
Abstract
The reduction of 4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones with aqueous hydrazine hydrate selectively involved the C1=O carbonyl group to give the corresponding 4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones within a few hours. The reduction products were condensed with aldehydes and acetone to afford new 1-[(het)arylmethylidene]- and 1-(propan-2-ylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones in 59–78% yield. The reaction of 4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones with N,N-dimethylformamide dimethyl acetal, followed by transamination with primary amines led to the formation of 1-{[(het)arylamino]methylidene}-4H-pyrrolo[3,2,1-ij]quinolin-2-ones in 65–83% yield. The synthesized compounds were evaluated for their anticoagulant activity by measuring inhibition of blood coagulation factors Xa and XIa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Organic compounds containing a pyrrolo[3,2,1-ij]quinoline fragment often exhibit high biological activity. For example, pyrrolo[3,2,1-ij]quinoline derivatives were reported as antibacterial [1–4] and antitumor agents [5], diuretics [6–7], aldosterone synthase inhibitors [8], melatonin receptor agonists and antagonists [9], and compounds promising for the treatment of human lymphoma [10], diabetes [11], asthma [12], epilepsy, and obesity [13].
We recently found that some 4H-pyrrolo[3,2,1-ij]quinolin-2-one derivatives displayed inhibitory activity against blood coagulation factors Xa and XIa [14–18]. Therefore, search for new blood coagulation factors among derivatives of 4H-pyrrolo[3,2,1-ij]quinolin-2-ones is of significant interest. Target-oriented synthesis of differently substituted 4H-pyrrolo[3,2,1-ij]quinolin-2-ones opens wide possibilities for functional diversification of this scaffold, which could considerably affect the type and strength of physiological action of the resulting compounds.
Thus, the present work was aimed at synthesizing 4H-pyrrolo[3,2,1-ij]quinolin-2-one derivatives functionalized at the 1-position and evaluating their in vitro inhibitory activity against blood coagulation factors Xa and XIa.
RESULTS AND DISCUSSION
4H-Pyrrolo[3,2,1-ij]quinolin-2-ones can be synthesized by cyclization of 1- and 8-substituted quinolines [19–26]. One of the most widely used methods is based on the intramolecular Friedel–Crafts alkylation of haloacetyl derivatives of quinoline. For example, 1,2,5,6-tetrahydro-4H-pyrrolo[3,2,1-ij]quinolin-2-one was synthesized by cyclization of 1-chloroacetyl-1,2,3,4-tetrahydroquinoline in the presence of aluminum chloride in o-dichlorobenzene [21–22]. However, the reaction with 1-haloacetyl-2,2,4-trimethyl-1,2-dihydroquinoline under these conditions can be complicated by concurrent alkylation at the 4-position. On the other hand, selective reduction of 4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones with aqueous hydrazine hydrate is a convenient one-step method of synthesis of 4H-pyrrolo[3,2,1-ij]quinolin-2-ones. This procedure was used previously to obtain derivatives of 2,3-dihydro-1H-indol-2-one and its N-alkyl analogs [27–30].
Like isatins, 4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones 1a–1d were selectively reduced with hydrazine hydrate in water on heating under reflux for 2 h to give 67–82% of 4H-pyrrolo[3,2,1-ij]quinolin-2-ones 2a–2d (Scheme 1). Compounds 2a–2d are white or pale yellow solids readily soluble in chloroform, acetone, isopropyl alcohol, and N,N-dimethylformamide. Unlike initial compounds 1a–1d, the 1H NMR spectra of 2a–2d showed a singlet at δ ~3.43–3.45 ppm due to methylene protons on C1. In the 13C NMR spectra of 2b and 2d, the C1=O signal at δC 160 ppm disappeared, and C1H2 signal appeared at δC 56 ppm.

1.
4H-Pyrrolo[3,2,1-ij]quinolin-2-ones 2a–2d possess an active methylene group, which makes it possible to involve them in well-known condensations with various carbonyl compounds by analogy with [27–29, 31–34], so that new 1-substituted derivatives could be obtained. We examined reactions of 4H-pyrrolo[3,2,1-ij]quinolin-2-ones 2a–2c with aromatic and heterocyclic aldehydes, as well as with acetone. The reactions were carried out by heating the reactants in boiling ethanol in the presence of piperidine as base catalyst for 2–5 h. After standard workup, we isolated 1-[(het)arylmethylidene]- and 1-(propan-2-ylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones 4a–4g in good yields (59–78%; Scheme 2). Compounds 4a–4g were isolated as bright red or orange powders.

2.
The structure of 4a–4g was confirmed by 1H NMR spectra. The spectra of 4a–4e lacked methylene proton signal at δ ~3.4 ppm, but a singlet appeared in the region δ 7.58–8.15 ppm due to the exocyclic C=CH proton together with additional aromatic protons at δ 7.1–8.8 ppm. In the spectra of 4f and 4g we observed new singlets at δ 2.27–2.29 ppm corresponding to protons of the isopropylidene group on C1.
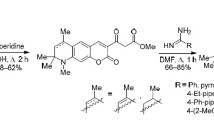
N,N-Dimethylformamide dimethyl acetal (DMF-DMA) as a one-carbon building block is widely used in the synthesis of heterocyclic systems. The reaction of pyrrolo[3,2,1-ij]quinolin-2-ones with DMF-DMA in boiling o-xylene readily afforded the corresponding 1-(dimethylamino)methylidene derivatives which underwent transamination with primary amines to produce 1-{[(het)arylamino]methylidene}-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones 5a–5f. The reactions with amines were carried out in o-xylene under reflux in the presence of acetic acid (Scheme 3).

3.
The reaction with amines was not selective, and most transamination products 5a–5f were isolated as mixtures of Z and E isomers (according to the HPLC/MS and 1H NMR data). Apart from signals typical of initial pyrrolo[3,2,1-ij]quinolin-2-ones 2b and 2c, the 1H NMR spectra of 5a–5f showed signals corresponding to the aromatic part of the amine moiety, the CH=C1 proton resonated as a singlet at δ 7.2–7.3 ppm, and the NH proton appeared as a doublet at δ 9– 11 ppm. Taking into account the obvious possibility for stabilization of the Z isomer by intramolecular hydrogen bonding between the NH proton and C2=O carbonyl oxygen, we presumed that the major isomer has Z configuration. Depending on the substituents, the Z/E ratio ranged from 3:1 to 6:1.
With the goal of finding lead compounds, pyrrolo[3,2,1-ij]quinolin-2-ones 4a–4g and 5a–5f were evaluated for their in vitro inhibitory activity against blood coagulation factors Xa and XIa. Contrary to our expectations, most of the tested compounds showed no anticoagulant activity. Only compounds 4a and 5b moderately inhibited factor Xa (Table 1). These results prompted us to plan further studies aimed at rational molecular design of effective anticoagulants in the series of pyrrolo[3,2,1-ij]quinolin-2-one derivatives.
EXPERIMENTAL
The 1H and 13C NMR spectra were recorded on Bruker DRX-500 and Agilent MR 400+ spectrometers (500 and 400 MHz for 1H and 125 and 101 MHz for 13C, respectively), using DMSO-d6 as solvent and tetramethylsilane as internal standard. HPLC/MS analyses were carried out with an Agilent Infinity 1260 liquid chromatograph equipped with an Agilent 6230 TOF mass-selective detector; HPLC conditions: Poroshell 120 EC-C18 column, 4.6×50 mm, particle size 2.7 μm; eluent A: 0.1% formic acid in acetonitrile; eluent B: 0.1% formic acid in water; gradient elution: (1) A/B 50:50, 3.5 min; (2) A/B (50–100):(50–0), 1.5 min; flow rate 0.4 mL/min; column temperature 28°C; positive electrospray ionization, capillary voltage 3.5 kV; fragmentor voltage +191 V; OctRF +66 V. The melting points were measured on a Stuart SMP30 melting point apparatus. The progress of reactions and the purity of the initial reactants and isolated products were monitored by TLC on Silica gel 60 F254 plates (Merck) using chloroform–methanol (10:1) as eluent; visualization was done under UV light and by treatment with iodine vapor. Initial compounds 1a–1d were synthesized according to the procedure described in [35]. Commercially available reagents were purchased from Acros Organics and VEKTON.
8-R1-4,4,6-Trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones 2a–2d (general procedure). Hydrazine hydrate (64%), 10 mL, was added to 10 mmol of 4,4,6-tetramethyl-4H-pyrrolo[3,2,1-ij]quinoline-1,2-dione 1a–1d, and the mixture was refluxed at 115°C for 1–3 h. After completion of the reaction, the mixture was cooled, and excess hydrazine hydrate was removed under reduced pressure. The residue was treated with water, and the precipitate was filtered off, washed with water, dried, and recrystallized from hexane–ethyl acetate (4:1).
4,4,6-Trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (2a) was synthesized from 2.27 g of 1a. Yield 1.60 g (75%), white powder, mp 117–119°C. 1H NMR spectrum (400 MHz), δ, ppm: 1.55 s (6H, 4-CH3), 1.93 d (3H, 6-CH3, J = 1.2 Hz), 3.41 s (2H, CH2), 5.29 d (1H, 5-H, J = 1.2 Hz), 6.82 d.d (1H, 9-H, J = 7.3, 1.2 Hz), 6.85 d.d (1H, 9-H, J = 7.2, 1.1 Hz), 7.03 t (1H, 8-H, J = 7.5 Hz). Mass spectrum: m/z 214.1232 [M + H]+. C14H15NO. Calculated: M + H 214.1227.
4,4,6,8-Tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (2b) was synthesized from 2.41 g of 1b. Yield 1.52 g (67%), white powder, mp 107–109°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.58 s (6H, 4-CH3), 1.95 s (3H, 6-CH3), 2.24 s (3H, 8-CH3), 3.44 s (2H, CH2), 5.32 s (1H, 5-H), 6.87 s (1H, 7-H), 6.88 s (1H, 9-H). 13C NMR spectrum (125 MHz), δC, ppm: 16.9, 20.9, 27.2, 36.4, 56.1, 117.9, 121.2, 121.7, 124.1, 124.8, 130.1, 130.5, 138.0, 174.6. Mass spectrum: m/z 228.1379 [M + H]+. C15H17NO. Calculated: M + H 228.1384.
8-Methoxy-4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (2c) was synthesized from 2.57 g of 1c. Yield 1.99 g (82%), off-white crystals, mp 112–115°C. 1H NMR spectrum (400 MHz), δ, ppm: 1.54 s (6H, 4-CH3), 1.97 s (3H, 6-CH3), 3.44 s (2H, CH2), 3.73 s (3H, CH3O), 5.32 s (1H, 5-H), 6.89 s (1H, 7-H), 7.06 s (1H, 9-H). Mass spectrum: m/z 244.1336 [M + H]+. C15H17NO2. Calculated: M + H 244.1333.
8-Ethyl-4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (2d) was synthesized from 2.55 g of 1d. Yield 0.1.88 g (78%), off-white powder, mp 135–137°C. 1H NMR spectrum (400 MHz), δ, ppm: 1.13 t (3H, CH2CH3, J = 7.6 Hz), 1.56 s (6H, 4-CH3), 1.94 d (3H, 6-CH3, J = 1.2 Hz), 2.51 q (2H, CH2CH3, J = 7.7 Hz), 3.43 s (2H, CH2), 5.30 d (1H, 5-H, J = 1.2 Hz), 6.84 s (1H, 7-H), 6.89 s (1H, 9-H). 13C NMR spectrum (101 MHz), δC, ppm: 16.7, 17.4, 27.6, 28.7, 36.8, 56.5, 118.3, 120.6, 122.1, 123.4, 125.3, 130.5, 137.7, 138.6, 175.1. Mass spectrum: m/z 242.1544 [M + H]+. C16H19NO. Calculated: M + H 242.1540.
1-Ylidene-4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2-ones 4a–4g (general procedure). 4H-Pyrrolo[3,2,1-ij]quinolin-2-one 2a–2c, 2.4 mmol, was dissolved in ethanol, 2.4 mmol of the corresponding aldehyde or acetone and a catalytic amount of piperidine were added, and the mixture was refluxed for 2–5 h. After completion of the reaction, the mixture was cooled, and the precipitate was filtered off, dried, and recrystallized from isopropyl alcohol.
1-[(1H-Indol-3-yl)methylidene]-4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (4a) was synthesized from 0.51 g of 2a. Yield 0.53 g (65%), yellow powder, mp 234–236°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.71 s (6H, 4-CH3), 2.0 d (3H, 6-CH3, J = 1.3 Hz), 5.41 d (1H, 5-H, J = 1.5 Hz), 6.95 t (1H, 8-H, J = 7.5 Hz), 6.99 d.d (1H, Harom, J = 6.7 Hz), 7.24–7.26 m (2H, Harom), 7.52–7.55 m (1H, Harom), 7.72 d.d (1H, Harom, J = 7.4, 1.1 Hz), 8.15 s (1H, C=CH), 8.16–8.19 m (1H, Harom), 9.44 d (1H, Harom, J = 2.9 Hz), 12.1 s (1H, NH). Mass spectrum: m/z 341.1645 [M + H]+. C23H20N2O. Calculated: M + H 341.1649.
4,4,6,8-Tetramethyl-1-(thiophen-2-ylmethylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (4b) was synthesized from 0.54 g of 2b. Yield 0.58 g (75%), orange powder, mp 200–202°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.63 s (6H, 4-CH3), 1.99 d (3H, 6-CH3, J = 1.4 Hz), 2.33 s (3H, 8-CH3), 5.43 d (1H, 5-H, J = 1.5 Hz), 6.99 s (1H, Harom), 7.32 d.d (1H, Harom, J = 5.1, 3.7 Hz), 7.78 s (1H, C=CH), 7.81– 7.82 m (2H, Harom), 8.00 d (1H, Harom, J = 5.0 Hz). Mass spectrum: m/z 322.1264 [M + H]+. C20H19NOS. Calculated: M + H 322.1261.
4,4,6,8-Tetramethyl-1-(pyridin-3-ylmethylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (4c) was synthesized from 0.54 g of 2b. Yield 0.45 g (59%), orange powder, mp 136–138°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.63 s (6H, 4-CH3), 1.98 s (3H, 6-CH3), 2.31 s (3H, 8-CH3), 5.39 s (1H, 5-H), 6.92 s (1H, Harom), 7.36 s (1H, Harom), 7.48 d.d (1H, Harom, J = 7.8, 5.1 Hz), 7.75 s (1H, C=CH), 8.58 d (1H, Harom, J = 4.8 Hz), 8.83–8.85 m (1H, Harom), 9.15 d (1H, Harom, J = 1.9 Hz). Mass spectrum: m/z 317.1647 [M + H]+. C21H20N2O. Calculated: M + H 317.1649.
1-(2,4-Dimethoxybenzylidene)-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (4d) was synthesized from 0.54 g of 2b. Yield 0.60 g (67%), yellow powder, mp 166–168°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.62 s (6H, 4-CH3), 1.97 d (3H, 6-CH3, J = 1.2 Hz), 2.2 s (3H, 8-CH3), 3.869 s (3H, CH3O), 3.873 s (3H, CH3O), 5.41 d (1H, 5-H, J = 1.5 Hz), 6.69–6.72 m (1H, Harom), 6.70 s (1H, Harom), 6.90 s (1H, Harom), 7.17 s (1H, Harom), 7.64 s (1H, C=CH), 7.66 d.d (1H, Harom, J = 6.5, 2.7 Hz). Mass spectrum: m/z 376.1905 [M + H]+. C24H25NO3. Calculated: M + H 376.1908.
1-(3-Bromobenzylidene)-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (4e) was synthesized from 0.54 g of 2b. Yield 0.69 g (73%), red powder, mp 120–122°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.63 s (6H, 4-CH3), 1.97 d (3H, 6-CH3, J = 1.3 Hz), 2.17 s (3H, 8-CH3), 5.43 d (1H, 5-H, J = 1.5 Hz), 6.95 s (1H, Harom), 7.08 s (1H, Harom), 7.50 t (1H, Harom, J = 7.9 Hz), 7.58 s (1H, C=CH), 7.68– 7.70 m (1H, Harom), 7.71 d (1H, Harom, J = 7.8 Hz), 7.88 s (1H, Harom). Mass spectrum: m/z 394.0806 [M + H]+. C22H20BrNO. Calculated: M + H 394.0802.
4,4,6,8-Tetramethyl-1-(propan-2-ylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (4f) was synthesized from 0.54 g of 2b. Yield 0.50 g (78%), yellow powder, mp 182–184°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.62 s (6H, 4-CH3), 1.98 s (3H, 6-CH3), 2.17 s (3H, 8-CH3), 2.27 s (6H, CH3), 5.41 s (1H, 5-H), 6.91 s (1H), 7.04 s (1H, Harom). Mass spectrum: m/z 268.1895 [M + H]+. C18H21NO. Calculated: M + H 268.1897.
8-Methoxy-4,4,6-trimethyl-1-(propan-2-ylidene)-4H-pyrrolo[3,2,1-ij]quinolin-2-one (4g) was synthesized from 0.58 g of 2c. Yield 0.48 g (71%), yellow powder, mp 163–165°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.60 s (6H, 4-CH3), 1.97 s (3H, 6-CH3), 2.29 s (6H, CH3), 3.75 s (3H, CH3O), 5.40 s (1H, 5-H), 6.65 d (1H, 7-H, J = 1.95 Hz), 6.93 d (1H, 9-H, J = 1.9 Hz). 13C NMR spectrum (125 MHz), δC, ppm: 17.1, 22.2, 24.8, 27.1, 55.8, 107.1, 109.1, 118.1, 120.4, 122.9, 124.7, 130.6, 131.1, 155.0, 155.4, 166.9. Mass spectrum: m/z 284.1649 [M + H]+. C18H21NO2. M + H 284.1646.
1-{[(Het)arylamino]methylidene}-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-ones 5a–5f (general procedure). A mixture of 2.4 mmol of pyrroloquinolin-2-one 2b or 2c and 2.5 mmol of DMF-DMA in 10 mL of o-xylene was refluxed for 1 h. The corresponding amine, 2.4 mmol, and 1–2 drops of acetic acid were added, and the mixture was refluxed until the reaction was complete. The precipitate was filtered off, dried, and recrystallized from petroleum ether with addition of isopropyl alcohol.
1-{[(1H-Benzo[d]imidazol-2-yl)amino]methylidene}-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (5a) was synthesized from 0.54 g of 2b. Yield 0.68 g (77%), a mixture of Z and E isomers at a ratio of 3:1, yellow powder, mp 232–234°C. 1H NMR spectrum (500 MHz), δ, ppm: [1.63]Footnote 1 1.68 s (6H, 4-CH3), [1.97] 1.99 s (3H, 6-CH3), 2.29 [2.32] s (3H, 8-Me), [5.34] 5.40 s (1H, 5-H), [6.74] 6.79 s (1H, Harom), 7.09–7.12 m (2H, Harom), 7.30 s (1H, C=CH), [7.35] 7.41 d (1H, Harom, J = 7.0 Hz), 7.44 d (1H, Harom, J = 7.1 Hz), [7.60] s (1H, Harom), [8.22] s (1H, Harom), [8.31] s (1H, Harom), 8.52 d (1H, Harom, J = 11.4 Hz), 11.14 d (1H, NH, J = 11.8 Hz), 11.9 s (1H, NHBim). Mass spectrum: m/z 371.1872 [M + H]+. C23H22N4O. Calculated: M + H 371.1867.
4,4,6,8-Tetramethyl-1-{[(pyridin-2-yl)amino]methylidene}-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (5b) was synthesized from 0.54 g of 2b. Yield 0.64 g (81%), Z/E ratio 4:1, yellow powder, mp 178–180°C. 1H NMR spectrum (400 MHz), δ, ppm: [1.59] 1.64 s (6H, 4-Me), 1.96 d (3H, 6-Me), 2.27 [2.32] s (3H, 8-Me), [5.33] 5.36 d (1H, 5-H), 6.73 [6.76] s (1H, Harom), 7.05 d.d (1H, Harom, J = 8.0 Hz), 7.23 s (1H, C=CH), 7.35 d (1H, Harom, J = 8.0 Hz), [7.68] s (1H, Harom), 7.77 t (1H, Harom, J = 4.0 Hz), 8.31 [8.33] d.d (1H, Harom, J = 8.0, 4.0 Hz), [8.56] d (1H, Harom, J = 12.0 Hz), 8.72 d (1H, Harom, J = 8.0 Hz), [9.74] d (1H, NH, J = 12.0 Hz), 10.86 d (1H, NH, J = 8.0 Hz). Mass spectrum: m/z 332.1763 [M + H]+. C21H21N3O. Calculated: M + H 332.1758.
1-[(2-Methoxyanilino)methylidene]-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (5c) was synthesized from 0.54 g of 2b. Yield 0.72 g (83%), yellow powder, mp 208–210°C. 1H NMR spectrum (500 MHz), δ, ppm: 1.65 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.3 Hz), 2.29 s (3H, 8-Me), 3.93 s (3H, OCH3), 5.37 d (1H, 5-H, J = 1.5 Hz), 6.70 s (1H, Harom), 7.01–7.06 m (2H, Harom), 7.11 d.d (1H, Harom, J = 7.9, 1.6 Hz), 7.27 s (1H, C=CH), 7.64 d.d (1H, Harom, J = 7.8, 1.8 Hz), 8.60 d (1H, Harom, J = 12.8 Hz), 10.88 d (1H, NH, J = 12.8 Hz). Mass spectrum: m/z 361.1907 [M + H]+. C23H24N2O2. Calculated: M + H 361.1912.
1-[(4-Methoxyanilino)methylidene}-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one (5d) was synthesized from 0.54 g of 2b. Yield 0.66 g (77%), Z/E ratio 6:1, yellow powder, mp 264–266°C. 1H NMR spectrum (500 MHz), δ, ppm: [1.61] 1.65 s (6H, 4-Me), [1.97] 1.98 d (3H, 6-Me, J = 1.2 Hz), 2.29 [2.33] s (3H, 8-Me), [3.80] 3.81 s (3H, OCH3), [5.34] 5.37 d (1H, 5-H, J = 1.4 Hz), 6.64 [6.67] d.d (1H, Harom, J = 8.2, 2.3 Hz), 6.71 [6.76] s (1H, Harom), 6.95 [6.97] d.d (1H, Harom, J = 8.0, 2.2 Hz), 7.04 t (1H, Harom, J = 2.2 Hz), [7.25] 7.27 s (1H, C=CH), 7.28–7.29 m (1H, Harom), [7.63] s (1H, Harom), [7.75] d (1H, Harom, J = 13.6 Hz), 8.56 d (1H, Harom, J = 12.6 Hz), [9.3] 10.68 d (1H, NH, J = 12.6 Hz). Mass spectrum: m/z 361.1909 [M + H]+. C23H24N2O2. Calculated: M + H 361.1912.
1-[(4-Acetylanilino)methylidene]-4,4,6,8-tetramethyl-4H-pyrrolo[3,2,1-ij]quinolin-2-one (5e) was synthesized from 0.54 g of 2b. Yield 0.67 g (75%), Z/E ratio 5:1, yellow powder, mp 208–210°C. 1H NMR spectrum (500 MHz), δ, ppm: [1.62] 1.66 s (6H, 4-Me), 1.98 s (3H, 6-CH3), 2.30 [2.34] s (3H, 8-CH3), 2.54 s [3H, C(O)CH3], [5.36] 5.38 s (1H, H-5), 6.75 [6.80] s (1H, Harom), 7.3 s (1H, C=CH), [7.50] 7.52 d (2H, Harom, J = 7.9 Hz), [7.7] s (1H, Harom), [7.81] d (1H, Harom, J = 13.8 Hz), 7.97 [7.99] d (2H, Harom, J = 8.3 Hz), 8.63 d (1H, Harom, J = 12.3 Hz), [9.56] 10.84 d (1H, NH, J = 12.3 Hz). Mass spectrum: m/z 373.1915 [M + H]+. C24H24N2O2. Calculated: M + H 373.1912.
1-[(Benzylamino)methylidene]-8-methoxy-4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinolin-2-one (5f) was synthesized from 0.58 g of 2c. Yield 0.64 g (74%), light yellow powder, mp 152–154°C. 1H NMR spectrum (400 MHz), δ, ppm: 1.65 s (6H, 4-CH3), 1.98 d (3H, 6-CH3, J = 1.3 Hz), 3.87 s (3H, OCH3), 4.42 s (2H, CH2), 5.37 d (1H, 5-H, J = 1.5 Hz), 6.73 s (1H, C=CH), 7.03–7.05 m (1H, Harom), 7.07 t (1H, Harom, J = 1.9 Hz), 7.11 d.d (1H, Harom, J = 7.4, 1.7 Hz), 7.17 d.d (1H, Harom, J = 7.9, 1.6 Hz), 7.64 d.d (1H, Harom, J = 7.8, 1.8 Hz), 8.60 d (1H, Harom, J = 12.8 Hz), 10.82 d (1H, NH, J = 12.8 Hz). Mass spectrum: m/z 361.1910 [M + H]+. C23H24N2O2. Calculated: M + H 361.1912.
The inhibitory activity of the synthesized compounds against blood coagulation factors Xa and XIa was evaluated by measuring the kinetics of hydrolysis of enzyme-specific substrates in the presence of tested compounds. In the case of factor Xa, low-molecular-weight specific chromogenic substrate S2765 (Z-D-Arg-Gly-Arg-pNA·2HCl, Chromogenix, Instrumentation Laboratory Company, Lexington, MA 02421, USA) was used, and S2366 (pyroGlu-Pro-Arg-pNA·HCl, Chromogenix, Instrumentation Laboratory Company, Lexington, MA 02421, USA) was used for factor XIa.
Each well of a 96-well microplate was charged with a buffer solution containing 140 mM NaCl, 20 mM HEPES, 0.1% PEG (6000) (pH 8.0), factor Xa or XIa was added to a final concentration of 2.5 or 0.8 nM, respectively, and S2765 or S2366 (final concentration 200 μM), compound to be tested (30 μM), and DMSO (≤2%) were then added. The rate of formation of 4-nitroaniline was determined by measuring the absorbance of the final solution at λ 405 nm using a THERMOmax Microplate Reader (Molecular Devices Corporation, Sunnyvale, California). The initial rate of substrate cleavage was estimated from the initial slope of the kinetic curve for the formation of 4-nitroaniline (pNA). The rate of substrate cleavage by the enzyme in the presence of an inhibitor was normalized with respect to the rate of substrate cleavage in the absence of inhibitor. The results are summarized in Table 1. The data were processed using GraphPad Prism (GraphPad, San Diego, CA 92108, USA) and OriginPro 8 (OriginLab Corporation, Northampton, MA 01060, USA).
CONCLUSIONS
Selective reduction of the C2=O carbonyl group of pyrrolo[3,2,1-ij]quinoline-1,2-diones afforded 4H-pyrrolo[3,2,1-ij]quinolin-2(1H)-one derivatives as convenient building blocks for the design of hybrid molecules, and their further functionalization pathways were studied. Primary screening of the newly synthesized functionally substituted 4H-pyrrolo[3,2,1-ij]quinolin-2-ones for their in vitro inhibitory activity against blood coagulation factors Xa and XIa showed that most of them possess no anticoagulant properties. Only compounds 4a and 5b showed a moderate inhibitory activity against factor Xa.
Notes
Values in brackets refer to the E isomer.
REFERENCES
Miles, T.J., Hennessy, A.J., Bax, B., Brooks, G., Brown, B.S., Brown, P., Cailleau, N., Chen, D., Dabbs, S., Davies, D.T., Esken, J.M., Giordano, I., Hoover, J.L., Jones, G.E., Sukmar, S.K.K., Markwell, R.E., Minthorn, E.A., Rittenhouse, S., Gwynn, M.N., and Pearson, N.D., Bioorg. Med. Chem. Lett., 2016, vol. 26, p. 2464. https://doi.org/10.1016/j.bmcl.2016.03.106
Schrader, K.K., Avolio, F., Andolfi, A., Cimmino, A., and Evidente, A., J. Agric. Food Chem., 2013, vol. 61, p. 1179. https://doi.org/10.1021/jf304586j
Tsuji, K., Tsubouchi, H., and Ishikawa, H., Chem. Pharm. Bull., 1995, vol. 43, p. 1678. https://doi.org/10.1248/cpb.43.1678
Ishikawa, H., Uno, T., Miyamoto, H., Ueda, H., Tamaoka, H., Tominaga, M., and Nakagawa, K., Chem. Pharm. Bull., 1990, vol. 38, p. 2459. https://doi.org/10.1248/cpb.38.2459
Al-Said, N.H., Shawakfeh, K.Q., and Abdullah, W.N., Molecules, 2005, vol. 10, p. 1446. https://doi.org/10.3390/10121446
Ukrainets, I.V., Golik, M.Y., Sidorenko, L.V., Korniyenko, V.I., Grinevich, L.A., Sim, G., and Kryvanych, O.V., Sci. Pharm., 2018, vol. 86, p. 31. https://doi.org/10.3390/scipharm86030031
Ishichi, Y., Sasaki, M., Setoh, M., Tsukamoto, T., Miwatashi, S., Nagabukuro, H., Okanishi, S., Imai, S., Saikawa, R., Doi, T., and Ishihara, Y., Bioorg. Med. Chem., 2005, vol. 13, p. 1901. https://doi.org/10.1016/j.bmc.2005.01.022
Yin, L., Hu, Q., and Hartmann, R.W., J. Med. Chem., 2013, vol. 56, p. 460. https://doi.org/10.1021/jm301408t
Tsotinis, A., Panoussopoulou, M., Eleutheriades, A., Davidson, K., and Sugden, D., Eur. J. Med. Chem., 2007, vol. 42, p. 1004. https://doi.org/10.1016/j.ejmech.2007.01.005
Matesic, L., Locke, J.M., Vine, K.L., Ranson, M., Bremner, J.B., and Skropeta, D., Tetrahedron, 2012, vol. 68, p. 6810. https://doi.org/10.1016/j.tet.2012.06.049
Layek, M., Reddy, A.M., Rao, A.V.D., Alvala, M., Arunasree, M.K., Islam, A., Mukkanti, K., Iqbal, J., and Pal, M., Org. Biomol. Chem., 2011, vol. 9, p. 1004. https://doi.org/10.1039/C0OB00771D
Paris, D., Cottin, M., Demonchaux, P., Augert, G., Dupassieux, P., Lenoir, P., Peck, M.J., and Jasserand, D., J. Med. Chem., 1995, vol. 38, p. 669. https://doi.org/10.1021/jm00004a013
Isaac, M., Slassi, A., O’Brien, A., Edwards, L., MacLean, N., Bueschkens, D., Lee, D.K.H., McCallum, K., De Lannoy, I., Demchyshyn, L., and Kamboj, R., Bioorg. Med. Chem. Lett., 2000, vol. 10, p. 919. https://doi.org/10.1016/S0960-894X(00)00141-4
Ilin, I., Lipets, E., Sulimov, A., Kutov, D., Shikhaliev, Kh., Potapov, A., Krysin, M., Zubkov, F., Sapronova, L., Ataullakhanov, F., and Sulimov, V., J. Mol. Graphics Modell., 2019, vol. 89, p. 215. https://doi.org/10.1016/j.jmgm.2019.03.017
Sulimov, V.B., Gribkova, I.V., Kochugaeva, M.P., Katkova, E.V., Sulimov, A.V., Kutov, D.C., Shikhaliev, Kh.S., Medvedeva, S.M., Krysin, M.Yu., Sinauridze, E.I., and Ataullakhanov, F.I., BioMed Res. Int., 2015, vol. 2015, article ID 120802. https://doi.org/10.1155/2015/120802
Medvedeva, S.M., Potapov, A.Yu., Gribkova, I.V., Katkova, E.V., Sulimov, V.B., and Shikhaliev, Kh.S., Pharm. Chem. J., 2018, vol. 51, p. 975. https://doi.org/10.1007/s11094-018-1726-4
Novichikhina, N., Ilin, I., Tashchilova, A., Sulimov, A., Kutov, D., Ledenyova, I., Krysin, M., Shikhaliev, Kh., Gantseva, A., Gantseva, E., Podoplelova, N., and Sulimov, V., Molecules, 2020, vol. 25, article no. 1889. https://doi.org/10.3390/molecules25081889
Novichikhina, N.P., Skoptsova, A.A., Shestakov, A.S., Potapov, A.Y., Kosheleva, E.A., Kozaderov, O.A., Ledenyova, I.V., Shikhaliev, Kh.S., Podoplelova, N.A., and Panteleev, M.A., Russ. J. Org. Chem., 2020, vol. 56, p. 1550. https://doi.org/10.1134/S1070428020090080
Hardtmann, G.E., US Patent no. 4015005A, 1977.
Kajino, H., Michida, M., Takahashi, Y., and Kuwahara, Y., US Patent Appl. Pub. no. 2016/0137644 A1, 2016.
Zhuravleva, Yu.A., Zimichev, A.V., Zemtsova, M.N., and Klimochkin, Yu.N., Russ. J. Org. Chem., 2011, vol. 47, p. 617. https://doi.org/10.1134/S1070428011040270
Zemtsova, M.N., Golovko, Yu.A., Gruzd, Yu.A., Kulemina, S.V., Baimuratov, M.R., and Klimochkin, Yu.N., Russ. J. Org. Chem., 2021, vol. 57, p. 793. https://doi.org/10.1134/S1070428021050055
McAllister, L.A., McCormick, R.A., James, K.M., Brand, S., Willetts, N., and Procter, D.J., Chem. Eur. J., 2007, vol. 13, p. 1032. https://doi.org/10.1002/chem.200601429
Bass, R.J., Koch, R.C., Richards, H.C., and Thorpe, J.E., J. Agric. Food Chem., 1981, vol. 29, p. 576. https://doi.org/10.1021/jf00105a036
Axon, J., Boiteau, L., Boivin, J., Forbes, J.E., and Zard, S.Z., Tetrahedron Lett., 1994, vol. 35, p. 1719. https://doi.org/10.1016/0040-4039(94)88328-9
Nakamura, S., Kozuka, M., Bastow, K.F., Tokuda, H., Nishino, H., Suzuki, M., Tatsuzaki, J., Natschke, S.L.M. Kuo, S.-C., and Lee, K.-H., Bioorg. Med. Chem., 2005, vol. 13, p. 4396. https://doi.org/10.1016/j.bmc.2005.04.078
Singh, G., Kalra, P., Arora, A., Singh, A., Sharma, G., Sanchita, S., and Satija, P., New J. Chem., 2018, vol. 42, p. 16902. https://doi.org/10.1039/C8NJ02884B
Lozinskaya, N.A., Babkov, D.A., Zaryanova, E.V., Bezsonova, E.N., Efremov, A.M., Tsymlyakov, M.D., Anikina, L.V., Zakharyascheva, O.Yu., Borisov, A.V., Perfilova, V.N., Tyurenkov, I.N., Proskurnina, M.V., and Spasov, A.A., Bioorg. Med. Chem., 2019, vol. 27, p. 1804. https://doi.org/10.1016/j.bmc.2019.03.028
Zhang, C., Xu, J., Zhao, X., and Kang, C., J. Chem. Res., 2017, vol. 41, p. 537. https://doi.org/10.3184/174751917X15040891974776
Crestini, C. and Saladino, R., Synth. Commun., 1994, vol. 24, p. 2835. https://doi.org/10.1080/00397919408010603
Jeankumar, V.U., Alokam, R., Sridevi, J.P., Suryadevara, P., Matikonda, S.S., Peddi, S., Sahithi, S., Alvala, M., Yogeeswari, P., and Sriram, D., Chem. Biol. Drug Des., 2014, vol. 83, p. 498. https://doi.org/10.1111/cbdd.12265
Sun, L., Tran, N., Tang, F., App, H., Hirth, P., McMahon, G., and Tang, C., J. Med. Chem., 1998, vol. 41, p. 2588. https://doi.org/10.1021/jm980123i
Sharma, P., Thummuri, D., Reddy, T.S., Senwar, K.R., Naidu, V.G.M., Srinivasulu, G., Bharghava, S.K., and Shankaraiah, N., Eur. J. Med. Chem., 2016, vol. 122, p. 584. https://doi.org/10.1016/j.ejmech.2016.07.019
Spencer, J., Chowdhry, B.Z., Hamid, S., Mendham, A.P., Male, L., Coles, S.J., and Hursthouse, M.B., Acta Crystallogr., Sect. C, 2010, vol. 66, p. o71. https://doi.org/10.1107/S0108270109054134
Lescheva, E.V., Medvedeva, S.M., and Shikhaliev, Kh.S., J. Org. Pharm. Chem., 2014, vol. 12, p. 15. https://doi.org/10.24959/ophcj.14.798
ACKNOWLEDGMENTS
The high-resolution mass spectra were recorded at the joint research equipment center at the Voronezh State University.
Funding
This study was performed under financial support by the Russian Science Foundation (project no. 18-74-10097, https://rscf.ru/project/18-74-10097).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare the absence of conflict of interest.
Additional information
Translated from Zhurnal Organicheskoi Khimii, 2022, Vol. 58, No. 9, pp. 965–974 https://doi.org/10.31857/S0514749222090051.
Rights and permissions
About this article

Cite this article
Novichikhina, N.P., Ashrafova, Z.E., Ledenyova, I.V. et al. Synthesis and Anticoagulant Activity of New Functionalized 4H-Pyrrolo[3,2,1-ij]quinolin-2-ones. Russ J Org Chem 58, 1225–1232 (2022). https://doi.org/10.1134/S1070428022090056
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070428022090056