
Abstract
The title peptide, oxytocin trisulfide (3), was required on a multi-100 mg scale, as part of an analytical method validation process for the clinically and commercially important product “Oxytocin Injection,” which is a sterile isotonic injectable formulation of oxytocin (2). We report here that previous chemistry from our academic laboratory can be successfully scaled up in the biotechnology sector to indeed provide pure 3 (overall yield 6–7%, > 95% purity). The scaled-up synthesis also gave rise, somewhat unexpectedly, to modest levels of oxytocin tetrasulfide (4), and even higher homologues. Protocols for synthesis, purification, and characterization [by HPLC, mass spectrometry and amino acid analysis] are described and discussed, along with possible mechanistic explanations for our findings.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The neurohypophyseal hormone oxytocin (Scheme 1) occupies a special place in the history of peptide science. Its discovery and synthesis, by Vincent du Vigneaud and coworkers, was recognized by the 1955 Nobel Prize in Chemistry (du Vigneaud et al. 1953; du Vigneaud 1956, 1964; Ragnarsson 2007). Synthesis of oxytocin (2) has been an important checkpoint in the development of innovative methods of solution and solid-phase peptide synthesis, particularly those focused on the efficient creation of its heterodectic intramolecular disulfide bridge (reviewed by Andreu et al. 1994, Annis et al. 1997, Góngora-Benítez et al. 2014). Furthermore, the medicinal chemistry and pharmacology of oxytocin and its analogues has been a fertile research area for seven decades (reviewed by Manning et al. 2012, Wiśniewski 2019).

Chemical structures of oxytocin (2), its linear (reduced) form (1), and its trisulfide (3) and tetrasulfide (4) variants
Starting in the early 1960s, medicinal treatments for which oxytocin was the active pharmaceutical ingredient (API) have been approved for clinical use–specifically to induce or improve uterine contractions (when this is medically indicated due to maternal or fetal concerns) during childbirth (vaginal delivery), as well as to continue uterine contractions as a means to control postpartum bleeding or hemorrhaging.Footnote 1,Footnote 2 Clinically approved formulations of oxytocin, under the brand name PITOCIN and the generic name “Oxytocin Injection” (a sterile isotonic injectable formulation of 2), represent the first use of a synthetic peptide for therapeutic purposes (Lau and Dunn 2018). Oxytocin and related drugs are of critical importance to women’s health, as postpartum hemorrhaging is still the most common cause of maternal death in the world (Owen et al. 2021). Innovative medicinal chemistry and formulation strategies are improving access to oxytocic drugs in low to middle income geographies, as demonstrated through development of heat-stable formulations that do not require the cold supply chain that often limits access and quality (Fabio et al. 2015; Malm et al. 2018; Widmer et al. 2018).
Starting in 1984 and picking up momentum in the mid-to-late 1990’s, our Minnesota laboratory developed several routes to prepare novel trisulfideFootnote 3 analogues of oxytocin and deamino-oxytocin, in what seemed at the time to be an academic exercise (Mott et al. 1986; Chen and Barany 1996; Chen et al. 1997). The new variants were characterized in several ways that confirmed their structures, and biological studies were carried out as well. The practical relevance of this work came into focus with several reports of peptide synthesis by-products (Parmentier et al. 1994; Moutiez et al. 1997) and small recombinant proteins (Andersson et al. 1996; Canova-Davis et al. 1996; Pristatsky et al. 2009; Kshirsagar et al. 2012) and monoclonal antibodies (Gu et al. 2010), all of which include a trisulfide bridge (reviewed by Nielsen et al. 2011). Relatedly, we extended our synthetic expertise to confirm the formation of a trisulfide by-product in the commercial production of a somatostatin analogue called “lanreotide” (or “somatuline”), a peptide drug that has been used clinically for treatment of acromegaly (Chen et al. 1999).
About a dozen years ago, we were approached by Teva Parenteral Medicines, a commercial manufacturer then based in Irvine, California, to assist them in a challenge they had encountered with respect to their Oxytocin Injection product. Thus, in the process of conducting regulatory stability studies of their formulation as mandated by the United States of America Food and Drug Administration (FDA), Teva Parenteral Medicines analytical chemists discovered a chromatographically resolvable degradant that was suspected, on the basis of a mass 32 amu higher than expected, to be oxytocin trisulfide (3). A contemporaneous publication (Hawe et al. 2009) reinforced their suspicion. Authentic material in excess of 100 mg was required in order to definitively establish the identity of this degradant, and as a reference compound for further studies that were expected in the regulatory process; this included determination of the relative response factor of the trisulfide in the analytical method used by Teva Parenteral Medicines.
In the present contribution, we report on the larger-scale streamlined synthesis, purification, and characterization of oxytocin trisulfide (3) in good yield and excellent purity, and describe technical improvements and new observations made along the way. In particular, we provide convincing evidence for the formation, during synthesis directed at 3, of tetrasulfide analogue 4, and note the presence of even higher homologues as unexpected by-products.
Materials and Methods
General
All solvents and reagents used were of peptide synthesis, HPLC, or LC-MS grade, as appropriate, from established suppliers such as American Bioanalytical, J.T. Baker, Mallinckrodt, EMD Millipore (Supelco), and Sigma-Aldrich. Nα-9-Fluorenylmethyloxycarbonyl (Fmoc) l-amino acid derivatives with appropriate side-chain protection, as well as Nα-(tert-butyloxycarbonyl)-l-S-acetamidomethylcysteine [Boc-l-Cys(Acm)-OH], were from Novabiochem and other high-quality suppliers. 4-(2′,4′-Dimethoxyphenyl-Fmoc-aminomethyl)phenoxy (Rink amide) polystyrene (PS) resin was from Agilent Technologies. Isopropyl xanthic anhydride (5) was prepared by the procedure given in Schroll and Barany (1986), and was a starting material for in-house (Minnesota) preparation of the Cys building block 9 (see later). 1H and 13C nuclear magnetic resonance (NMR) spectra were acquired on a VI-300 Inova instrument from Varian.
Analytical reversed-phase high performance liquid chromatography (HPLC) was conducted on a Gilson instrument [model 231 sample injector, 401 syringe dilutor, 155 UV/vis detector set at 220 and 280 nm], with H2O (A) and CH3CN (B) mobile phases, each containing 0.1% trifluoroacetic acid (TFA), on YMC C18 columns (4.6 × 150 mm, 5.0 μm particle size, 12 nm pore size). Elution was with a gradient of A:B from 99:1 to 50:50 over 25 min, 0.8 mL/min flow rate. Liquid chromatography-electrospray ionization mass spectrometry (LC/ESI-MS) data were obtained on a Gilson HPLC instrument, with the same chromatography conditions but a shorter column (4.6 × 50 mm, 3.0 μm particle size), coupled to MS detection on a Thermo quadrupole ion trap (LCQ) with a standard ESI source (Stafford 2002).
In parallel, ultraperformance liquid chromatography-electrospray ionization mass spectrometry (UPLC/ESI-MS) was carried out with a Waters Acquity UPLC coupled to a Waters Synapt G2 quadrupole time-of-flight (QTOF) mass spectrometer, with H2O (A) and CH3CN (B) mobile phases, each containing 0.1% formic acid, using a Waters Acquity UPLC BEH C18 (2.1 × 50 mm, 1.7 μm particle size) column at 35 °C. Peptides were eluted using the following gradient elution system at a flow rate of 0.4 mL/min: isocratic at A:B 97:3 for 3 min, then linear from 97:3 to 3:97 over the next 7 min, then isocratic at 3:97 for 3 min, then linear to 97:3 for 1 min, total run time 20 min. Electrospray ionization mass spectra were acquired over the range m/z 150–1500 every 0.1 s during the chromatographic separations. Simultaneous to this, high energy (MSe) spectra (trap collision energy ramped 15 to 45 eV) were acquired every 0.1 s. Lockspray calibration spectra (leucine enkephalin, m/z 556.2771) were collected every 10 s during the analysis to assure accurate mass measurement. The following instrumental parameters were used for MS detection: Capillary: 2.8 kV; Sample Cone: 35 V; Extraction Cone: 4 V; Source temperature: 100 °C; desolvation temperature: 350 °C; cone gas: 0 L/h; desolvation gas: 800 L/h.
Preparative chromatography was run on a Gilson instrument [model 306 pumps, 811 C dynamic mixer, 805 manometric module, 215 fraction collector, 155 UV/vis detector set at 220 and 280 nm], using a YMC ODS AQ (C18;19 × 230 mm, 10 μm particle size, 12 nm pore size) column, at a flow rate of 15 mL/min. The column was equilibrated for 15 min with A:B (99:1), after which the crude peptide (∼ 125 mg) was loaded in A:B (9:1), and then eluted with a linear gradient from 9:1 to 3:2 over 90 min.
Amino acid analysis (AAA)Footnote 4 involved gas-phase hydrolysis of the purified peptide and quantitation of the amino acids by ion-exchange chromatography on a Hitachi model L8900, with ninhydrin post-column derivatization, against an analytical standard, according to manufacturer’s protocols. Thus, a portion of the purified peptide 3 was weighed on an analytical balance and dissolved volumetrically in water; a known volume of this solutions (~ 10 µg of peptide) was placed in a clean glass microfuge tube and dried by centrifugal evaporation. The microfuge tube was placed in a glass vessel that was loaded into a hydrolysis vessel on an Eldex “Pico Tag” hydrolysis workstation with 6 n HCl containing 2% phenol. Following three cycles of evacuation and nitrogen blanketing, the vessel was evacuated one final time and heated at 110 °C for 18 h. The peptide hydrolysate was dissolved in a known volume of citrate buffer and analyzed by cation exchange chromatography with pH step gradients and quantitated by post-column derivatization with ninhydrin against an amino acid standard. Peptide concentration was determined as an average of the amino acid concentrations of all the amino acids except for Cys, which was destroyed during hydrolysis; the side-chain amide groups of Asn and Gln were hydrolyzed during HCl treatment and so these residues were quantified as Asp and Glu, respectively. Net peptide content was calculated based on the gross weight concentration of the peptide sample versus the experimentally determined value above.
(Chlorocarbonyl)Disulfanyl Chloride (6)
Following Schroll and Barany (1986), sulfuryl chloride (25 mL, 41.5 g, 0.4 mol) was added to a suspension of isopropyl xanthic anhydride (5, 19.9 g, 83 mmol) in petroleum ether (100 mL), and the reaction mixture was brought to reflux whereupon it became homogeneous. The reaction proceeded for 3 h, after which concentration in vacuo [rotary evaporator; bath temperature not to exceed 40 °C] provided an orange liquid (23.9 g), which was immediately purified by short path vacuum distillation (4.4 g, 33%); bp 29–41 °C (0.47–0.54 mm). 13C NMR (75 MHz, CDCl3): δ 163.8. Purity of the title product was confirmed by an analytical N-methylaniline assay (Barany et al. 1983; Schroll and Barany 1986), which provided N,N’-dimethyl-N,N’-diphenylcarbamoyl)disulfane that was pure as judged by its 1H NMR spectrum (CDCl3) which matched that reported in the literature (Barany and Mott 1984; Schroll and Barany 1986; Henley et al. 2015; diagnostic singlets of equal height at δ 3.38 and 3.35).
N α-(tert-Butyloxycarbonyl)-S-[(N-methyl-N-phenylcarbamoyl)disulfanyl]-l-cysteine [Boc-Cys(Ssnm)-OH] (9)
The title compound was prepared according to Chen et al. (1997), who started with Nα-(tert-butyloxycarbonyl)-S-(acetamidomethyl)-l-cysteine (Boc-Cys(Acm)-OH, 7), and carried out a series of steps (3 mmol scale) culminating in a flash chromatography purification, resulting in a 49% yield of a white solid, mp 104–107 °C (Scheme 2). For this work, the literature procedure was reproduced at several scales (i.e., 2 to 10 mmol), with comparable results, but the product was obtained as an oil. This oil, analyzed by HPLC just prior to its use in SPPS, showed a purity of 87%. It solidified after several days under high vacuum (< 0.1 mm) to a weighable foam.
Linear Protected Oxytocin Rink Amide Polystyrene Resin
The first eight residues of oxytocin were assembled, in C ⟶ N order, on a Rink amide polystyrene resin (4.0 g, 0.6 mmol/g; 24 × 0.1 mmol = 2.4 mmol). Fmoc-amino acids with appropriate side-chain protection (Scheme 2) were coupled via O-benzotriazole-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HBTU) protocols (30 min) in N,N-dimethylformamide [6-fold excess of amino acid derivative, 5.4-fold excess each of 1-hydroxybenzotriazole (HOBt) and HBTU; 12-fold excess of N-methylmorpholine (NMM); these conditions corresponded to an activated amino acid concentration of ∼ 0.25 m. A small portion of the resulting octapeptide resin, H-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Cys(Trt)-Pro-Leu-Gly–Rink amide–PS, was cleaved [TFA–iPr3SiH–phenol–H2O (92.5:2.5:2.5:2.5); Solé and Barany 1992] and shown by subsequent LC-MS analysis to be the desired octapeptide (> 95%). Next, the linear synthesis was completed [using essentially all of the peptide-resin which included 2.4 mmol peptide], by adding Boc-Cys-(Ssnm)-OH (9, 1.50 g, 3.6 mmol, 1.5 equiv.) along with HOBt (540 mg, 3.6 mmol) and N,N’-diisopropylcarbodiimide (DIPCDI) (0.53 mL, 3.6 mmol) in DMF (30 mL) for 3 h; concentration of activated Boc-Cys(Ssnm)-OH was ∼ 0.12 m). An additional portion of DIPCDI (0.2 mL, 1.2 mmol) was added after 3 h, meaning that the total coupling time was 4 h. At the end of this period, a negative ninhydrin test (Kaiser et al. 1970) indicated that acylation was complete.
Oxytocin Trisulfide (3)
The protected linear nonapeptide-resin (see above) was rinsed with DMF (5 ×) and CH2Cl2 (5 ×), and then treated with TFA–iPr3SiH–phenol–H2O (92.5:2.5:2.5:2.5; “Reagent B,” 200 mL) for 2 h (Solé and Barany 1992). The cleaved resin was removed by filtration, and the TFA-based solution was added to cold ether–hexane (1:1; 1600 mL) so as to precipitate the crude peptide. The precipitate was collected by centrifugation, and then taken up essentially immediately in CH3CN–H2O (1:1, with 0.1% TFA; pH ∼ 3; ∼ 200 mL). LC-MS analysis less than 1 h after this solution was created showed that cyclization was complete, with a mixture of 2, 3, and 4 being produced in an approximate ratio of 1:6:2 by integration of peaks in the LC trace (detection based on UV absorbance at 220 nm). The bulk reaction was lyophilized to provide a white powder (1.5 g, 64%) that was purified by preparative HPLC (conditions given earlier) in twelve separate injections of ~ 125 mg each. The fractions containing oxytocin (disulfide) (2), oxytocin trisulfide (3), and oxytocin tetrasulfide (4) were pooled separately, and lyophilized. This provided desired 3 (166 mg, 6.7% yield, > 95% purity), along with disulfide 2 (308 mg, 12.7% yield, ∼ 90% purity) and a highly impure fraction (80 mg) containing tetrasulfide 4 admixed with other co-products. Quantitative amino acid analysis of the purified oxytocin trisulfide (3) showed Asx, 1.01; Glx, 1.01; Pro, 0.98; Gly, 1.01; Ile, 1.00; Leu, 1.01; Tyr, 0.98; and a net peptide content of 76.4%. ESI-MS: m/z calcd 1039.42; found 1039.24 [(3) + H]+.
Results and Discussion
Synthetic Organosulfur Chemistry
The first task requiring specialized expertise was the production of (chlorocarbonyl)disulfanyl chloride (6). Although there have been several previously reported routes to this key reagent (Böhme et al. 1981, Barany and Mott 1984, Schroll and Barany 1986, Chen et al. 1997, Tobón et al. 2011), we opted for the method discovered by Schroll and Barany (1986) that involves careful chlorination of isopropyl xanthic anhydride (5) (Scheme 2). The 6 thus obtained was redistilled, and then, according to the procedure of Chen et al. (1997), was reacted with Boc-Cys(Acm)-OH (7), followed shortly thereafter by a quench with N-methylaniline, to provide the needed Boc-Cys(Ssnm)-OH (9) building block.

Organosulfur chemistry in support of controlled synthesis of peptide trisulfides
Linear Solid-Phase Peptide Synthesis
An overall route first used by Chen et al. (1997) was followed, with some changes in the protection schemes and other details (Scheme 3). More specifically, we used triphenylmethyl (trityl = Trt) in place of 2,4,6-trimethoxybenzyl (Tmob), to protect the side-chains of Asn, Gln, and the internal Cys residue, and we used a 4-(Fmoc-aminomethyl-2′,4′-dimethoxyphenyl)phenoxy polystyrene resin (Rink amide − PS) (0.45 mmol/g) whereas Chen et al. (1997) worked with a 5-(4-Fmoc-aminomethyl-3,5-dimethoxyphenoxy)valeryl poly(ethylene glycol) − polystyrene (PAL − PEG − PS) graft support. Fmoc solid-phase peptide synthesis (SPPS) chemistry with primarily HBTU coupling protocols on an Abimed/Gilson AMS 422 instrument at the New England Peptide facility was used to assemble Boc-Cys(Ssnm)-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Cys(Trt)-Pro-Leu-Gly–Rink amide − PS (Scheme 3, top line). This was done several times at varying scales.

Solid-phase synthesis and solution cyclization to produce oxytocin trisulfide (3), via intermediate 11. Upon completion of the deprotection/cleavage step, the cleaved resin was removed by filtration and the filtrate was added to cold Et2O–hexane (1:1, 8 volumes) to precipitate crude peptide, which was collected by centrifugation and then dissolved in CH3CN–H2O (1:1) containing 0.1% TFA (overall pH ∼ 3)
Cleavage of Peptide-Resin and Spontaneous Cyclization
The peptide-resin (Scheme 3) was cleaved with “Reagent B” [TFA–iPr3SiH–phenol–H2O (92.5:2.5:2.5:2.5)] (Solé and Barany 1992), and rather rapid cyclization to the trisulfide occurred spontaneously as the peptide was taken up in CH3CN–H2O (1:1) containing 0.1% TFA (net pH ~ 3). Concomitantly, other oxytocin species formed, so that the crude product comprised regular oxytocin (disulfide) (2), oxytocin trisulfide (3), and oxytocin tetrasulfide (4) in an approximate ratio of 1:6:2 (based on integration of peaks on an HPLC trace with UV detection).
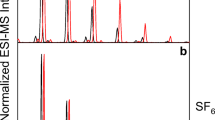
Identifications of peaks were confirmed by ultraperformance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC/QTOF-MS) (Fig. 1). High resolution electrospray ionization mass spectra detected protonated molecular ions, [2 + H]+, [3 + H]+, and [4 + H]+ in the mixture at m/z 1007.4427, 1039.4150, and 1071.3849 respectively. These measured values correspond to the theoretical molecular formulas of (2: C43H66N12O12S2), (3: C43H66N12O12S3), and (4: C43H66N12O12S4), with measurement errors of 1.0 ppm, 0.8 ppm, and 2.7 ppm, respectively. In addition, measurement of the doubly-charged ion at m/z 552.1862 corresponds to the theoretical molecular formula of oxytocin pentasulfide (C43H66N12O12S5), with a measurement error of 4.7 ppm; the corresponding singly-charged protonated molecular ion was not observed.

UPLC/QTOF-MS analysis of a “crude” synthetic oxytocin trisulfide (3) mixture, prior to purification. Buffers and elution conditions are listed in the “General” section of “Materials and Methods.” a Total ion current (TIC) chromatogram for the separation of the pre-purification mixture. b Extracted ion chromatogram of m/z 1007.44, corresponding to detection of disulfide 2. c Extracted ion chromatogram of m/z 1039.42, corresponding to detection of trisulfide 3. d Extracted ion chromatogram of m/z 1071.38 corresponding to detection of tetrasulfide 4. e High resolution mass spectrum of 3, detected in the synthetic mixture, illustrating detection of [3 + H]+ and [3 + 2 H]2+ ions. Theoretical [3 + H]+ = 1039.4159. The measured [3 + H]+ of 1039.4150 corresponds to determination of 3 [molecular formula C43H66N12O12S3]with a measurement error of 0.8 ppm. As detailed in “Results and Discussion,” corresponding spectra for 2 and 4 were used to confirm their identities
Purification and Characterization of Oxytocin Trisulfide (3)
The desired oxytocin trisulfide (3) was obtained after careful reversed-phase preparative HPLC, in an overall isolated yield of about 6.4% based on starting resin, and in excellent purity [> 95%, by analytical HPLC, as shown in Fig. 2, with the very minor by-products clearly identified as reduced oxytocin (1), oxytocin (disulfide) (2), and oxytocin tetrasulfide (4)].Footnote 5 The structure of 3 was confirmed by ESI-MS and amino acid analysis (AAA). The lyophilized product comprised 76% of 3, as quantified by AAA, with the remaining material likely to be counterions from the final purification (trifluoroacetate) plus residual water, as is typical for most peptides.

Analytical HPLC chromatogram, with UV detection at 220 nm, of oxytocin trisulfide (3) after purification and lyophilization. Buffers and elution conditions are listed in the “General” section of “Materials and Methods.” The target compound is peak C, which comprises 96.0% of the total material. The following impurities were identified by ESI-MS: Peak A: Oxytocin (2, 1.7%, m/z = 1007.4); Peak B: Linear (reduced) oxytocin (1, 1.5%. m/z = 1009.2); Peak D: Oxytocin tetrasulfide (4, 0.5%, m/z = 1070.9)
Perspective on Scaled-up Oxytocin Trisulfide Synthesis
In our earlier work (Chen et al. 1997), we investigated multiple directed routes to title peptide 3, focusing on (i) appropriate orthogonally removable pairs of protecting groups for the two cysteine residues (N-terminal and internal); (ii) which cysteine residue would serve as the nucleophile versus which could be the electrofugal moiety, and (iii) the relative merits of on-resin cyclization (taking advantage of pseudo-dilution which favors intramolecular reactions) versus carrying out this critical step in solution.
For the work reported herein, we focused on a strategy involving attack by the internal cysteine (originally protected by S-Trt) on a terminal S-Ssnm moiety, with the cyclization reaction taking place in solution, after acidolytic release of the peptide from the support. By having Ssnm protect the sulfhydryl side-chain of N-terminal cysteine in the oxytocin sequence, we did not have to be concerned about maintaining protection for further deprotection/coupling cycles of Fmoc chemistry, and we were able to use Boc to protect the N-terminal α-amino group. Just as (N-methyl-N-phenylcarbamoyl)sulfenyl (Snm) (Schroll and Barany 1989) is somewhat preferable over (methoxycarbonyl)sulfenyl (Scm) (Brois et al. 1970; Kamber 1973; Hiskey et al. 1975; Mullen et al. 2010], so too is Ssnm somewhat preferable over (methoxycarbonyl)disulfanyl (Sscm) (Mott and Barany 1984, Chen et al. 1997).
The fact that oxytocin trisulfide formation did not require weakly basic catalysis, e.g., from a weak tertiary amine [compare to conditions reported by Mott and Barany (1984)], but already occurred under the acidic conditions of the crude peptide workup, can be attributed to the added facility of any cyclization that is intramolecular. Even so, not just desired trisulfide formed, but also extraneous disulfide and tetrasulfide species were present; this appears to be unavoidable and is discussed in the next section. While for the present study, we did not explicitly look for parallel or antiparallel dimers, these are known to be potential by-products of disulfide-forming reactions, and could very well have formed here as well. However, their amounts were likely to be lower since directed approaches seem to offer more control than co-oxidation type methods.
We should note that non-directed solution-phase approaches to trisulfide formation have been reported; such approaches take advantage of electrophilic “S2+” equivalents that react with free sulfhydryl groups. One suitable reagent, N,N’-thiobisphthalimide (TBPI), which by now is commercially available, can be applied to peptide bis(thiols) in dilute solution (< 0.1 mm)Footnote 6 to form cyclic peptide trisulfides (Lundin et al. 1994; Moutiez et al. 1997). Furthermore, sequential addition of two peptide free thiols to TBPI has been described for production of unsymmetrical trisulfide peptides (Lundin et al. 1994). For completeness, we refer to the elegant work of Erlanson and Wells (1998) where commercially available bis(tetrabutylammonium)hexasulfide was used to convert peptide bis(thiols) to a mixture of the corresponding cyclic di-, tri-, tetra-, and pentasulfides.
Possible Mechanisms to Explain Formation of Disulfide (2) and Tetrasulfide (4)
The directed route chosen in this study led to a facile synthesis of oxytocin trisulfide (3) in acceptable yield (6–7%, overall from starting resin), but the corresponding disulfide 2 and tetrasulfide 4 species also formed as by-products, among others,Footnote 7 despite our use of optimized reaction protocols. These findings were already anticipated based on our earlier comprehensive academic-scale publication (Chen et al. 1997). The simplest way to account for the past and present results is to invoke disproportionation, a common issue with even simple model disulfides and higher polysulfides, let alone peptides.
Another paper from our laboratory (Chen and Barany 1996) provided a case study for how trisulfides could form in experiments that were designed to create disulfides. Related, we need to explain a foundational observation for the current studies, namely the formation of a disulfide (and even a tetrasulfide) as degradants from what started as pure trisulfide [see Hawe 2009 as already cited, along with an invaluable investigation by Wiśniewski et al. 2013, and mechanistic proposals reviewed by Yang 2016]. What actually happens in these disparate cases might be more nuanced, and we hope that the speculations that follow will stimulate rigorous mechanistic studies.
First, it is not entirely clear at which point in our synthetic route (Scheme 3) the target trisulfide actually forms. It is not out of the question that considerable cyclization already occurs within the highly acid cleavage medium [“Reagent B,” which is mostly TFA, along with scavengers], but it is also plausible that the reaction requires the slightly aqueous acid milieu provided once the crude cleaved peptide product becomes dissolved.
With the goal of finding some common underlying mechanism to explain formation of both disulfide and tetrasulfide by-products from an acyl trisulfane intermediate, i.e., the Cys(Ssnm)-containing oxytocin derivative 11, we propose multifurcated pathways (Scheme 4). Thus, in addition to the expected, well-precedented nucleophilic attack of the Cys6 sulfhydryl onto the central sulfur atom of the Cys1(Ssnm) group which leads to 3 (Scheme 2), the attack could occur on alternative sulfur atoms of the trisulfane [Scheme 4, paths (a) and (b)].
In path (a), the Cys6 thiol attacks the Cys1 β-sulfur, leading to the peptide disulfide 2 along with loss of the dithiocarbamic acid PhN(Me)(C = O)SSH; this latter transient species could continue to decompose to N-methylaniline, carbonyl sulfide (COS), and elemental sulfur [S0]. In path (b), the Cys6 thiol attacks the Cys1 carbonyl-proximal sulfur, in a process that results in peptide tetrasulfide 4, along with loss of carbon monoxide (CO) and N-methylaniline.
As a possible precedent for our proposed path (b), heterocyclic disulfane systems such as 1,2,4-dithiazolidinones react with phosphorus nucleophiles on carbonyl-proximal sulfur (Goerdeler and Nandi 1975; Goerdeler et al. 1981; Ponomarov et al. 2012). Under strong acid conditions, intermediate 11 is likely to be protonated (i.e., 12), which would increase the electrophilicity of the carbamoyl trisulfane and lead to less selectivity in reactions at the three sulfur centers. The overall increased electrophilicity of protonated intermediate 12 could also lead to a reduction in energy differences in the three possible S–S bond forming reactions, leading to partitioning of the reaction through the desired (expected) route along with the less desired paths (a) and (b).

Potential mechanisms for formation of disulfide (2) and tetrasulfide (4) by-products during the conversions of Cys(Ssnm) intermediate 11 that are directed towards oxytocin trisulfide (3)
Our proposal (Scheme 4) is not intended to be the only possible mechanistic explanation for the observed results. For example, the (S0)n species (elemental sulfur) formed in any of several ways [e.g., path (a) in Scheme 4, or decomposition of 11] could react with free cysteine residues to insert one or more sulfur atoms. Additionally, the presence of iPr3SiH as a scavenger during the acidolytic cleavage step provides a reducing environment. As a result, sulfur–sulfur bonds in any of the intermediates or products could result again in (S0)n generation, with subsequent production of higher order polysufanes (reviewed by Steudel 2002, Lindahl and Xian 2023).
The reducing power of iPr3SiH-containing TFA-based cleavage cocktails with respect to S–S bonds has been invoked recently by Yang et al. (2020) as a factor in the formation of free thiol species from resin-bound disulfide-containing peptides. The aforementioned hypothesis is consistent with our finding of linear (reduced) oxytocin (1) as one of the by-products in the crude cleaved peptide (nominally 3) and even as a minor impurity to final purified oxytocin [see Fig. 2; note the presence of 1 (peak B) in what is mostly 3 (peak C)].
Conclusions
Notwithstanding the intellectual stimulation of trying to explain some interesting observations in peptide sulfur chemistry, our primary goal was to fulfill a practical need from our partnering sponsor. Using state-of-the-art methods, the Minnesota–New England Peptide team was able to synthesize significant amounts of authentic pure oxytocin trisulfide (3). Upon receipt of this material by the in-house analytical team at Teva Parenteral Medicines, it was confirmed to co-elute with the degradant observed in their Oxytocin Injection stability studies (data not shown). This result validated the identification of the aforementioned degradant. As trisulfide motifs continue to emerge in surprising ways in vitro and in vivo, we hope that our results will inform future work in this area.
Notes
Information from the United States of America Food and Drug Administration (FDA) website. See https://labels.fda.gov/ in general, and specifically https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/018261s031lbl.pdf (PITOCIN label).
Further biological indications of the hormone, as yet not harnessed clinically, have been reviewed (Magon and Kalra 2011).
In the nomenclature of contemporary organosulfur chemistry, compounds with two are more linearly connected sulfurs are referred to as (poly)sulfanes (suffix “ane”), but the “ide” suffix is so widely entrenched historically that we continue to name the range of peptide derivatives described throughout this article as di-, tri-, tetra-, etc., -sulfides.
The AAA protocol used adheres to guidance from the United States Pharmacopeia (USP) for quantitation of peptide and protein concentration following hydrolysis to constituent amino acids; USP < 1052>.
Two reviewers correctly pointed out an apparent inconsistency between the distribution of products in the crude material and the isolated yields after purification. We believe that the isolation of pure oxytocin (disulfide) (2) was highly efficient [and matches the expected amount], whereas the isolation of pure trisulfide (3) was not nearly as efficient because we placed a premium on obtaining highly pure material over maximizing the yield. It is also possible that in the leadup to purification, some of the 3 that had been produced may have lost a sulfur atom, hence raising the yield of 2 while lowering the yield of 3.
The requirement for highly dilute concentrations during literature solution cyclizations should be contrasted to the effective concentration for the cyclization method described in this paper, which can be estimated as two orders of magnitude higher, i.e., 7 to 10 mm.
Higher polysulfides are almost surely present, albeit in diminishing amounts. For the present work, we provide strong support for the presence of oxytocin pentasulfide, based on UPLC/QTOF-MS of the crude product mixture.
References
Andersson C, Edlund PO, Gellerfors P, Hansson Y, Holmberg E, Hult C, Johansson S, Kördel J, Lundin R, Mendel-Hartvig I, Norén B, Wehler T, Widmalm G, Öhman J (1996) Isolation and characterization of a trisulfide variant of recombinant human growth hormone formed during expression in Escherichia coli. Int J Pept Prot Res 47:311–321. https://doi.org/10.1111/j.1399-3011.1996.tb01360.x
Andreu D, Albericio F, Solé NA, Munson MC, Ferrer M, Barany G (1994) Formation of disulfide bonds in synthetic peptides and proteins. In: Pennington MW, Dunn BM (eds) Methods in Molecular Biology, vol 35. Humana Press, Totowa, pp 91–169. https://doi.org/10.1385/0-89603-273-6:91
Annis I, Hargittai B, Barany G (1997) Disulfide bond formation. In: Fields GB (ed) Solid-phase peptide synthesis. Meth Enzymol 289:198–221. https://doi.org/10.1016/s0076-6879(97)89049-0
Barany G, Mott AW (1984) Chemistry of bis(alkoxycarbonyl)polysulfanes and related compounds. J Org Chem 49:1043–1051. https://doi.org/10.1021/jo00180a018
Barany G, Schroll AL, Mott AW, Halsrud DA (1983) A general strategy for elaboration of the dithiocarbonyl functionality, –(C=O)SS–: application to the synthesis of bis(chlorocarbonyl)disulfane and related derivatives of thiocarbonic acids. J Org Chem 48:4750–4761. https://doi.org/10.1021/jo00172a056
Böhme H, Brinkmann M, Steudel H-P (1981) Gewinnung und umsetzungen von chlordisulfanylderivaten der kohlensäure. Liebigs Ann Chem 1981:1244–1251. https://doi.org/10.1002/jlac.198119810711
Brois SJ, Pilot JF, Barnum HW (1970) New synthetic concepts in organosulfur chemistry. I. New pathway to unsymmetrical disulfides. The thiol-induced fragmentation of sulfenyl thiocarbonates. J Am Chem Soc 92:7629–7631. https://doi.org/10.1021/ja00729a042
Canova-Davis E, Baldonado IP, Chloupek RC, Ling VT, Gehant R, Olson K, Gillece-Castro BL (1996) Confirmation by mass spectrometry of a trisulfide variant in methionyl human growth hormone biosynthesized in Escherichia coli. Anal Chem 68:4044–4051. https://doi.org/10.1021/ac9605915
Chen L, Barany G (1996) N-dithiasuccinoyl (Dts)-glycine: a novel oxidation reagent for the formation of intramolecular disulfide bridges under mild conditions. Lett Pept Sci 3:283–292. https://doi.org/10.1007/bf00127662
Chen L, Zoulíková I, Slaninová J, Barany G (1997) Synthesis and pharmacology of novel analogues of oxytocin and deaminooxytocin: directed methods for the construction of disulfide and trisulfide bridges in peptides. J Med Chem 40:864–876. https://doi.org/10.1021/jm9607156
Chen L, Skinner SR, Gordon TD, Taylor JE, Barany G, Morgan BA (1999) Isolation, characterization and synthesis of a trisulfide related to the somatostatin analog lanreotide. In: Tam JP, Kaumaya PTP (eds) Peptides—Frontiers of Peptide Science: Proceedings of the Fifteenth American Peptide Symposium. Kluwer Academic Publishers, Dordrecht, pp 275–276. https://doi.org/10.1007/0-306-46862-X_114
du Vigneaud V (1956) Trail of sulfur research: from insulin to oxytocin. Science 123:967–974. https://doi.org/10.1126/science.123.3205.967
du Vigneaud V (1964) A trail of sulfur research: from insulin to oxytocin (Nobel Lecture, December 12, 1955). Nobel lectures–chemistry–including presentation speeches and laureates’ biographies, 1942–1962. Elsevier, Amsterdam, pp 446–465. https://www.nobelprize.org/uploads/2018/06/vigneaud-lecture.pdf
du Vigneaud V, Ressler C, Swan JM, Roberts CW, Katsoyannis PG, Gordon S (1953) The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J Am Chem Soc 75:4879–4880. https://doi.org/10.1021/ja01115a553
Erlanson DA, Wells JA (1998) Facile synthesis of cyclic peptides containing di-, tri-, tetra-, and pentasulfides. Tetrahedron Lett 39:6799–6802. https://doi.org/10.1016/s0040-4039(98)01461-0
Fabio K, Curley K, Guarneri J, Adamo B, Laurenzi B, Grant M, Offord R, Kraft K, Leone-Bay A (2015) Heat-stable dry powder oxytocin formulations for delivery by oral inhalation. AAPS PharmSciTech 16:1299–1306. https://doi.org/10.1208/s12249-015-0314-0
Goerdeler J, Nandi K (1975) Über thioacyl-isocyanate, XI. Verfahren zu ihrer herstellung aus dithiazolonen. Chem Ber 108:3066–3070. https://doi.org/10.1002/cber.19751080926
Goerdeler J, Tiedt M-L, Nandi K (1981) Über thioacyl-isocyanate, XV. Reaktionen mit nucleophilen C‐verbindungen. Chem Ber 114:2713–2722. https://doi.org/10.1002/cber.19811140805
Góngora-Benítez M, Tulla-Puche J, Albericio F (2014) Multifaceted roles of Disulfide Bonds. Peptides as therapeutics. Chem Rev 114:901–926. https://doi.org/10.1021/cr400031z
Gu S, Wen D, Weinreb PH, Sun Y, Zhang L, Foley SF, Kshirsagar R, Evans D, Mi S, Meier W, Pepinsky RB (2010) Characterization of trisulfide modification in antibodies. Anal Biochem 400:89–98. https://doi.org/10.1016/j.ab.2010.01.019
Hawe A, Poole R, Romeijn S, Kasper P, van der Heijden R, Jiskoot W (2009) Towards heat-stable oxytocin formulations: analysis of degradation kinetics and identification of degradation products. Pharm Res 26:1679–1688. https://doi.org/10.1007/s11095-009-9878-2
Henley MJ, Schroll AL, Young VG, Barany G (2015) Crystal structures of (N-methyl-N-phenylamino)(N-methyl-N-phenylcarbamoyl)sulfide and the corresponding disulfane. Acta Crystallogr E Cryst Commun 71:1371–1374. https://doi.org/10.1107/S2056989015018289
Hiskey RG, Muthukumaraswamy N, Vunnam RR (1975) Sulfur-containing polypeptides XVII. The S-carbomethoxysulfenyl derivative as a protective group for cysteine. J Org Chem 40:950–953. https://doi.org/10.1021/jo00895a600
Kaiser E, Colescott RL, Bossinger CD, Cook PI (1970) Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem 34:595–598. https://doi.org/10.1016/0003-2697(70)90146-6
Kamber B (1973) Die gezielte synthese offenkettiger asymmetrischer cystinpeptide mittels thiol-induzierter fragmentierung von sulfenylthiocarbonanten. Insulinfragmente mit intakter disulfidbrücke A20-B19. Helv Chim Acta 56:1370–1381. https://doi.org/10.1002/hlca.19730560420
Kshirsagar R, McElearney K, Gilbert A, Sinacore M, Ryll T (2012) Controlling trisulfide modification in recombinant monoclonal antibody produced in fed-batch cell culture. Biotech Bioengin 109:2523–2532. https://doi.org/10.1002/bit.24511
Lau JL, Dunn MK (2018) Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg Med Chem 26:2700–2707. https://doi.org/10.1016/j.bmc.2017.06.052
Lindahl S, Xian M (2023) Recent development of polysulfides: chemistry and biological applications. Curr Opin Chem Biol 75:e102325. https://doi.org/10.1016/j.cbpa.2023.102325
Lundin RHL, Norén BE, Olof Edlund P (1994) A convenient method for the synthesis of peptide trisulfides. Tetrahedron Lett 35:6339–6342. https://doi.org/10.1016/S0040-4039(00)73427-7
Magon N, Kalra S (2011) The orgasmic history of oxytocin: love, lust, and labor. Indian J Endocr Metab 15:S156–S161. https://doi.org/10.4103/2230-8210.84851
Malm M, Madsen I, Kjellström J (2018) Development and stability of a heat-stable formulation of carbetocin for the prevention of postpartum haemorrhage for use in low and middle-income countries. J Pep Sci 24:e3082. https://doi.org/10.1002/psc.3082
Manning M, Misicka A, Olma A, Bankowski K, Stoev S, Chini B, Durroux T, Mouillac B, Corbani M, Guillon G (2012) Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J Neuroendocrinol 24:609–628. https://doi.org/10.1111/j.1365-2826.2012.02303.x
Mott AW, Barany G (1984) A new method for the synthesis of unsymmetrical trisulfanes. Synthesis. https://doi.org/10.1055/s-1984-30922
Mott AW, Słomczyñska U, Barany G (1986) Formation of sulfur-sulfur bonds during solid-phase peptide synthesis: application to the synthesis of oxytocin. In: Castro B, Martinez J (eds) Forum Peptides Le Cap d’Agde 1984. Les Impressions Dohr, Nancy, pp 321–324
Moutiez M, Lippens G, Sergheraert C, Tartar A (1997) Vasopressin trisulphide: synthesis, NMR study and affinity studies with V1 and V2 subtypes receptors. Bioorg Med Chem Lett 7:719–724. https://doi.org/10.1016/S0960-894X(97)00050-4
Mullen DG, Weigel B, Barany G, Distefano MD (2010) On-resin conversion of Cys(Acm)-containing peptides to their corresponding Cys(Scm) congeners. J Pept Sci 16:219–222. https://doi.org/10.1002/psc.1223
Nielsen RW, Tachibana C, Hansen NE, Winther JR (2011) Trisulfides in proteins. Antiox Redox Signal 15:67–75. https://doi.org/10.1089/ars.2010.3677
Owen MD, Cassidy AL, Weeks AD (2021) Why are women still dying from obstetric hemorrhage? A narrative review of perspectives from high and low resource settings. Int J Obstetric Anesthesia 46:102982. https://doi.org/10.1016/j.ijoa.2021.102982
Parmentier B, Moutiez M, Tartar A, Sergheraert C (1994) Preparation of trisulfide derivatives of cystine and their formation as by-products during peptide synthesis. Tetrahedron Lett 35:3531–3534. https://doi.org/10.1016/S0040-4039(00)73228-X
Ponomarov O, Laws AP, Hanusek J (2012) 1,2,4-Dithiazole-5-ones and 5-thiones as efficient sulfurizing agents of phosphorus(III) compounds—a kinetic comparative study. Org Biomol Chem 10:8868–8876. https://doi.org/10.1039/c2ob26460a
Pristatsky P, Cohen SL, Krantz D, Acevedo J, Ionescu R, Vlasak J (2009) Evidence for trisulfide bonds in a recombinant variant of a human IgG2 monoclonal antibody. Anal Chem 81:6148–6155. https://doi.org/10.1021/ac9006254
Ragnarsson U (2007) The Nobel trail of Vincent du Vigneaud (historical essay). J Pept Sci 13:431–433
Schroll AL, Barany G (1986) Novel symmetrical and mixed carbamoyl and amino polysulfanes by reactions of (alkoxydichloromethyl)polysulfanyl substrates with N-methylaniline. J Org Chem 51:1866–1881. https://doi.org/10.1021/jo00360a039
Schroll AL, Barany G (1989) A new protecting group for the sulfhydryl function of cysteine. J Org Chem 54:244–247. https://doi.org/10.1021/jo00262a051
Solé NA, Barany G (1992) Optimization of solid-phase synthesis of [Ala8]-dynorphin A. J Org Chem 57:5399–5403. https://doi.org/10.1021/jo00046a022
Stafford G (2002) Ion trap mass spectrometry: a personal perspective. J Am Soc Mass Spectrom 13:589–596. https://doi.org/10.1016/S1044-0305(02)00385-9
Steudel R (2002) The chemistry of organic polysulfanes R–Sn–R (n > 2). Chem Rev 102:3905–3946. https://doi.org/10.1021/cr010127m
Tobón YA, Cozzarín MV, Wang WG, Ge MF, Della Védova CO, Romano RM (2011) Vibrational and valence photoelectron spectroscopies, matrix photochemistry, and conformational studies of ClC(O)SSCl. J Phys Chem A 115:10203–10210. https://doi.org/10.1021/jp204789h
Widmer M, Piaggio G, Nguyen TMH, Osoti A, Owa OO, Misra S, Coomarasamy A, Abdel-Aleem H, Mallapur AA, Qureshi Z, Lumbiganon P, Patel AB, Carroli G, Fawole B, Goudar SS, Pujar YV, Neilson J, Hofmeyr GJ, Su LL, de Carvalho JF, Pandey U, Mugerwa K, Shiragur SS, Byamugisha J, Giordano D, Gülmezoglu AM (2018) Heat-stable carbetocin versus oxytocin to prevent hemorrhage after vaginal birth. Obstet Gynecol Survey 73:613–614. https://doi.org/10.1097/OGX.0000000000000616
Wiśniewski K (2019) Design of oxytocin analogs. In: Goetz G (ed) Cyclic peptide design, vol 2001. Springer, New York, pp 235–271. https://doi.org/10.1007/978-1-4939-9504-2_11
Wiśniewski K, Finnman J, Flipo M, Galyean R, Schteingart CD (2013) On the mechanism of degradation of oxytocin and its analogues in aqueous solution. Pept Sci 100:408–421. https://doi.org/10.1002/bip.22260
Yang Y (2016) Cys disulfide-related side reactions in peptide synthesis. In: Yang Y (ed) Side reactions in peptide synthesis. Tsinghua University Press, London, pp 299–310. https://doi.org/10.1016/B978-0-12-801009-9.00013-6
Yang Y, Hansen L, Badalassi F (2020) Investigation of on-resin disulfide formation for large-scale manufacturing of cyclic peptides: a case study. Org Proc Res Dev 24:1281–1293. https://doi.org/10.1021/acs.oprd.0c00168
Acknowledgements
We thank Teva Parenteral Medicines, Inc. for sponsorship of this research. We are grateful to Brandon C. Smith of Biosyn for help in retrieving data from a dozen years ago and for insightful current discussions, and appreciate the contributions of Kristin M. Coari and Yi Z. Wang to experiments aimed at the efficient creation of compound 6. Finally, we thank Kaeli Hammer for assistance with graphics.
Author information
Authors and Affiliations
Contributions
Experimental design and supervision, RPH and GB; Organosulfur chemistry, PTG and AMS; Peptide synthesis, KD and RPH; Peptide purification and analytical characterization, MAB and RPH; UPLC/ESI-MS, JJD; Writing, GB and RPH; Background and literature review, RPH, AB, and GB. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
None of the authors have competing interests that are relevant to the content of this article. None of the authors have any relevant financial interests to disclose. In the early 2010s, although not overlapping with the duration of the sponsored support to his research program at the University of Minnesota, GB consulted for Teva Parenteral Medicines, and received appropriate remuneration.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hammer, R.P., Butrie, M.A., Davidson, K. et al. Scaled-up Synthesis and Characterization of Oxytocin Trisulfide. Int J Pept Res Ther 30, 5 (2024). https://doi.org/10.1007/s10989-023-10580-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-023-10580-9