
Abstract
This work is devoted to the large-scale solid-phase synthesis (SPS) of Atosiban, Mpa1-D-Tyr(OEt)-Ile-Thr-Asn-Cys6-Pro-Orn-Gly-NH2 cyclic 1,6 disulfide, the only clinically used oxytocin receptor antagonist. The conditions have been selected for the closure of the disulfide bond (S–S) in the Atosiban molecule both in the solution and solid phase with the minimal formation of by-products. A comparative assessment of the formation of the S–S bond was carried out under various conditions. The formation of by-products during the closure of the disulfide bond has been studied both in solution and on the polymer support. The developed technique allows for the synthesis of Atosiban on an enlarged scale (10–20 mmol) involving the cyclization of a protected intermediate with the formation of the S–S bond during solid-phase synthesis with the minimal formation of by-products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The disulfide bond is one of the structure-forming elements in the molecules of many biologically active peptides, i.e., peptide hormones, such as Oxytocin, Vasopressin, Somatostatin, and Insulin [1, 2], neurotransmitters, growth factors, etc. Disulfide bridges play an important role in the biological effects of many peptide drugs [3]. Examples of these compounds are vasopressin and somatostatin receptor agonists, such as Terlipressin, Felipressin, Desmopressin, Ornipressin, Octreotide, Lanreotide, and Pasireotide. These peptides that contain one disulfide bond are produced on an industrial scale [4–7]. To date, there is only one clinically used oxytocin receptor antagonist, Atosiban, which is used to prevent premature birth and also produced on an industrial scale [8, 9]. The peptide drugs, such as Linaclotide and Plekanatide act as guanylate cyclase agonists and contain three and two S–S bonds in their structure, respectively [10].
Currently, there are quite a large number of ways to create disulfide bridges in peptides. Atmospheric oxygen, potassium ferricyanide, dimethyl sulfoxide, and hydrogen peroxide are used for the oxidation of thiol precursors [11]. The synthesis of complex peptides is carried out in buffers, which simulate physiological conditions by using a glutathione system, i.e., 5-mM reduced and 0.1-mM oxidized glutathione in various ratios [12]. In the synthesis of natural peptides with several disulfide bonds, soft oxidizers were successfully used for the spontaneous formation of the S–S bridges. Conotoxins with two disulfide bridges were synthesized during spontaneous cyclization of thiol precursors with atmospheric oxygen, which led to the predominant formation of natural disulfide isomers [13]. An alternative approach is a direct conversion of protected (Trt, Acm, Tmob, Mob, etc.) linear peptide precursors into cyclic disulfides. For this purpose, iodine is most often used in various solvents [14]; thallium (III) trifluoroacetate [15], or sulfoxides in the presence of chlorosilanes [16] are used less often.
The synthesis of natural peptides or their analogs that contain intramolecular disulfide bridges is still a rather difficult task [17]. The difficulty lies in the fact that the cyclization of a corresponding linear precursor, regardless of the classical or solid-phase method of its preparation, should be carried out in very dilute solutions to avoid the intermolecular aggregation and the formation of side disulfide dimers and oligomers at the stage of the disulfide bond closure in the peptide. The working concentrations of peptides during cyclization, as a rule, are 10–4–10–5 М, i.e., 0.1–1.0 mg/mL [14]. The concentration of cyclization reaction mixtures before the release of the target product is a rather long process, which may lead to the formation of by-products because of noncompliance with the pH or temperature regime. The presence of residual amounts of oxidizer in the mixture can promote deeper oxidation of sulfur, thus resulting in the formation of corresponding sulfoxide by-products. Atosiban is allowed for practical use in our country as a medicinal product, which dictates the need to develop a large-scale scheme for its production. We encountered some problems in the synthesis of Atosiban at the stage of creating the disulfide bond.
The goal of this study was to develop an optimal method for the large-scale synthesis of Atosiban, to compare different conditions for the S–S bond formation, and to study the impurities formed during the synthesis in the solution and on the solid support.
RESULTS AND DISCUSSION
The structure of cyclic 1,6-disulfide Atosiban (I) is Mpa1-D-Tyr(OEt)-Ile-Thr-Asn-Cys6-Pro-Orn-Gly-NH2. This antagonist was synthesized by the replacement of four amino acid residues in the Oxytocin molecule. Tyrosine, cysteine, leucine, and glutamine were replaced by an alkylated analog D-Tyr (OEt), deaminocysteine (Mpa), ornithine, and threonine, respectively. These modifications were performed to obtain the oxytocin receptor antagonist with increased proteolytic resistance [3].
Various methods of industrial production of Atosiban are known with a preference for the synthesis in solution, according to the literature data [9, 18]. In this work, the solid-phase method was chosen for the synthesis of this peptide because we consider it more technologically advanced. Acid-labile protective groups in combination with Nα-Fmoc protection were used to block the functional groups of the side chains of amino acids, i.e., But for threonine, Boc for ornithine, Trt for asparagine. The cysteine residue was protected by the Acm or Trt group, and the mercaptopropionic acid residue was protected by the Trt group. The solution of 5% 4-MePip/2% DBU/DMF was used to remove Fmoc-protection [19]. The DIC/HOBt method was used to create the peptide bond. The resulting linear precursor of Atosiban, Mpa-D-Tyr(OEt)-Ile-Thr-Asn-Cys-Pro-Orn-Gly-NH2, was obtained with a purity of 76% according to HPLC data. The fundamental stage in the synthesis of Atosiban is cyclization, and we studied the closure of the disulfide bridge both in solution and on the polymer.
Closure of S–S bridge in solution. There are some references in the literature that the introduction of organic solvents into the reaction mixtures at the cyclization stage contributes to the formation of intramolecular disulfides [20]. Our data on the synthesis of Octreotide [21] show that in this case, the cyclization of the thiol precursor at the concentration of 10–20 mg/mL occurs in methanol without the formation of noticeable amounts of dimers. We performed a series of experiments to obtain Atosiban in aqueous or water-organic solutions (H2O/isopropanol, H2O/dioxane, H2O/isopropanol/CH3CN) using increasing concentrations of linear SH-peptide (1.0–20.0 mg/mL) at pH 7.0–8.0 (Table 1). The Ellman test and HPLC were used to control the completeness of cyclization [22]. The use of organic solvents as the components of the reaction mixture during cyclization made it possible to increase the concentration of the initial dithiol (II).
We were not able to avoid the formation of products with the intermolecular S–S bonds, i.e., parallel and antiparallel dimers (III) and (IV), under the conditions used for cyclization (Table 1, Fig. 1). These products were isolated by HPLC, and their structure was confirmed by mass spectrometry. The ESI(+) spectrum of the fraction that consisted of a mixture of peptides (III) and (IV) contained a single peak of the molecular ion (1988.6) corresponding to the mass of dimeric products. Further, we took into account the total amount of peptides (III) and (IV) when evaluating the results of cyclization. The content of by-products at the concentration of the initial SH compound in the range 1–10 mg/mL barely changed and amounted to 10–14% (Table 1). The content of disulfide dimers (III) and (IV) increased to 18.2% with an increase in the concentration of the initial compound (II) to 20 mg/mL (Table 1). The dimers appeared to have lower solubility than Atosiban and significantly complicate the isolation of the target product. The best results for the S–S-bridge formation in solution were obtained at the peptide concentration of 10 mg/mL when an H2O/isopropyl alcohol mixture was used as a solvent. The oxidation occurred for 15 min. After the completion of the reaction, the pH of the reaction mixture was adjusted to 4 with acetic acid, and the target peptide, which has HPLC purity of 73.7%, was then isolated by preparative HPLC.

Profile of the analytical HPLC and the ESI(+) spectra of products when forming the S–S bridge in Atosiban in solution by the action of H2O2 in an H2O/i-PrOH mixture (1 : 1) for 15 min. (I), Atosiban; (II), linear SH precursor of Atosiban; (III–IV), dimers.
It is worth noting that trifluoroacetic acid cannot be used at the stage of acidification of the reaction mixture because residual H2O2 can promote the formation of the corresponding sulfoxide. The isolated by-products of this type had a peak of the molecular ion (1010) in the ESI(+) mass spectrum corresponding to Atosiban sulfoxide. The yield of Atosiban acetate after the disulfide bond closure in the solution was 31.5% relative to the starting amino acid attached to the polymer support. The results prompted us to develop the optimal technique for the formation of the disulfide bond on the polymer support.
Formation of S–S bond on the polymer support. An important aspect of the formation of the S–S-bridge on the solid phase is the pseudodilution phenomenon. The swelling of the peptidyl polymer in a certain solvent increases the distance between neighboring peptide chains, thus mimicking dilution, which leads to weakening of the interaction between the molecules. We assumed that the solid-phase cyclization of Atosiban would help to overcome the problem of high dilutions, which was observed during oxidation in solution, reduce the formation of by-products, and simplify the procedure of isolation of the product. As a rule, the formation of the S–S bridge in various peptides requires the selection of special conditions [11].
Since Atosiban does not contain the tryptophan residue in the molecule, I2 was chosen as an oxidizer when creating the S–S bond on the polymer. Iodine is highly soluble in organic solvents, which ensure good solvation of the peptidyl polymer, and provides direct conversion of the cysteine-protected peptide to cyclic disulfide [11]. To date, there is a fairly large number of works on the study of the disulfide bridges formation on the polymer support [14, 23, 24]. However, no systematic studies have been performed to evaluate the influence of the amount of oxidizer, cyclization time, and solvent on the composition of the formed impurities in the synthesis of Atosiban. We carried out a series of experiments on the solid-phase synthesis of Atosiban including the stage of the disulfide bridge formation (Table 2).
Using two different protections of the cysteine residue, we synthesized two peptidyl polymers on the Rink amide resin:
Trt-Mpa-D-Tyr(OEt)-Ile-Thr(But)-Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-P and
Trt-Mpa-D-Tyr(OEt)-Ile-Thr(But )-Asn(Trt)-Cys(Acm)-Pro-Orn(Boc)-Gly-P.
The conversion of the protected peptide derivative into a cyclic intermediate on the solid support was carried out by the action of iodine (3–35 equiv.) in organic solvents, such as DMF, dioxane, and acetic acid at room temperature for 1–5 h (Table 2).
The best results were obtained when using DMF as a solvent. The use of dioxane and acetic acid under the same conditions (10-fold excess of iodine, 1 h) led to a large amount of linear SH-peptide and dimers. The content of the linear SH-precursor of Atosiban (II) and dimers unexpectedly increases in all cases with an increase in the amount of the oxidizer and the time of cyclization (Table 2). When using Trt protection and a three-fold excess of I2, the most complete formation of the S–S bridge was observed for 1 h at room temperature. In this case, the content of the SH-peptide and dimers was less than 7% and no other by-products were observed. The purity of Atosiban was more than 85%.
We have chosen the HPLC conditions for high resolution of peaks corresponding to the SH and SS forms of the peptide and by-products when analyzing the reaction mixtures after the synthesis of Atosiban (I) (Fig. 2). It is worth noting that the same conditions but using the Acm protection for cysteine led to the formation of a sufficiently large amount of SH-peptide (II) in the reaction mixture. We identified the product (II) (Fig. 2) as the directionally synthesized SH-precursor of Atosiban (II). In the ESI(+) mass spectrum, we observed the molecular ion (997.2) corresponding to SH-peptide (II). The most noticeable changes in the signals of cysteine protons were observed in the 1H NMR spectra of Atosiban (I) and peptide (II). The signal of the amide proton of the linear precursor of Atosiban (I) shifted in a stronger field compared to the signal of amide protons of Atosiban (I), i.e., αNH Cys (7.94 ppm) in peptide (II) and αNH, Cys (8.44 ppm) in Atosiban. The same trend is observed for βCH2 protons, i.e., βCH2, Cys (2.60 and 2.75 ppm) in peptide (II) and βCH2, Cys (3.02 and 2.85 ppm) in Atosiban. The signals of the amide protons of tyrosine, threonine, and ornithine in the peptide (II) also changed, i.e., αNH, D-Tyr(OEt) (8.15 ppm), αNH, D-Tyr(OEt) (8.39 ppm) in Atosiban; αNH, Thr (7.78 ppm) in peptide II, αNH, Thr (7.20 ppm) in Atosiban; αNH, Orn (8.17 ppm) in peptide (II), αNH, Orn (8.08 ppm) in Atosiban. It is worth noting the change in the signals of protons at the β-carbon atom in isoleucine, i.e., βCH2, Ile (1.70 ppm) in the peptide (II) and βCH2, Ile (1.85 ppm) in Atosiban. Thus, the 1H NMR spectrum and the ESI(+) mass spectrum of the by-product coincide with the spectra of the presynthesized sulfhydryl derivative.

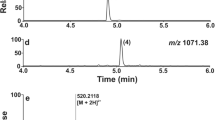
Profile of the analytical HPLC and the ESI(+) spectra of products when forming the S–S bridge in Atosiban on solid phase by the action of 3 equiv. of I2 in DMF for 1 h. (I), Atosiban; (II), linear SH precursor of Atosiban; (III–IV), dimers.
When treating the cyclization reaction mixture with H2O2, the product corresponding to peptide (II) is converted into Atosiban (I).
To date, there is no unambiguous idea in the literature on the mechanism of the S–S bridge formation by the action of I2 [17]. We assume that the essence of the reaction can be explained by Scheme 1. When the protected peptide derivative interacts with I2, monoiodine derivative (Ib) is initially formed, followed by the intramolecular conversion into cyclic product (Id) at a high rate. At the same time, a certain amount of a diiodine derivative (Ic) is formed due to the excess of iodine. This is supported by the fact that the amount of the linear product (II) with free SH groups also increases with increasing the amount of iodine (Table 2). In this case, the diiodine derivative (Ic), apparently, is not converted into product (Id), otherwise, we would mostly obtain the cyclic product. The content of the SH-peptide in the reaction mixture was 63.73%, 43.8%, and 3.71% with 40-fold, 10-fold, and 3-fold excess of I2, respectively (Table 2). This situation does not change fundamentally in the case of the Acm protective group on cysteine (Table 2). We assume that the rate of intramolecular closure of the cycle in the case of the trityl derivative is higher than in the case of the Acm derivative.

Scheme 1 . Scheme of cyclization of Atosiban (I) in solution and on the solid phase.
The content of the dimers in all cases is lower after cyclization in DMF compared to that in solution (Table 2).
The yield of Atosiban acetate (I) at the closure of the disulfide bond on the polymer was 50%.
Comparison of the efficiency of the S–S bridge closure on the solid phase and in solution. The time of the S–S bridge formation in solution (not more than 15 min) is lower than on the solid phase. Moreover, the completeness of the reaction in the solution can be monitored by the Ellman test and HPLC. Nevertheless, there are more advantages of oxidation on the solid support. First, there is no problem of solubility; second, the treatment of the reaction mixture is significantly simplified because several washings of the peptidyl polymer followed by filtration make it possible to completely remove the excess of the oxidizer; third, we managed to achieve the almost complete absence of dimers during cyclization on the polymer.
A decrease in the polymer capacity in the solid-phase synthesis will probably minimize the formation of dimers. The main advantage of cyclization on the polymer is an increase in the total yield of Atosiban (I) relative to the starting amino acid (50% compared to 31.5% in solution).
The technique developed was successfully used for the synthesis of Atosiban (I) on an enlarged scale (10–20 mmol).
EXPERIMENTAL
We used derivatives of L and D amino acids, trityl-3-mercaptopropionic acid (Trt-Mpa), DMF, NMM, HOBt, TBTU, TIS, DTNB (Ellman’s reagent), dichloromethane, and trifluoroacetic acid (Fluka, Switzerland); acetic acid, metallic iodine, and ascorbic acid (extra pure grade) (Reakhim, Russia). Acetonitrile (CH3CN; Carl Roth GmbH, Germany) was used for HPLC.
Analytical HPLC was performed on a Knauer 1001A chromatograph (Germany) on a Kromasil 100-5 ODS column (Sweden) (4.6 × 250 mm; the particle size, 5 µm; the pore size, ~100 Å). Buffer A (0.05 M KH2PO4, pH 3.0) and buffer B (70% acetonitrile in buffer A) were used as eluents; elution was performed at a rate of 1 mL/min in the concentration gradient of buffer B in buffer A (20–80%) for 30 min with detection at 220 nm. Preparative HPLC of Atosiban was performed using a Knauer 1001 chromatograph (Germany) on a 50 × 250 mm column with a sorbent particle size of 10 µm. Buffer A (0.01-M aqueous solution of ammonium acetate, pH 4.5, containing 3% acetonitrile) and buffer B (70% acetonitrile in buffer A) were used as eluents. Elution was performed at a rate of 20 mL/min from 100% of buffer A in the concentration gradient of buffer B (0.5%/min). The fractions corresponding to the target compound were combined, concentrated in a vacuum, and lyophilized.
The 1H NMR spectra were recorded on a WH-500 spectrometer (500 MHz; Bruker, Germany) in DMSO-d6 at 300 K; the concentration of peptides was 2–3 mg/mL; chemical shifts (δ, ppm) were measured relative to tetramethylsilane. The signals were assigned to certain groups of protons of the amino acid residues using the differential double resonance method. The mass spectra were recorded on an Amazon mass spectrometer (Bruker, Germany) by electrospray ionization (ESI) in the positive ion registration mode (capillary voltage, 3500 V; the mass scanning range; m/z, 70–2200; the spray gas, nitrogen; the interface temperature, 100°C.). A sample in a CH3CN–water mixture was injected with a syringe.
Solid-phase synthesis of Atosiban (I). Synthesis of Trt-Mpa-D-Tyr(OEt)-Ile-Thr(But)-Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-polymer (Ia). The synthesis of the peptide was carried out in a manual mode on the Rink amide resin (8 g, 5.44 mmol) (Novabiochem, Great Britain) with the capacity for the amino groups of 0.68 mmol/g. The Fmoc protection was removed from the α-amino group by the successive action of a 5% solution of 4-methylpiperidine and 2% solution of 1,8-diazobicyclo[5.4.0.]undec-7-en (DBU) in DMF for 5 and 10 min, respectively. The amino acid chains were extended by standard procedures of a one-stage cycle, which included a 30-min activation of the attached amino acid (16.32 mmol) in the presence of the equimolar amount of DIC and HOBt in an NMP–DMF mixture (1 : 1). The synthesis cycle included all the necessary washings of the peptidyl polymer by DMF and a ninhydrin test to evaluate the residual amino groups [18]. Aliquots of peptidyl polymer (Ia) with a peptide content of ~0.5 mmol were used for the test syntheses of Atosiban (I). The conditions and results of this synthesis are presented in Tables 1 and 2. The main part of the nonapeptidyl polymer (Ia) with a peptide content of 3.6 mmol was used to obtain the target product (I). To assess the quality of the intermediate nonapeptide, a sample of Nα-free peptidyl polymer (Ia) was treated with a deprotection mixture TFA/TIS/H2O (90 : 5 : 5 v/v/v) for 1 h. After precipitation of the product with diethyl ether, the content of the main compound in the sample was 89% according to HPLC data.
Synthesis of Mpa1-D-Tyr(OEt)-Ile-Thr-Asn-Cys6-Pro-Orn-Gly-NH2 cyclic 1,6-disulfide (I). (а) The S–S closure in solution. The solid-phase synthesis of nonapeptidyl-polymer (Ia) was carried out according to the above method starting with the Rink amide resin (0.48 mmol/g of the amino groups). Resulting peptidyl-polymer (Ia) (28.5 g) was suspended in a cooled mixture (4°С) that contained TFA (200 mL), deionized water (10 mL), TIS (10 mL), and DTT (10 g), followed by stirring for 2 h. The polymer was filtered, washed successively with the deprotection mixture (2 × 30 mL) and СН2Сl2/TFA mixture (1 : 1, 2 × 30 mL), followed by evaporation of the filtrate to an oily state. The product was precipitated with cooled diethyl ether, filtered, washed with diethyl ether (2 × 30 mL) and ethyl acetate (2 × 30 mL), and dried at room temperature. The crude product of the solid-phase synthesis (7.1 g) was dissolved in an i‑PrOH/CH3CN/H2O mixture (2 : 1 : 5) (800 mL), followed by the addition of the 2% aqueous solution of NH4OH (3 mL, pH 8.0–9.0) and 3% aqueous solution of H2O2 (5 mL). The completeness of the formation of the disulfide bond was examined by the Ellman test and HPLC. At the end of cyclization, AcOH was added to the reaction mixture to adjust the pH value to 4.0. Organic solvents were evaporated in a vacuum, and the product was purified by HPLC. The yield of Atobisan acetate was 2.39 g (31.5% relative to the starting amino acid).
The ESI(+) mass spectrum: m/z (Irel, %): 994.54 (100) [M]+. Purity (HPLC): 99.66%. The 1H NMR spectrum is presented in Supplementary Information.
(b) The S–S closure on the solid phase. The iodine solution (10.86 mmol, 100 mL DMF) was added to the suspension of nonapeptidyl polymer (Ia) (10 g, 3.62 mmol) in DMF (400 mL), and the reaction mixture was vigorously mixed for 2 h. The peptidyl polymer was filtered and washed with DMF (3 × 100 mL). The excess of iodine was removed by 10% ascorbic acid in a DMF/H2O (2 : 1) mixture (2 × 100 mL), peptidyl polymer was filtered, washed with DMF (3 × 100 mL) and dichloromethane (3 × 100 mL), and dried. The peptide was removed from the polymer in one stage by treating the peptidyl polymer with a mixture (85 mL) that contained 90% TFA, 5% deionized water, and 5% TIS at room temperature for 1.5 h. The polymer was filtered and washed with the deprotection mixture (2 × 40 mL). The filtrate was evaporated to an oily state, and the product was precipitated by diethyl ether (85 mL). The precipitate was filtered and washed with diethyl ether (2 × 40 mL). The product (3 g) was a white powder. The content of the main compound in the sample was 85.51% according to HPLC data. The crude product was dissolved in water (150 mL), and the pH value of the solution was adjusted with 2.5% ammonia to 6.5–7.5. While stirring, 3% H2O2 (0.6 mL) was added to the homogeneous solution under stirring. The completeness of the formation of the disulfide bond was examined by the Ellman test [22] and HPLC. AcOH was added to the solution to adjust the pH value to 4.0–5.0, and the product was purified using HPLC. The yield of Atosiban acetate was 1.9 g (50% relative to the starting amino acid).
CONCLUSIONS
The method developed allows one to obtain technical Atosiban with a purity of more than 85% and the content of dimeric products of less than 5%, which opens up the possibility of introducing this technique into industrial production.
REFERENCES
Myers, R.D., Peptides, 1994, vol. 15, pp. 367–381. https://doi.org/10.1016/0196-9781(94)90025-6
Kondo, F., Okada, S., Miyachi, A., Kurita, M., Tsuji, K., and Harada, K., Anal. Bioanal. Chem., 2012, vol. 7, pp. 1783–1791. https://doi.org/10.1007/s00216-011-5635-6
Schteingart, C.D. and Lau, J.L., Annu. Rep. Med. Chem., 2017, vol. 50, pp. 543–586.
Avdeev, D.V., Sidorova, M.V., Ovchinnikov, M.V., Moiseeva, NI., Osipov, V.N., Balaev, A.N., and Khachatryan, D.S., Russ. J. Bioorg. Chem., 2019, vol. 45, pp. 248–252. https://doi.org/10.1134/S1068162019040034
Kyncl, J. and Rudinger, J., J. Endocrinol., 1970, vol. 48, pp. 157–165. https://doi.org/10.1677/joe.0.0480157
Gajjar, K., Martin-Hirsch, P.P., Bryant, A., and Owens, G.L., Cochrane Database Syst. Rev., 2016, vol. 7, pp. 1–81. https://doi.org/10.1002/14651858.CD006120.pub4
Mannucci, P.M., Haemophilia, 2000, vol. 6, pp. 60–67.
Ronald, F.L. and Ronald Kam, K.Y., Expert Rev. Obstet. Gynecol., 2008, vol. 3, pp. 163–174. https://doi.org/10.1586/17474108.3.2.163
Andersson, L., Blomberg, L., Flegel, M., Lepsa, L., Bo, N., and Verlander, M., Biopolymers, 2000, vol. 55, pp. 227–250. https://doi.org/10.1002/1097-0282(2000)55:3<227::AID-BIP50>3.0.CO;2-7
Musaimi, O.Al., Shaer, D.Al., de la Torre, B.G., and Albericio, F., Pharmaceuticals, 2018, vol. 11, pp. 1–10. https://doi.org/10.3390/ph11020042
Kudryavtseva, E.V., Sidorova, M.V., and Evstigneeva, R.P., Usp. Khim., 1998, vol. 67, pp. 611–630.
Rabenstein, D.L. and Yeo, P.L., Org. Chem., 1994, vol. 59, pp. 4223–4229. https://doi.org/10.1021/jo00094a039
Zhmak, M.N., Kasheverov, I.E., Utkin, Yu.N., Tsetlin, V.I., Vol’pina, O.M., and Ivanov, V.T., Russ. J. Bioorg. Chem., 2001, vol. 27, pp. 67–71. https://doi.org/10.1023/A:1011319101676
Andreu, D., Albericio, F., Sole, N.A., Munson, M.C., Ferrer, M., and Barany, G., Methods Mol. Biol., 1994, vol. 35, pp. 91–169. https://doi.org/10.1385/0-89603-273-6:91
Fujii, N., Otaka, A., Funakoshi, S., Bessho, K., Watanabe, T., Akaji, K., and Yajima, H., Chem. Pharm. Bull., 1987, vol. 35, pp. 2339–2347.
Koide, T., Otaka, A., Suzuki, H., and Fujii, N., Synlett, 1991, vol. 345, p. 1. https://doi.org/10.1002/chin.199205259
Góngora-Benitez, M., Tulla-Puche, J., and Albericio, F., Chem. Rev., 2014, vol. 114, pp. 901–926. https://doi.org/10.1021/cr400031z
Bray, B.L., Nat. Rev., 2003, vol. 2, pp. 586–593. https://doi.org/10.1038/nrd1133
Sidorova, M.V., Palkeeva, M.E., Azmuko, A.A., Ovchinnikov, M.V., Molokoedov, A.S., Bushuev, V.N., and Pisarenko, O.I., Russ. J. Bioorg. Chem., 2019, vol. 45, pp. 18–26. https://doi.org/10.1134/S106816201901014X
Moroder, L., Besse, D., Musiol, H.-J., Rudolph-Boehner, S., and Sideler, F., Biopolymers, 1996, vol. 40, pp. 207–234. https://doi.org/10.1002/(sici)1097-0282(1996)40:2<207::aid-bip2>3.0.co;2-#
Sidorova, M.V., Molokoedov, A.S., Az’muko, A.A., Kudryavtseva, E.V., Krause, E., Ovchinnikov, M.V., and Bespalova, Zh.D., Russ. J. Bioorg. Chem., 2004, vol. 30, pp. 101–110. https://doi.org/10.1023/B:RUBI.0000023093.05123.31
Ellman, G.L., Arch. Biochem. Biophys., 1959, vol. 82, pp. 70–77. https://doi.org/10.1016/0003-9861(59)90090-6
Albericio, F., Hammer, R.P., Garcia-Echeverria, C., Molins, M.A., Chang, J.L., Munson, M.C., Pons, M., Giralt, E., and Barany, G., Int. J. Pept. Protein Res., 1991, vol. 37, pp. 402–413. https://doi.org/10.1111/j.1399-3011.1991.tb00755.x
Garcia-Echeverria, C., Albericio, F., Pons, I.M., Barany, G., and Giralt, E., Tetrahedron Lett., 1989, vol. 30, pp. 2441–2444. https://doi.org/10.1016/S0040-4039(01)80422-6
Funding
The work was supported by the Joint-stock Company “Obninsk Chemical Pharmaceutical Company” (JSC “OCPC”).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
This article does not contain any studies with the use of humans or animals as objects of research.
Conflict of Interest
The authors state that there is no conflict of interest.
Additional information
Translated by A. Levina
Abbreviations: Boc, tert-butyloxycarbonyl; But, tert-butyl; DBU, 1,8 -diazabicyclo[5.4.0]-undec-7-en; DCM, dichloromethane; DIC, N,N'-diisopropylcarbodiimide; DMF, dimethylformamide; DMSO , dimethylsulfoxide; D-Tyr(OEt)-OH, D-2-amino-3-(4-ethoxyphenyl)propanoic acid; DTT, dithiotreitol; Fmoc, 9-fluorenylmethyloxycarbonyl; ESI, electrospray chemical ionization at atmospheric pressure; HOBt, 1-hydroxybenzotriazole; MePip, 4-methylpiperidine; Mpa, 3-mercaptopropionic acid; i-PrOH, isopropanol; TFA, trifluoracetic acid; TIS, triisopropylsilane; Trt, trityl.
Corresponding author: phone: +7 (915) 051-25-55.
Supplementary Information
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Avdeev, D.V., Ovchinnikov, M.V., Dudkina, Y.S. et al. Optimal Method for Disulfide Bond Closure in the Synthesis of Atosiban—Antagonist of Oxytocin Receptors. Russ J Bioorg Chem 47, 1241–1248 (2021). https://doi.org/10.1134/S1068162021060042
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162021060042