancer cells. When drugs with different effects are combined, each drug can be used at its optimal dose, thereby reducing nonspecific toxicity. Secondly, through limited drug exposure, the emergence of multidrug resistance (MDR) is reduced [20]. Pharmaceutical company investment is constantly increasing in cancer research and cancer therapies account for more than 30% of all preclinical and phase I clinical development, with 21 new molecular entities being launched and reaching patients in the last 2 years alone [21]. As a result, most of the new anticancer drugs approved by the US Food and Drug Administration (FDA) since 2000 are targeted therapy [22]. Seven out of nine anticancer drugs approved in the last year are target therapy drug [23]. Major advances have been made in recent years in the design and development of small molecule inhibitors useful as cancer therapeutics by targeting MDM2–p53 interaction and several compounds have moved into advanced preclinical development or clinical trials [19, 24].
2 Targeting Interaction of p53 and MDM2 as a New Cancer Therapeutic Strategy
It is well established that p53 is a tumor suppressor protein that plays a central role in the regulation of cell cycle, apoptosis, DNA repair, and senescence [25, 26]. It has been described as “the guardian of the genome” because it is essential for conserving genomic stability by preventing genome mutation [27]. p53 inhibits tumor development by its ability to respond to stress signals by triggering cell cycle arrest and cell death by apoptosis in response to anticancer therapy [28, 29]. Normally, the life-span of p53 is short, and its activity and levels are tightly regulated by a protein called MDM2 (also called HDM2 in humans). In unstressed cells, p53 levels are generally kept low through a continuous degradation of p53 by MDM2. p53 and MDM2 form an autoregulatory feedback loop in which p53 stimulates the expression of the MDM2 protein, which in turn is the negative regulator of p53 [30]. MDM2 regulates p53 by binding to p53 transactivation domain, it inhibits p53 transcriptional activity, promotes degradation, and favors the export of p53 from the nucleus to cytoplasm where it acts as ubiquitin ligase and covalently attaches ubiquitin to p53 and thus marks p53 for degradation by the proteasome [31] (Fig. 1). Thus, in the presence of MDM2, the p53 protein is inactivated and does not stimulate the expression of genes involved in apoptosis, DNA repair, or cell cycle arrest.

Regulation of p53 by MDM2: p53 stimulates the expression of MDM2; MDM2, in turn, inhibits p53 activity. A broad range of DNA-damaging agents or deregulated oncogenes induce p53 activation. DNA damage promotes phosphorylation of p53 and MDM2, thereby preventing their interaction and stabilizing p53. Blockade of the MDM2–p53 interaction would lead to p53 accumulation, thus restoring the tumor suppressor function
However, deregulation of the MDM2/p53 balance leads to malignant transformation of cells. More than 50% of human cancers including sarcomas, gliomas, hematological malignancies, melanomas, and carcinomas [32] express mutant p53 which becomes inactive as a result of partial folding [33] while in the other 50% of tumors retaining wild-type p53, p53 becomes non-functional by enhanced degradation by MDM2 [34]. Often tumors cells acquire alternative mechanisms for p53 inactivation by overexpression of MDM2 protein leading to the inhibition of p53 function [35]. In case of tumors with overexpressed MDM2, p53 is constantly degraded thus favoring tumor growth [36]. So, the inhibition of MDM2 and restoration of p53 activity in cancer cells could lead to rapid and impressive regression of tumors [37]. Several different therapeutic approaches have been attempted with the goal of restoring p53 function [38]; these approaches target MDM2 in tumors, e.g., by use of antisense oligonucleotides to lower the cellular level of MDM2 [39] or use of compounds inhibiting ubiquitin ligase activity of MDM2 thus preventing p53 degradation [40]. The mechanism of induction of p53 activation by MDM2 inhibitors is different from that induced by radiation and traditional chemotherapy drugs. Both radiation and chemotherapy drugs induce p53 accumulation by post-translational modifications of p53; in contrast, MDM2 inhibitors induce accumulation and activation of p53 in cancer and normal cells without inducing DNA damage or requiring p53 phosphorylation [36].
As MDM2 antagonists are potent activators of p53-dependent cell cycle arrest and apoptosis, there may be a strong selective pressure for loss of p53 function, either by p53 gene mutation or by epigenetic changes; inhibition of MDM2 can also lead to stabilization of mutant forms of p53 [41]. Thus, the treatment with MDM2 inhibitors as single agents may lead to tumor relapse or secondary tumor formation [42]. To avoid this situation, MDM2 antagonists are given in combination with agents that induce tumor cell apoptosis independently of p53, or with agents that are able to reactivate mutant p53 and so combination chemotherapy has been the standard treatment for most advanced solid and hematological cancers. Clinical trials of MDM2 inhibitors as a combination of non-genotoxic p53 activators or MDM2 antagonists with clinically used chemotherapeutic agents [43] such as cisplatin, doxorubicin, or cytarabine have been shown to augment efficacy in the preclinical studies [44] and are currently being tested in several phase I clinical trials (ClinicalTrials.gov: NCT01773408 and NCT01605526).
3 Small Molecules as Inhibitors of MDM2–p53 Interaction
Inhibition of p53–MDM2 interaction using small molecules has been proposed as a novel approach for treating cancer [45]. Increasing knowledge of the relationship between p53 and MDM2 has led to the development of potential small molecule inhibitors that block the p53–MDM2 interaction and activate p53 for the treatment of cancer and other human diseases [46] (Fig. 1). A high resolution crystal structure of the p53–MDM2 complex revealed that the p53–MDM2 interaction is mediated by a well-defined hydrophobic surface pocket in MDM2 and three key hydrophobic amino acid residues in p53, namely Phe19, Trp23, and Leu26, called the FWL motif [47]. It is believed that Leu22 is also important for the p53–MDM2 complex, and it could assist in the development of molecules with higher binding affinity than the p53’s N-terminal transactivation domain peptide [28]. The transcriptional activity of p53 is essential for its tumor suppressive function but the genes that need to be induced to achieve this represent only a small subset of the total library of p53 responsive genes. Thus restoration of the p53 pathway in human tumor cells that retain the wild-type gene represents an ideal selective target for cancer drug development. The p53 pathway is induced not just by DNA damage but also by ribosomal stress and oncogene-induced stress [48]. These signals act through MDM2 to change the levels and activity of p53 and block the p53–MDM2 interaction which is sufficient to induce tumor regression [17] and trigger significant ligand-induced changes in MDM2 [49]. While inducing p53 through ionizing radiation or systemic DNA damage is highly toxic to normal tissue, systemic treatment with small molecule inhibitors of p53–MDM2 interaction is only toxic to tumor tissue and not to normal tissue. This is because these inhibitors are only fully active in cells in which the p53–MDM2 interaction is weakened by tumor-specific stress signals. Likewise, selective inhibitors of the p53 rapidly palliate the cellular damage caused by an acute response to ischemia, chemotherapy, or radiation [50].
The relatively compact binding pocket in MDM2 facilitates the design of non-peptide, drug-like small molecule inhibitors to block this interaction as a means to reactivate p53 in cells harboring wild-type p53 [18]. This constitutes the basis of numerous structure-based studies designed to identify inhibitors of the MDM2–p53 interaction [51]. In search of new biological probes and drugs, identification of biologically relevant fragments derived from biologically validated natural products has been recently recognized as a powerful strategy [52]. Many naturally derived biomolecules have been found to inhibit the p53–MDM2 interaction. Some of them have been shown to decrease MDM2 activity in vitro and in vivo [53]. These newly identified natural MDM2 inhibitors include a plethora of diverse chemical frameworks, ranging from flavonoids, steroids, and sesquiterpenes to alkaloids. Peptides based on the amino acid sequence around the FWL motif were among the first tested [54] and have been used with varying success [55]. Out of numerous synthetic small molecules, small peptides, peptidomimetics, and natural products which were revealed to target the p53–MDM2 pathway, the most promising developments for p53 reactivation are from the non-peptide small molecule inhibitors [45]. Two major approaches developed to design optimal small molecule MDM2 inhibitors, using information technology-based computational power, are the straightforward in silico compound selection and structure-based de novo design [56]. The compounds were designed to mimic the interactions provided by the important amino acid side chains within the p53 peptide to disrupt the p53–MDM2 binding [57]. Extensive chemical modifications after virtual screening of compound libraries led to the discovery of several classes of highly potent and specific small molecule inhibitors of the MDM2–p53 interaction with suitable physiochemical and pharmacological properties for clinical development [8, 24] (Fig. 2).

Structure-based design of MDM2 inhibitors
To date, over 20 small molecule p53 pathway modulators with a diverse array of scaffolds have been discovered, and around seven candidate compounds with potent inhibitory activity and superior pharmacokinetics (PK) profiles have entered clinical development [50]. Thus, it is highly desirable to design novel small molecule inhibitors with potent activity and an improved ADME (absorption, distribution, metabolism, excretion) profile [56, 58]. Various representative MDM2 inhibitors are shown in Fig. 2.
In 2004, cis-diphenyl substituted imidazoline-containing compounds called Nutlins were identified as the first class of potent, non-peptide, specific inhibitors of the MDM2–p53 interaction by high-throughput screening [59]. After extensive modifications by scientists from Hoffmann-La Roche, RG7112, a Nutlin-3 derivative (A in Fig. 2) with an IC50 value of 90 nM, was the first MDM2 inhibitor in this class to advance to phase I clinical trials. The results from phase I trials of Nutlin treatment in patients with MDM2-amplified liposarcoma showed clear evidence of p53 activation and cell growth inhibition but only limited tumor volume reduction [60]. Patients with liposarcoma demonstrated an increase in intratumoral p53, p21, and macrophage-inhibitory cytokine 1 concentrations, an increase in MDM2-mRNA expression, and a small decrease in Ki-67-positive cells in the treated as compared to the pretreated samples. Clinically, the results were modest; however, 40% of the patients experienced serious adverse events, the majority of which were hematological in nature [61].
Another class of potent p53–MDM2 inhibitors bearing a piperidinone scaffold has been successfully designed and optimized using the structure-based de novo design strategy by scientists from Amgen [62]. After optimization, AMG-232 (B in Fig. 2) emerged as a potent inhibitor and was selected for clinical development [63]. It has a remarkable biochemical HTRF (homogeneous time-resolved fluorescence) IC50 of 0.1 nM and cellular potency with an IC50 of 16 nM as well as favorable pharmacokinetic properties [64]. In efficacy studies, using human tumor xenograft models in mice, AMG-232 effectively inhibits tumor growth in the SJSA-1 osteosarcoma model with an ED50 of 9.1 mg/kg with daily oral administration and demonstrates complete tumor regression in 10 of 12 animals treated with 60 mg/kg daily. It also effectively and dose-dependently inhibits tumor growth in the HCT-116 xenograft model with twice daily dosing with an ED50 of 16 mg/kg and achieves tumor stasis (100% tumor growth inhibition) without tumor regression. Preclinical data supports further evaluation of AMG-232 in clinical trials as both monotherapy and in combination with standard-of-care cytotoxics [65].
Benzodiazepine-based MDM2 inhibitors, reported by Johnson & Johnson in 2005, were discovered by a high-throughput screening of 338,000 compounds from combinatorial libraries [66]. The best compound of this class (C in Fig. 2) had an MDM2 binding IC50 of 394 nM and an IC50 of 1.1 μM in the MCF-7 cell line in a BrdU incorporation assay and was 50-fold more selective over the MDA-MB-213 cell line with mutated p53 [67].
Despite these significant efforts, as a result of low cell permeability, rapid in vivo clearance, and low bioavailability, this class of MDM2 inhibitors did not show strong antitumor activity in animal models of human cancer and did not progress into clinical development [17] (Fig. 2).
Scientists from Amgen also reported a class of chromenotriazolopyrimidine compounds as MDM2 inhibitors [68]. One extensively modified compound (D in Fig. 2) that binds to MDM2 with an IC50 of 350 nM showed moderate cellular activity, microsomal stability, high oral bioavailability (54%), and slow clearance in rodents [69].
Some terphenyl-based compounds were identified as alpha-helical mimetics of p53 and inhibitors of the MDM2–p53 interaction. The most potent inhibitor in this class (E in Fig. 2) has a K i value of 182 nM for MDM2 [70].
In 2005, researchers from the University of Newcastle reported the design of MDM2 inhibitors based on the isoindolinone scaffold by computational docking studies and extensive modifications led to compound F (Fig. 2) which binds to MDM2 with an IC50 of 0.17 μM in an ELISA assay and cell growth inhibition (GI50 = 5.2 μM) in the SJSA-1 cell line [71].
Hoffmann-La Roche designed and synthesized a pyrrolidine-containing compound (G in Fig. 2) as an MDM2 inhibitor that binds to MDM2 with an IC50 of 6 nM [72]. It displays potent cell growth inhibitory activity in cancer cell lines containing wild-type p53 (average IC50 = 30 nM) and displays greater than 100-fold selectivity over cancer cell lines containing mutated p53. Furthermore, it showed good microsomal stability and good PK properties in mice. In addition, it has high oral bioavailability (80%), good systemic exposure, metabolic stability, and also exhibited potent induction of p53 activation in vivo. Compound G is currently in phase I clinical trials for the treatment of patients with solid tumors, acute myelogenous leukemia, or advanced malignancies as a single agent and in combination with other chemotherapeutics (ClinicalTrials.gov identifier for RG7388: NCT01773408, NCT01462175, and NCT01901172).
Merck scientists have reported a class of piperidine-containing compounds as MDM2 inhibitors with IC50 values as low as 41 nM [73]. Novartis has explored several small molecular scaffolds such as 3-imidazolylindoles, substituted dihydroimidazole derivatives, tetrasubstituted heteroaryl compounds, and substituted isoquinolinones and quinazolinones as inhibitors of MDM2 and/or MDMX [74]. Though many of these new MDM2 inhibitors were capable of inhibiting tumor growth to a great extent in xenograft models of human cancer, most of these MDM2 inhibitors were unable to achieve complete tumor regression and none of them showed enough antitumor activity in animal models of human cancer to progress into clinical development [74].
Spiro-oxindoles are a newly identified class of efficacious p53–MDM2 inhibitors developed by the Wang group at the University of Michigan using computational techniques and have been progressively patented in recent years as MDM2 antagonists [75]. Many spiro-oxindole-based modifiers of the p53–MDM2 interaction have gained regulatory approval over the last few years and are actively being developed by several renowned companies like Ascenta Therapeutics, Hoffmann-La Roche, Sanofi-Aventis, and Daiichi Sankyo. The first candidate from Sanofi, SAR405838 (H in Fig. 2), has completed a phase I trial for patients with advanced cancer prescreening with wild-type p53 for its safety, tolerability, pharmacokinetics, and biological activity [74].
Here, we present a comprehensive account of diverse spiro-oxindole derivatives which are active as potent selective inhibitors of p53–MDM2 interaction, with special emphasis on spiro-pyrrolidinyl oxindoles (MI series), which are the most sought after.
4 Spiro-oxindoles as Novel Class of Selective and Potent MDM2 Inhibitors
Spirocyclic oxindoles or spiro-oxindoles constitute a biologically active class of anticancer agents in which the spirocyclic ring is fused at the 3-position of the oxindole core with varying degree of substitution around it (Fig. 3).

Spiro-oxindole pharmacophore
Although very simple in structure, these derivatives have demonstrated excellent p53–MDM2 antagonistic activity with more than 400 compounds reported in various patent applications by Hoffmann-La Roche in the last few years [76]. Spiro(oxindole-3,3′-pyrrolidine) emerged as the most potent class of MDM2 inhibitors with high binding affinities to MDM2, cellular potencies, and significantly optimized pharmacokinetic properties. They effectively induced complete tumor regression in the SJSA-1 osteosarcoma xenograft model in mice and have high specificity over other proteins [77]. Various synthetic spiro-oxindoles were identified through structure-based drug design [78]. Chemical modification at the C3 position of spiro-oxindole, with a rigid heterocyclic ring system, is an effective approach to study the modulation of p53 activity through p53–MDM2 inhibition [79]. Apart from pyrrolidine rings, compounds with other heterocyclic ring systems like isoxazolines, thiazolidines, piperidines, and thiazepine were also designed and tested as inhibitors of p53–MDM2 interaction in recent years for cancer therapy [80] and these are discussed in later sections. The complete tumor regression achieved by spiro-oxindoles boosts their therapeutic potential for the treatment of human cancers with MDM2 genes [72, 81].
5 Molecular Mechanism of p53 Reactivation by Spiro-oxindoles
Analysis of the crystal structure of p53–MDM2 complex showed that the interaction between p53 and MDM2 was primarily mediated by three hydrophobic residues, namely Phe19, Trp23, and Leu26 of p53 peptide and a small deep hydrophobic cleft in MDM2. Since the indole ring of the Trp23 residue of p53 protein is buried deep inside a hydrophobic cavity in MDM2 and its NH group forms a hydrogen bond with the backbone carbonyl in MDM2, Trp23 seems to be the most critical site for binding of p53 to MDM2 [57]. The X-ray structure of the p53–MDM2 complex and mutagenesis and alanine scanning of p53 peptides suggested that a fourth residue, Leu22, also plays an important role in the overall interaction between p53 and MDM2. The binding to MDM2 of wild-type p53-based peptides and a number of high-affinity mutant peptides have also been well established [82, 83].
Spiro-oxindoles which were more potent and selective than Nutlins were identified by Wang and co-workers as a new class of small molecule inhibitors using a structure-based de novo design strategy [84]. Initial compounds were designed to target the p53–MDM2 interaction with different hydrophobic groups with different stereochemistry and were docked into the MDM2 binding cleft using the GOLD program [84, 85]. The oxindole ring was found to be a perfect mimic for Trp23’s indole in both the hydrogen bond formations and the potential hydrophobic interaction with MDM2 in this class of compounds. Substructure searches of natural products containing oxindole rings allowed the identification of two alkaloids, spirotryprostatin A and alstonisine, which contained the spiro-pyrrolidinyl oxindole core as a scaffold to design possible MDM2 inhibitors (Fig. 4). But the computational modeling studies of these natural alkaloids showed that as a result of steric hindrance these specific compounds docked poorly into the MDM2 pocket [75]. However, the spiro(oxindole-3,3′-pyrrolidine) core structure emerged as the starting point in which the oxindole ring inserts into the binding cavity occupied by Trp23 indole in p53; two additional hydrophobic groups were installed on the rigid spiropyrrolidine ring to mimic the side chain of Phe19 and Leu26 [86]. Structure-based optimization to capture the additional interaction between Leu22 in p53 and MDM2 was done by using a 6-halo-oxindole moiety which is characterized by the presence of an –NH group able to bind MDM2 through a hydrogen bond and an oxo-group pointing outside into the water (solvent) (Fig. 5).

Resemblance of spiro-oxindole alkaloids and Trp23 in p53 protein

Similarity of three key residues of p53 peptide involved in the MDM2–p53 interaction with spiro-oxindole lead compound
The cellular mechanism of p53 activation by spiro-oxindole was consistent with its in vitro biochemical binding assays indicating that these potent MDM2 inhibitors are capable of blocking the p53–MDM2 protein–protein interaction in cells [87]. They induce accumulation of p53 protein but do not increase the transcription of the p53 gene in either tumor or normal cells with wild-type p53. Instead, they induce transcription of the p53-targeted genes for p21 and MDM2, increasing their protein levels [88]. These molecules bind MDM2 with high affinity, which leads to the activation of the p53 pathway and subsequent inhibition of cell growth in cancer cell lines expressing wild-type TP53 [38]. The high-resolution structure of spiro-oxindole bound to MDM2 gave interesting insight regarding how substituents of a small molecule that bind to the three subpockets of the p53–MDM2 complex should be optimized for effective binding to MDM2 or MDMX [89]. Further computational docking studies and the reported experimental data done on small molecules in this scaffold confirmed that the occupancy of the three subpockets of Phe19, Trp23, and Leu26 is necessary to achieve good inhibitor binding [90] and that the 6-chloro-oxindole group in the Trp23 subpocket is well suited to bind MDM2 with the phenyl group occupying the Phe19 pocket and the aliphatic hydrophobic tert-butyl group occupying the Leu26 pocket.
6 Various Classes of Spiro-oxindoles
6.1 Spiro-pyrrolidinyl-Based Inhibitors
The spiro(oxindole-3,3′-pyrrolidine) core is now well established as the most potent inhibitor to MDM2 with exceedingly high specificity and K i values of less than 1 nM [90]. The MI series, originally designed by the Wang group at the University of Michigan, has been further developed by Ascenta Therapeutics and licensed by Sanofi-Aventis [74]. A review on strategies for the enantioselective synthesis of various spiro-oxindoles was reported by Ball-Jones et al. [91].
Progressive development of various spiro-pyrrolidine oxindole derivatives to date is illustrated in Table 1. It is observed that the presence of a rigid heterocyclic motif at the C3 position, a halogen atom at position 5 or 6 of the spiro-oxindole system, and variations in the side chain of the amide linkage greatly influence the p53 activity through inhibition of p53–MDM2 protein interaction.
Initial compound MI-5 (V) was designed using a fluorescence polarization binding assay and prepared using an efficient procedure involving a 1,3-dipolar cycloaddition via a multistep synthesis in high stereoselectivity [75]. Briefly, spiro(oxindole-3,3′-pyrrolidine) (IV) was synthesized by asymmetric 1,3-dipolar cycloaddition of the (E)-3-aryl-1,3-dihydro-indol-2-one (I) with isopropyl aldehyde (II) and (5R,6S)-5,6-diphenylmorpholine (III) in toluene. Intermediate compound (IV) on treatment with dimethylamine in THF afforded compound (V) in overall good yield with controlled absolute configuration of the stereocenters of the products (Fig. 6).

Multistep synthesis of spiro-pyrrolidine oxindole derivatives
Computational modeling studies predicted that the oxindole nucleus of compound (V) inserts into the Trp23 pocket in p53 and the spiro-pyrrolidine ring fits the side chain of Phe19 and Leu26 [92]. The phenyl group occupies the Phe19 pocket, but the isopropyl group fitted poorly into the binding pocket occupied by Leu26. Compound MI-17 was designed and found to be 100 times more potent than the initial compound as the 3-chloro substituent fits perfectly into the MDM2 binding pocket [84]. Though this chemical optimization achieved high binding affinities they were still significantly less potent than most peptide-based inhibitors. Wang then attempted to modify this class of compounds to also fit into p53’s Leu22 pocket. Since Leu22 is not buried deeply into the MDM2 cleft and is partially exposed to solvent, the addition of a polar moiety was explored to introduce pharmacophores to improve the physiochemical properties of the compounds such as aqueous solubility. Computational studies of the spiro-oxindole scaffold were done using the Leu22 as an additional pharmacophore site. Design of MI-43 closely mimics Leu22 at the two-carbon linker between the morpholine ring and the 5′-carbonyl group. In addition, the oxygen of the morpholine ring seems to be in close proximity to the charged amine group in Lys90; the positively charged Lys90 is thought to have a charge–charge interaction with the carbonyl group in p53’s Glu17 [87]. Co-crystal analysis showed that the oxindole group occupies the Trp23 pocket and also forms a hydrogen bond to MDM2’s Leu54 [93]. MI-43 showed high selectivity towards cancerous cells possessing wild-type p53. It inhibited cell growth of LNCaP human prostate cancer cells with an IC50 value of 0.83 μM, but showed higher IC50 values of 22.5 μM in the human prostate cancer cell line and 10.5 μM in healthy human prostate epithelial cells with wild-type p53. The efficacy of MI-43 was tested against two pairs of human lung cancer lines differing in the p53 status: adenocarcinoma A549 (p53 wild-type) and H522 (p53-null) and non-small cell lung carcinoma H460 (p53 wild-type) and H1299 (p53-null). Interestingly, MI-43 is much less toxic to normal fetal lung fibroblast MRC5 cells. Finally, when used in combination, MI-43 sensitized chemoresistant A549 cells to etoposide-induced apoptosis [94]. Despite its potent in vitro antitumor activity, specificity, and well-defined mechanism of action, MI-61 showed a significantly decreased activity (K i > 10 µM), indicating that the substituent on the oxindole core played an important role in its binding affinity to MDM2 [95]. MI-43 lacks the desirable pharmacokinetic profile and is unsuitable for in vivo evaluations in animal models of human cancer. Further optimization of MI-43 led to the more potent inhibitor MI-63, which exhibited increased potency in LNCaP cytotoxicity assay with an IC50 of 0.28 μM in p53-null tumor cells [78]. Though MI-43 and MI-63 had shown great binding affinity and potent cytotoxic properties, they were poor candidates for in vivo evaluation because of their poor pharmacokinetic profiles [87] (Fig. 7).

Development of the MI series of spiro-oxindoles as MDM2 inhibitors
Some improved analogues like MI-122, MI-126, and M-142 were synthesized by alteration in the side chain and evaluated to be promising orally active inhibitors of the p53–MDM2 interaction [88]. MI-147, being the most potent compound in the series, was found to activate p53 at concentrations as low as 40 nM and selectively arrest cell growth in tumor cells with wild-type p53 with an IC50 value of 16.4 nM [88]. Furthermore, MI-147 showed good oral bioavailability and remarkable anticancer activity in the SJSA-1 xenograft model in an FP-based competitive binding assay (Table 1). To further improve the oral bioavailability and pharmacokinetic properties of MI-147, compound MI-219 was designed with higher binding affinity to MDM2 and is more than 1000-fold more potent than p53 peptide (Fig. 7). This compound effectively inhibited cell growth in SJSA-1, LNCaP, and 22Rv1 prostate cancer cell lines that express wild-type p53, with IC50 values in the range of 0.4–0.8 μM, but showed moderate activity in p53-deficient cell lines, confirming that the cell growth inhibitory activity is p53-dependent [38]. MI-219 has an oral bioavailability of 65% in rats. Immunohistochemical analysis showed that a single dose of MI-219 induced strong accumulation of p53 in the SJSA-1 tumor. Toxicity studies also showed that though it induces p53 activity in normal cells it does not induce apoptosis, thus confirming that only tumors remain vulnerable to p53 tumor suppressor functionality and showing the therapeutic potential of MI-219 in the treatment of pancreatic cancer and its molecular mechanism of action [88]. The efficacy of MI-219 was tested against a panel of lung cancer cell lines alone or in combination. When acting alone, MI-219 selectively inhibited growth of wild-type p53-containing lung cancer cells. MI-219 in combination with cisplatin induced cell growth inhibition and apoptosis in pancreatic cancer cells irrespective of their p53 mutational status [96]. While both γ radiation and irinotecan chemotherapy induce vigorous apoptosis in either single or repeated doses, MI-219 did not cause apoptosis or damage in either radiosensitive or radioresistant normal mouse tissues. MI-219 potently inhibited cell growth in cancer cells with wild-type p53 (IC50 values ca. 1 mM) and displayed more than 10-fold selectivity in tumor cells with mutated or deleted p53. Although MI-219 failed to achieve tumor regression, it achieved complete tumor growth inhibition in multiple xenograft models of human cancer. It also activated p53 in normal mouse tissues but caused no tissue damage even with repeated dosing [87]. The in vitro and in vivo data provided clear evidence of MI-219 being a potent and specific MDM2 inhibitor.
However, as a result of some shortcomings (listed in Table 1), optimization of MI-219 led to a more potent analogue, MI-319, in which the two phenyl groups adopt a trans configuration, which exhibited the best results for in vitro antiproliferative activity against a follicular small cleaved B cell lymphoma line and some wild-type p53 cell lines with good MDM2 binding affinity with an IC50 of 196 nM. Oral administration of MI-319 at 300 mg/kg for 14 days resulted in significant tumor growth inhibition without any observed toxicity to the animals [97]. In a mouse model for follicular small cleaved cell lymphoma (FSCCL), MI-319 was tolerated well by the animals, displayed effectiveness against FSCCL cells in blood, brain, and bone marrow, and achieved significant therapeutic impact by conferring a greater than 28% increase in host life-span (by 14.4 days) in the treatment group [98]. Thus, MI-319 exhibited potent activity and was pursued for treatment against follicular lymphoma and pancreatic cancer that retain wild-type p53 both in vitro and in vivo. Further optimization by side chain modification yielded compound MI-888. MI-888 has an excellent pharmacokinetic profile and is highly potent and selective in inhibition of tumor cell growth in cancer cell lines with wild-type p53 over those with mutated or deleted p53 (Fig. 7). MI-888 is capable of achieving complete and durable tumor regression in two animal models of human cancer, i.e., SJSA-1 osteosarcoma and acute lymphoblastic leukemia tumor xenograft models, upon daily oral administration [99]. When MI-888 was administered at 100 and 200 mg/kg via oral gavage daily for 3 weeks, rapid and complete tumor regression was achieved. After 4 days of dosing, tumor volume was decreased by 61 and 80% with the treatment at 100 and 200 mg/kg, respectively. Complete tumor regression was observed in all the animals after 7 days of dosing with MI-888. Even 26 days after the last dose, all mice treated with MI-888 at 200 mg/kg remained tumor-free [99]. There was no significant weight loss or other signs of toxicity for all the mice treated during the entire experiment.
A more potent analogue of MI-888 called MI-773 was designed by optimization of the amide side chain of MI-888 [74, 100]. The trans configuration of two substituted phenyl rings was found to be critical for improved MDM2 binding affinity [74]. The newly developed enantiomer MI-773-01 displayed 10 times higher binding affinity against MDM2 than MI-773 did. The antitumor activity of MI-773-01 (SAR405838) was more pronounced in a set of wild-type p53 xenograft models including osteosarcoma, human prostate, melanoma, colorectal tumor, and acute lymphoblastic leukemia [77]. Compound MI-773-01 (SAR405838) was advanced into phase I clinical trials by Sanofi in 2012 and results showed that the treatment of SJSA-1 osteosarcoma cell lines with SAR405838 in vitro leads to dose-dependent cell growth inhibition, cell cycle arrest, and robust apoptosis [86]. A second phase I trial was initiated in 2013 to evaluate MI-773-01 in combination with pimasertib, an allosteric inhibitor of MEK1/2 in patients with solid tumors (ClinicalTrials.gov identifiers for MI-773-01/SAR405838: NCT01636479 and NCT01985191). The phase I clinical trial results for MI-773-01 have not been published. Recent in vivo studies of SAR405838 demonstrated promising oral pharmacokinetics in mice, achieving dose-dependent and sustained p53 activation in extracted tumor tissues by successfully penetrating human xenografts and eliciting significant antitumor responses in a dose-dependent manner [101]. Thus, SAR405838 holds remarkable promise as a highly effective candidate therapeutic for patients suffering from dedifferentiated liposarcoma.
6.2 Spiro-piperidinone/Pyridinone-Based Inhibitors
The rigidity of the spiro-oxindole scaffold has provided many opportunities to design analogues with high affinity towards MDM2 [74]. In recent years, instead of using a five-membered pyrrolidine ring to project the hydrophobic side chains into the Leu and Phe pockets, a six-membered delta-lactam ring is utilized [102, 103] (Fig. 8). Researchers at Hoffmann-La Roche reported a high number of spiro-piperidine-based compounds in several patent applications [104, 105]. The ability of these compounds to inhibit the interaction between p53 and MDM2 proteins was measured by an HTRF assay in which recombinant GST-tagged MDM2 binds to a peptide that resembles the MDM2-interacting region of p53. The compounds were synthesized by heating substituted 3-benzylidene 2-oxindole with substituted 1-phenyl-3-trimethylsilyoxy--aza-1,3-butadiene in toluene under nitrogen at 110 °C, and most of the compounds in this series showed good MDM2 inhibitory activity with IC50 values less than about 10 µM [105]. Compounds 1 and 2 exhibited IC50 values of 0.48 and 0.37 µM, respectively [106]. The most potent compound has an m-chlorophenyl group on the pyridine ring and gave an IC50 value of 0.066 µM. Optimization of this series has been reported in successive patent applications by Zhang and co-workers from Hoffmann-La Roche [107]. Compounds 3 and 4 in this series are active in the submicromolar range with IC50 values of 0.05 and 0.034 µM, respectively [108] (Fig. 8). The potent spiro-pyridine oxindole derivative 5, in which the m-chlorophenyl ring is replaced with a substituted pyridine ring, showed good biological activity as an MDM2 antagonist with an IC50 around 0.1 μM [109]. Further reaction of 2 and similar compounds with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in toluene under heating at 110 °C afforded spiro-pyridine derivatives 6. The nitrogen can also be linked with the carbonyl to give a fused heterocyclic system [110] (Fig. 8). Compound 6 in which the m-chlorophenyl ring on the piperidine ring is replaced with other heteroaryl rings have shown very good activity as small molecule inhibitors of the MDM2–p53 interaction [109]. Thiazine-fused pyridine systems (e.g., compound 7) are also found to be promising derivatives of spiro-oxindoles with IC50 values in the sub-10 μM range [111].

Various spiro-oxindoles active as p53–MDM2 inhibitors
6.3 Spiro-cyclohexanone-Based Inhibitors
These compounds, in which the oxindole ring is fused with a cyclohexane ring, were prepared by Diels–Alder reaction of substituted 3-benzylidene-2-oxindoles with the respective silyl enol ether [108]. The ability of these compounds to inhibit the interaction between p53 and MDM2 protein was measured by an HTRF assay and the IC50 values of compounds of types 8 and 9 were found to be less than 10 µM (Fig. 8).
6.4 Spiro-thiazolidine-Based Inhibitors
5-Bromo-3′-(cyclohexane carbonyl)-1-methyl-2-oxospiro[indoline-3,2′-thiazolidine] (10) is the most potent compound in this series, inhibiting, in vitro, 30% of p53–MDM2 interaction and the cell growth of different human tumor cells at nanomolar concentrations [112] (Fig. 8). Docking studies confirmed the interactions of 10 with the well-known Trp23 and Phe19 clefts, thereby explaining the reasons for its binding affinity for MDM2. Compound 10 at 50 nM concentration is capable of inducing the accumulation of p53 protein, inducing significant apoptotic cell death without affecting the cell cycle progression. Research to identify more potent and selective spiro-thiazolidine-based derivatives and to better understand the mechanisms of their inhibition of tumor growth is currently underway. Some spiro(imidazo[1,5-c]thiazole-3,3′-indoline)-2,5,7-trione derivatives are also reported to inhibit cell growth in a panel of human tumor cells at submicromolar or micromolar concentrations [113]. In particular, compound 11 showed high efficacy in HEK-293 (kidney), M14 (melanoma), and U937 (leukemia) human cell lines with IC50 values of 0.44, 0.53, and 0.87 μM respectively. Compound 12, containing a trimethoxy benzyl group at the N-6 position, was found 3- to 5-fold less cytotoxic in all tested cell lines and showed a time-dependent activity different from that of 11. However, both these compounds induced a time-dependent increase in p53 expression, indicating that the activity profiles of derivatives might be regulated by this protein, thus demonstrating their ability to block the p53–MDM2 interaction and thereby restoring p53 activity.
6.5 Spiro-isoxazoline-Based Inhibitors
Spiro-isoxazoline oxindoles represent another class of inhibitors based on the spiro fusion of an isoxazoline ring to an oxindole scaffold (Fig. 8). The disruption of the p53–MDM2 complex by this class of compounds was demonstrated in a live-cell bimolecular fluorescence complementation assay [114]. The structure–activity relationship (SAR) study showed that the substituents on the three phenyl rings played an important role in the MDM2 inhibitory activity; compounds with no substituent on the phenyl rings were inactive. A chloro or bromo atom at position 6 of the oxindole moiety was beneficial to the activity and afforded the best cytotoxicity against HepG2 cells with IC50 values of 29.11 µM for compound 13 and 31.9 µM for compound 14. Different substituents like methoxy, nitro, and methyl at the para position of the phenyl ring yielded more active compounds 15. These derivatives were found to inhibit p53–MDM2 interaction in a cell-based bimolecular fluorescence complementation assay. Most of them showed IC50 values around 30 µM.
6.6 Spiro-azepanone-Based Inhibitors
Spirocyclohexane-2-oxindole derivatives 9 are converted to respective seven-membered spiro-azepine-2-oxindoles (e.g.16) via titanium chloride mediated ring expansion [115] (Fig. 8). These compounds are also found to possess IC50 values less than 10 µM. The ability of these compounds to inhibit the interaction between p53 and MDM2 proteins was measured by an HTRF assay in which recombinant GST-tagged MDM2 binds to a peptide that resembles the MDM2-interacting region of p53 [80].
Since most of these spiro-oxindole-based MDM2 inhibitors have only been disclosed in patents, little is known about their cellular activity, pharmacological properties, and mechanism of action.
7 Conclusions
The interaction between MDM2 and p53 represents a promising target for the development of novel anticancer drugs that could be targeted by small molecule inhibitors. Exploitation of spiro-oxindole scaffolds for restoration of p53 is a rapidly evolving area of cancer treatment by effective use of computational, structure-based design tools. Knowledge of the molecular mechanism of spiro-oxindole binding to MDM2 greatly helped in understanding how substituents on the small molecule should be optimized for effective binding to the three subpockets of the p53–MDM2 complex. Complete tumor regression achieved by spiro-oxindoles boosts their therapeutic potential for the treatment of human cancers. The anticancer potency of spiro-oxindoles mainly depends on the type of cyclic ring fused on the C3 position of oxindole core and the stereochemistry. Chemical modification at the C3 position of the spiro-oxoindole system is an effective approach to study the modulation of p53 activity through inhibition of p53–MDM2 protein interaction. Among them, spiro-pyrrolidinyl oxindoles are the most potent and selective inhibitors of p53–MDM2 interaction. The relative configuration of two substituted phenyl rings and structural modifications in the side chain play important roles in the optimization of the pharmacokinetic properties of the molecule. Small substituents attached at position 5 and/or 6 of the oxindole core also play a critical role in the resultant biological activity.
At present, many derivatives of spiro-oxindoles with very high affinities to MDM2 (K i < 1 nM) and optimal pharmacokinetic properties have been developed but there are numerous spiro-oxindoles with structural novelty reported in the literature that are not subjected to anticancer evaluation, thereby restricting the discovery of new anticancer agents. So, there is still a great need and opportunity for medicinal and organic chemists to further explore the biological activity of this scaffold through extensive SAR efforts. Compiling the data on p53 activator spiro-oxindoles is useful for both organic and medicinal chemists for further design and development of novel highly potent and selective anticancer agents.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) CA Cancer J Clin 65:87–108
WHO (2014) World cancer report, Chap 1.1. WHO, Geneva
Chabner BA, Roberts TG Jr (2005) Nat Rev Cancer 5:65–72
Lind MJ (2011) Principles of cytotoxic chemotherapy. Medicine 39(12):711–716
Rheingold SR, Neugut AI, Meadows AT (2003) Secondary cancers: incidence, risk factors, and management. Chapter 159. In: Kufe DW, Pollock RE, Weichselbaum RR et al (eds) Holland-Frei cancer medicine, 6th edn. BC Decker, Hamilton
Schreiber SL (2003) Chem Eng News 81:51–61
Bae YH, Mrsny RJ, Park K (2013) Cancer targeted drug delivery: an elusive dream. Springer, Berlin
Aggarwal S (2010) Nat Rev Drug Discov 9:427–428
Chessum N, Jones K, Pasqua E, Tucker M (2015) Prog Med Chem 54:1–63
Stratton MR (2011) Science 331:1553–1558
Collins I, Workman P (2006) Nat Chem Biol 2:689–700
Kummar S, Murgo AJ, Tomasze JE, Doroshow JH (2014) Therapeutic targeting of cancer cells: era of molecularly targeted agents. Elsevier Churchill Livingstone, Philadelphia
Gerber DE (2008) Am Fam Physician 77:311–319
Lacroix M (2014) Targeted therapies in cancer. Nova Sciences, Hauppauge. ISBN 978-1-63321-687-7
Tanner JE (2005) Cancer Metastasis Rev 24:585–598
Avendano C, Menendez JC (2015) Medicinal chemistry of anticancer drugs, 2nd edn. Elsevier, Amsterdam
Zhao Y, Bernard D, Wang S (2013) BioDiscovery 8(4):1–15
Dickens MP, Fitzgerald R, Fischer PM (2010) Semin Cancer Biol 20:10–18
Zhao Y, Aguilar A, Bernard D, Wang S (2015) J Med Chem 58:1038–1052
Vanneman M, Dranoff G (2012) Nat Rev Cancer 12:237–251
Masters GA, Krilov L, Bailey HH, Brose MS, Burstein H, Diller LR, Dizon DS, Fine HA, Kalemkerian GP, Moasser M, Neuss MN, O’Day SJ, Odenike O, Ryan CJ, Schilsky RL, Schwartz GK, Venook AP, Wong SL, Patel JD (2015) J Clin Oncol 33:786–809
Abramson R (2015) Overview of targeted therapies for cancer. My Cancer Genome. https://www.mycancergenome.org/content/molecular-medicine/overview-of-targeted-therapies-for-cancer/. Accessed 1 Jan 2016
Mullard A (2015) Nat Rev Drug Discov 14:77–81
Wang S, Zhao Y, Bernard D, Aguilar A, Kumar S (2012) Top Med Chem 8:57–80
Lane DP, Cheok CF, Lain S (2010) Cold Spring Harb Perspect Biol 2:a001222
Vogelstein B, Lane D, Levine AJ (2000) Nature 408:307–310
Moll UM, Petrenko O (2003) Mol Cancer Res 1:1001–1008
Fu T, Min H, Xu Y, Chen J, Li G (2012) Int J Mol Sci 13:9709–9740
Vu BT, Vassilev L (2011) Curr Top Microbiol Immunol 348:151–172
Picksley SM, Lane DP (1993) BioEssays 15:689–690
Wade M, Wang YV, Wahl GM (2010) Trends Cell Biol 20:299–309
Momand J, Wu HH, Dasgupta G (2000) Gene 242:15–29
Hainaut P, Hollstein M (2000) Adv Cancer Res 77:81–137
Vousden KH, Lu X (2002) Nat Rev Cancer 2:594–604
Riedinger C, McDonnell JM (2009) Future Med Chem 1:1075–1094
Shangary S, Wang S (2009) Annu Rev Pharmacol Toxicol 49:223–241
Chene P (2003) Nat Rev Cancer 3:102–109
Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S (2008) Proc Natl Acad Sci USA 105:3933–3938
Zhang R, Wang H, Agrawal S (2005) Curr Cancer Drug Targets 5:43–49
Herman AG, Hayano M, Poyurovsky MV, Shimada K, Skouta R, Prives C, Stockwell BR (2011) Cancer Discov 1:312–325
Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G (2008) Genes Dev 22:1337–1344
Hoelder S, Clarke PA, Workman P (2012) Mol Oncol 6:155–176
Azmi AS, Beck FW, Sarkar FH, Mohammad RM (2011) Curr Pharm Des 17:640–652
Zheng T, Wang J, Song X, Meng X, Pan S, Jiang H, Liu L (2010) J Cancer Res Clin Oncol 136:1597–1604
Wade M, Li YC, Wahl GM (2013) Nat Rev Cancer 13:83–96
Millard M, Pathania D, Grande F, Xu S, Neamati N (2011) Curr Pharm Des 17:536–559
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Science 274:948–953
Vazquez A, Bond EE, Levine AJ, Bond GL (2008) Nat Rev Drug Discov 7:979–987
Pujals A, Favre L, Gaulard P, Wiels J (2015) Activation of wild-type p53 by MDM2 inhibitors: a new strategy for lymphoma treatment. Blood Lymphat Cancer 5:93–100
Khoo KH, Verma CS, Lane DP (2014) Nat Rev Drug Discov 13:217–236
Wade M, Wahl GM (2009) Mol Cancer Res 7:1–11
Qin JJ, Wang W, Voruganti S, Wang H, Zhang WD, Zhang R (2015) Oncotarget 6:2623–2640
Qin JJ, Nag S, Voruganti S, Wang W, Zhang R (2012) Curr Med Chem 19:5705–5725
Bottger V, Bottger A, Howard SF, Picksley SM, Chene P, Garcia-Echeverria C, Hochkeppel HK, Lane DP (1996) Oncogene 13:2141–2147
Kritzer JA, Lear JD, Hodsdon ME, Schepartz A (2004) J Am Chem Soc 126:9468–9469
Zhang Q, Zeng SX, Lu H (2014) Subcell Biochem 85:281–319
Aeluri M, Chamakuri S, Dasari B, Guduru SK, Jimmidi R, Jogula S, Arya P (2014) Chem Rev 114:4640–4649
Carry JC, Garcia-Echeverria C (2013) Bioorg Med Chem Lett 23:2480–2485
Vassilev Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA (2004) Science 303:844–848
Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, To KH, Linn M, Podlaski F, Wovkulich P, Vu B, Vassilev LT (2013) Cancer Res 73:2587–2597
Ray-Coquard I, Blay JY, Italiano A, Le CA, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, Nichols GL, Bui BN (2012) Lancet Oncol 13:1133–1140
Rew Y, Sun D, De Gonzalez-Lopez TF, Bartberger MD, Beck HP, Canon J, Chen A, Chow D, Deignan J, Fox BM, Gustin D, Huang X, Jiang M, Jiao X, Jin L, Kayser F, Kopecky DJ, Li Y, Lo MC, Long AM, Michelsen K, Oliner JD, Osgood T, Ragains M, Saiki AY, Schneider S, Toteva M, Yakowec P, Yan X, Ye Q, Yu D, Zhao X, Zhou J, Medina JC, Olson SH (2012) J Med Chem 55:4936
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, De Gonzalez-Lopez TF, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu JJ, Lo MC, Long AM, Lucas B, McGee LR, McIntosh J, Mihalic J, Oliner JD, Osgood T, Peterson ML, Roveto P, Saiki AY, Shaffer P, Toteva M, Wang Y, Wang YC, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Yu M, Zhao X, Zhou J, Zhu J, Olson SH, Medina JC (2014) J Med Chem 57:1454–1472
Gonzalez AZ, Li Z, Beck HP, Canon J, Chen A, Chow D, Duquette J, Eksterowicz J, Fox BM, Fu J, Huang X, Houze J, Jin L, Li Y, Ling Y, Lo MC, Long AM, McGee LR, McIntosh J, Oliner JD, Osgood T, Rew Y, Saiki AY, Shaffer P, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Zhao X, Zhou J, Olson SH, Sun D, Medina JC (2014) J Med Chem 57:2963–2988
Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R (2015) Mol Cancer Ther 14:649–658
Raboisson P, Marugan JJ, Schubert C, Koblish HK, Lu T, Zhao S, Player MR, Maroney AC, Reed RL, Huebert ND, Lattanze J, Parks DJ, Cummings MD (2005) Bioorg Med Chem Lett 15:1857–1861
Marugan JJ, Leonard K, Raboisson P, Gushue JM, Calvo R, Koblish HK, Lattanze J, Zhao S, Cummings MD, Player MR, Schubert C, Maroney AC, Lu T (2006) Bioorg Med Chem Lett 16:3115–3120
Allen JG, Bourbeau MP, Wohlhieter GE, Bartberger MD, Michelsen K, Hungate R, Gadwood RC, Gaston RD, Evans B, Mann LW, Matison ME, Schneider S, Huang X, Yu D, Andrews PS, Reichelt A, Long AM, Yakowec P, Yang EY, Lee TA, Oliner JD (2009) J Med Chem 52:7044–7053
Beck HP, DeGraffenreid M, Fox B, Allen JG, Rew Y, Schneider S, Saiki AY, Yu D, Oliner JD, Salyers K, Ye Q, Olson S (2011) Bioorg Med Chem Lett 21:2752–2755
Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD (2005) Angew Chem Int Ed Engl 44:2704–2707
Hardcastle IR, Ahmed SU, Atkins H, Farnie G, Golding BT, Griffin RJ, Guyenne S, Hutton C, Kallblad P, Kemp SJ, Kitching MS, Newell DR, Norbedo S, Northen JS, Reid RJ, Saravanan K, Willems HM, Lunec J (2006) J Med Chem 49:6209–6221
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev LT, Graves B (2013) J Med Chem 56:5979–5983
Ma Y, Lahue BR, Shipps GW Jr, Brookes J, Wang Y (2014) Bioorg Med Chem Lett 24:1026–1030
Zak K, Pecak A, Rys B, Wladyka B, Domling A, Weber L, Holak TA, Dubin G (2013) Expert Opin Ther Pat 23:425–448
Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, Parrish DA, Deschamps JR, Wang S (2005) J Am Chem Soc 127:10130–10131
Chen L, Han X, Yang S, Zhang Z (2012) 3,3′-Spiroindolinone derivatives and their use for cancer. Patent No. EP2421866A1
Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, Stuckey JA, Meagher JL, Bai L, Liu L, Hoffman-Luca CG, Lu J, Shangary S, Yu S, Bernard D, Aguilar A, Dos-Santos O, Besret L, Guerif S, Pannier P, Gorge-Bernat D, Debussche L (2014) Cancer Res 74:5855–5865
Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S (2006) J Med Chem 49:3432–3435
Yu B, Yu DQ, Liu HM (2015) Eur J Med Chem 97:673–698
Weber L (2010) Expert Opin Ther Pat 20:179–191
Kamal A, Mohammed AA, Shaik TB (2012) Expert Opin Ther Pat 22:95–105
Czarna A, Popowicz GM, Pecak A, Wolf S, Dubin G, Holak TA (2009) Cell Cycle 8:1176–1184
Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J (2006) J Biol Chem 281:33030–33035
Ding K, Wang G, Deschamps JR, Parrish DA, Wang S (2005) Tetrahedron Lett 46:5949–5951
Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD (2003) Proteins 52:609–623
Wang S, Zhao Y, Sun W, Kumar S, Leopold L, Debussche L, Barriere C, Carry J-C, Amaning K (2014) Spiro-oxindole MDM2 antagonists. US 14/170,101 (US20140148494A1)
Shangary S, Ding K, Qiu S, Nikolovska-Coleska Z, Bauer JA, Liu M, Wang G, Lu Y, McEachern D, Bernard D, Bradford CR, Carey TE, Wang S (2008) Mol Cancer Ther 7:1533–1542
Yu S, Qin D, Shangary S, Chen J, Wang G, Ding K, McEachern D, Qiu S, Nikolovska-Coleska Z, Miller R, Kang S, Yang D, Wang S (2009) J Med Chem 52:7970–7973
Huang W, Cai L, Chen C, Xie X, Zhao Q, Zhao X, Zhou HY, Han B, Peng C (2016) J Biomol Struct Dyn 34:341–351
Aguilar A, Sun W, Liu L, Lu J, McEachern D, Bernard D, Deschamps JR, Wang S (2014) J Med Chem 57:10486–10498
Ball-Jones NR, Badillo JJ, Franz AK (2012) Org Biomol Chem 10:5165–5181
Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997) J Mol Biol 267:727–748
Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, Domling A, Holak TA (2010) Cell Cycle 9:1104–1111
Khan A, Lu H (2008) Cancer Biol Ther 7:853–855
Sun SH, Zheng M, Ding K, Wang S, Sun Y (2008) Cancer Biol Ther 7:845–852
Azmi AS, Aboukameel A, Banerjee S, Wang Z, Mohammad M, Wu J, Wang S, Yang D, Philip PA, Sarkar FH, Mohammad RM (2010) Eur J Cancer 46:1122–1131
Azmi AS, Philip PA, Aboukameel A, Wang Z, Banerjee S, Zafar SF, Goustin AS, Almhanna K, Yang D, Sarkar FH, Mohammad RM (2010) Curr Cancer Drug Targets 10:319–331
Mohammad RM, Wu J, Azmi AS, Aboukameel A, Sosin A, Wu S, Yang D, Wang S, Al-Katib AM (2009) Mol Cancer 8:115
Zhao Y, Yu S, Sun W, Liu L, Lu J, McEachern D, Shargary S, Bernard D, Li X, Zhao T, Zou P, Sun D, Wang S (2013) J Med Chem 56:5553–5561
Wang S, Sun W, Yu S et al (2011) Highly potent and optimized small-molecule inhibitors of MDM2 achieve complete tumor regression in animal models of solid tumors and leukemia. Abstract LB-204. AACR 102nd annual meeting, Orlando, FL
Bill KL, Garnett J, Meaux I, Ma X, Creighton CJ, Bolshakov S, Barriere C, Debussche L, Lazar AJ, Prudner BC, Casadei L, Braggio D, Lopez G, Zewdu A, Bid H, Lev D, Pollock RE (2016) Clin Cancer Res 22:1150–1160
Chen L, Ding Q, Liu J, Yang S, Zhang Z, Hoffmann-La RIU (2009) Spiroindolinone derivatives. US7495007 B2
Chen L, Han X, Yang S, Zhang Z (2010) Preparation of 3,3′-spiroindolinones for treatment of cancer. WO 2010121995 A1
Chen L, Han X, He Y, Yang S, Zhang Z. Hoffman-La Roche Inc, USA (2009) Spiroindolinone derivatives as interaction inhibitors between p53 and MDM2 proteins and their preparation, pharmaceutical compositions and use in the treatment of cancer. US 20090163512 A1
Ding Q, Liu JJ, Zhang Z. Zhang Z, Hoffmann La Roche, Switzerland (2007) Spiroindolinone derivatives. WO 2007104714 A1
Chen L, Ding Q, Liu JJ, Yang S, Zhang Z (2007) Preparation of spiroindolinone derivatives as antitumor agents. US 20070213341 A1
Ding Q, Jiang N, Yang S, Zhang J, Zhang Z (2009) Spiroindolinone derivatives. US 20090156610 A1
Liu JJ, Zhang Z. (2009) Spiroindolinone derivatives. US 7638548 B2
Chen L, Han X, Yang S, Zhang Z (2010) Spiroindolinone pyridine derivatives as inhibitors of MDM2–p53 protein interaction useful as potent and selective anticancer agents and preparation thereof. US 20100204257
Zhang J, Zhang Z Spiroindolinone pyridine derivatives as inhibitors of MDM2–p53 protein interaction useful as potent and selective anticancer agents and preparation thereof. US 20100210674
Liu J-J, Zhang Z, Hoffmann-La Roche (2008) Preparation of spiroindole pyridotriazinediones as anticancer drugs. WO 2008141975
Bertamino A, Soprano M, Musella S, Rusciano MR, Sala M, Vernieri E, Di Sarno V, Limatola A, Carotenuto A, Cosconati S, Grieco P, Novellino E, Illario M, Campiglia P, Gomez-Monterrey I (2013) J Med Chem 56:5407–5421
Gomez-Monterrey I, Bertamino A, Porta A, Carotenuto A, Musella S, Aquino C, Granata I, Sala M, Brancaccio D, Picone D, Ercole C, Stiuso P, Campiglia P, Grieco P, Ianelli P, Maresca B, Novellino E (2010) J Med Chem 53:8319–8329
Ribeiro CJ, Amaral JD, Rodrigues CM, Moreira R, Santos MM (2014) Bioorg Med Chem 22:577–584
Liu J-J, Tilley JW, Zhang Z (2008) 3,3-Spiroindolinone derivatives. US 12/101182 US20080287421 A1
Acknowledgements
AKG acknowledges the Department of Science and Technology (DST, New Delhi) for financial support under the Women Scientist Project Scheme (WOS-A).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Alpana K. Gupta, Mausumi Bharadwaj, Anoop Kumar, and Ravi Mehrotra declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Gupta, A.K., Bharadwaj, M., Kumar, A. et al. Spiro-oxindoles as a Promising Class of Small Molecule Inhibitors of p53–MDM2 Interaction Useful in Targeted Cancer Therapy. Top Curr Chem (Z) 375, 3 (2017). https://doi.org/10.1007/s41061-016-0089-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-016-0089-0