
Abstract
In this work binding properties and conformational changes in alpha lactalbumin (α-LA) upon interaction with sorafenib were investigated by spectrofluorimetry, UV-Vis spectroscopy, Fourier transform infrared spectroscopy and molecular modeling methods. The fluorescence spectroscopic results revealed that sorafenib could effectively quench the intrinsic fluorescence of alpha lactalbumin through a static quenching mechanism. Evaluation of the thermodynamic parameters (ΔH0 = –120.167 kJ mol–1, ΔS0 = –309.507 J mol–1 K–1) suggested that the binding process was spontaneous while hydrogen bonds and van der Waals forces played a major role in this interaction. The fluorescence, UV-absorption and FT-IR spectra showed that conformational changes occurred in alpha lactalbumin structure after interaction with sorafenib. The molecular docking studies showed one binding site in alpha lactalbumin which most of its interactions are hydrophobic. The value of calculated docking ΔG0 (–27.11232 kJ mol–1) (is in agreement with those obtained from fluorescence spectroscopy measurement. Finally, molecular dynamics simulation was performed on the best docked complex by considering the permanence and flexibility of α-LA–sorafenib complex in the binding site.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Whey proteins are globular proteins which constitute 20% of all milk proteins. alpha lactalbumin (α‑LA) type of whey protein is found in the milk of all mammals. It has molecular weight of 14.2 kDa and isoelectric point of 4.2 to 4.5 [1]. It has been reported that α-LA has many physiological functions, such as reduction of stress, opioid activity, antihypertensive action, regulation of cell growth and immunomodulation [2, 3]. In acidic pH and low ionic strength, α-LA can form a molten globule structure (incompletely folded tertiary structure). In this structure, the protein can bind several hydrophobic colorants and lipids [4]. Under normal conditions, the tertiary structure of a-LA is consist of a large domain (α) and a small domain (β) divided by a cleft. It made up of eight cysteines which form four disulfide bridges [5] and showed that the α-LA molecule has high-affinity binding site for Ca2+ and the weak binding of Zn2+. The removal of bound calcium greatly decreases the thermal stability of a-LA, however the protein retains essentially the same folded conformation [6]. Multimeric α-LA was shown to bind to the cell surface, go into the cytoplasm, and accumulate in cell nuclei, which is consistent with its ability to induce apoptosis. This multimeric form appears to exist in human milk [7].
Delavari and others discovered that The binding of vitamin D at the α-LA binding site is stable and has a hydrophobic interactions [8].
Some drugs that are used by the mother may have influence of on composition of mother’s milk. The milk volume may be decreased or increased by some drugs, furthermore many of drugs are transfer to the milk, resulting in exposure to the child [9]. Although the general opinion is to be conservative with drug use during lactation, the number of drugs that are usually compatible with breastfeeding is much larger than the number of drugs that have been correlated with adverse effects on infants [10]. For a number of drugs frequently used by lactating women, such as anti-cancer drugs, information about the effects on the infant is still limited, and use of these drugs during lactation thus warrants caution.
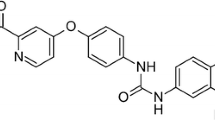
Sorafenib, which its chemical name is 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]-phenoxy]-N-methyl-pyridine-2-carboxamide, is a type of biological agent called a protein tyrosine kinase inhibitor (TKI). Tyrosine kinase is a protein which acts as a chemical messenger (an enzyme). There are a number of different tyrosine kinases that blocks cancer cells to grow. in two ways. It can stop signals that tell to cancer cells to grow and can also stops cancer cells blood vessels formation, which they need to grow. Treatment that stops the formation of blood vessels is called anti-angiogenesis treatment [11–13]. Until now, sorafenib is confirmed for the treatment of advanced renal cell carcinoma, hepatocellular carcinoma [14], and radioactive iodine resistant advanced thyroid carcinoma [15]. Furthermore, it has also been demonstrated that sorafenib has the preclinical and clinical activity against several other tumor types such as prostate cancer, non-small cell lung cancer and so on [16]. Many ligands such as nanoparticles, drugs bind reversibly to the human plasma protein. Thus, they are bounded in the form of complex with them [17, 18].
Sorafenib can bound to human plasma proteins extensively. This drug is widely distributed to tissues, mainly indicative of its lipophilic properties. In preclinical models, 27.3% of the administered dose was recovered in the milk within 32 hr and the total exposure of sorafenib and its metabolites in milk was higher than for maternal plasma with a milk-to plasma ratio of 4.9. These observations indicate that sorafenib administration should be used with caution in breastfeeding mothers [19].
The aim of this study was to analyze the thermodynamic and binding properties (including binding mechanism, binding constant, and the number of binding sites) of interaction of sorafenib with α-LA under physiological pH conditions. To study binding interactions of sorafenib with α-LA, fluorescence emission [20] and UV-Vis [21] spectroscopies, Fourier transform infrared spectroscopy (FT-IR), molecular docking [22] and molecular dynamics (MD) simulation [23] had been used in this work. This study is expected to provide important insight into further elucidating the transport process of sorafenib in breast feeding.
RESULTS AND DISCUSSION
Fluorescence spectroscopy. One method to study the ligand-protein interaction is based on the measure of changes in the fluorescence intensity of protein during titration with ligand or vice versa [32]. The intensity, the position of emission wavelength and the lifetime are some parameters that are used to characterize a fluorophore. Addition of sorafenib to α-LA decreases its intrinsic fluorescence intensity (Fig. 1). This fluorescence spectral changes are the result of solvent relaxation of fluorescent amino acids, in α-LA such as Trp and Tyr, and give us some important information on the protein ligand interactions.

The fluorescence quenching spectra of α-LA (10 μM) in the presence of different concentrations sorafenib under physiological condition (pH 7.4) at 298 K, λex = 280 nm.
In order to understand the mechanism of the fluorescence quenching, the Stern–Volmer equation is used (Eq. (2)). According to the following equation 1 the slope of this graph is KSV or kqτ0 [33]:
where F0 and F are the steady-state fluorescence intensities in the absence and in the presence of quencher (sorafenib), respectively, KSV is the Stern–Volmer quenching constant which is considered to be a measure of efficiency of fluorescence quenching, Kq is the quenching rate constant of the biomolecule, τ0 is the average lifetime of the biomolecule without quencher (τ0 =10−8 s), and [Q] is the concentration of the quencher. The variation in F0/F is plotted against the concentration of sorafenib to determine (Eq. (2)) whether the interaction is static or dynamic (Fig. 2). In this figure the Stern–Volmer plots for quenching of fluorescence emission of α-LA in the presence of sorafenib at three different temperatures.

Stern–Volmer plots for the quenching of α-LA by sorafenib at different temperatures.
Also linear Stern–Volmer plots indicate the presence of interaction between of fluorophore such as tryptophan accessible to the quencher.
The obtained values of KSV and Kq are listed in Table 1. As shown in this table, the values of KSV decreased by the increasing of temperature. which confirms that the quenching mechanism is static in system.
Binding constant and binding modes. When the quenching mechanism is confirmed as the static quenching, it can be assumed that there are several independent binding sites of ligand to α-LA. The binding constant (Kb) and number of binding sites (n) can be calculated by the following equation [34, 35]:
The values of Kb and n can be calculated from the intercept and slope of the plot of log[(F0 – F)/F] versus log[Q] (Fig. 3). According to this plot the value of Kb is 7.94 × 104 M–1 at 298 K, which suggesting that the binding interaction of sorafenib with α-LA is very strong.

The plot of log[(F0 – F)/F] versus log[Q] for quenching process of sorafenib with α-LA (10 μM) at 298, 303, 308 K, λex = 280 nm.
The values of Kb in investigated temperature are presented in Table 2. The values of n at the studied temperature range are near to 1, indicating that the existence of a single binding site on α-LA for sorafenib and suggesting that there is one high affinity binding site in the α-LA for sorafenib ( small deviation from 1 is the results of experimental uncertainty).
According to experimental data Kb indicating that the binding interaction sorafenib with α-LA is strong, Ksv indicating that is consistent with the high combined rate of sorafenib with α-LA also, The values of Stern–Volmer constant (KSV) and binding constant (Kb) show similar trend on.
In addition, the correlation coefficients (r2) in Fig. 3 three different temperatures are greater than 0.97, which indicated that the interaction of sorafenib with α-LA almost exactly match the site-binding model based on (Eq. (3)).
Thermodynamic parameters and binding forces. The interaction forces between drugs and biomacromolecule can be hydrophobic forces, electrostatic interactions, van der Waals interactions, hydrogen bonds formation, etc. The binding constant Kb of α-LA with sorafenib was measured at three different temperatures (298, 303, 308 K). To elucidate the energy changes during the interaction of sorafenib with α-LA, the thermodynamic parameters where calculated from van’t Hoff plots. If the enthalpy change (ΔH) during formation of complex does not vary significantly over studied temperature range, then the values of entropy change (ΔS) can be determined from the van’t Hoff equation [36] (Eq. (3)):
In the above equation Kb is the binding constant at corresponding temperature and R is the gas constant. The enthalpy changes (ΔH) is calculated from the slope and the entropy change (ΔS) from intercept of the van’t Hoff plot (Fig. 4). Then the free-energy change (ΔG) can estimated from the following equation (Eq. (5))

Van’t Hoff plots for the binding interaction of sorafenib with α-LA.
Based on the viewpoint of Ross and Subramanian [37], since both ΔH0 and ΔS0 are negative, therefore that the main force between sorafenib–α-LA complex is the van der Waals force and/or hydrogen bonding interaction [38]. On the other hands if both ΔH0 and ΔS0 are positive, the main interaction force is a hydrophobic interaction. Finally, if, ΔH0 is almost zero and ΔS0 is positive, the main interaction force is electrostatic forces. The van’t Hoff plot for the interaction of sorafenib with α-LA was shown in Fig. 4. The thermodynamic parameters were calculated and listed in Table 2 which indicated that the value of ΔG is negative, therefore that the interaction process is spontaneous and since the value of ΔS and ΔH are negative, therefore hydrogen bonding and van der Waals forces play the major role during the binding process.
Obtained value of ΔGbinding from the thermodynamic analysis of α-LA with sorafenib is –27.9343, ‒26.3868, –24.8392 kJ mol–1 and the binding constant Kb (7.94 × 104, 3.71 × 104, 1.78 × 104) was measured at three different temperatures (298, 303, 308 K), respectively, which indicated a stronge binding between sorafenib and α-LA, implicating that sorafenib can be stored and carried by the α-LA in breast milk.
The study provides an accurate and full basic data for explaining the binding mechanisms of sorafenib with α-LA and is helpful for understanding its effect on protein function during drug transportation process to mother’s milk and its toxicity for infants in vivo [39, 40].
Infrared spectroscopy. Infrared spectroscopy is used to investigate the secondary structures and dynamics of protein. The band frequencies as a result of amide I and II. vibrations in the IR region provide information about the secondary protein structure (i.e., the amide I band 3.407–3.517 cm−1 and amide II band 1.641cm−1). The information provided by amide I is more valuable due to its sensitivity to protein structure change than amide II. In Fig. 5 IR spectra provides information regarding the changes in α-LA structure during the titration with sorafenib. It is showing a shift in amide I peak from 3471.72 to 3483.29 cm−1 and a slight shift in amide II peak from 1544.70 to 1543.02 cm–1, by increasing the values of sorafenib to α-LA solution which suggesting a change in the secondary structure of α-LA after complex formation.

FT-IR spectra (free α-LA and α-LA + different concentration of sorafenib in aqueous solution.
UV-Vis spectroscopy. UV-Vis absorption spectroscopy is a very simple but effective method for exploring the structural change and understanding the complex formation [32]. The UV spectra of α-LA in the presence of sorafenib were shown in Fig. 6. The results revealed that there were two absorption bands for the α-LA–sorafenib complex. The absorption band near 210 nm reflect the framework conformation of α-LA and weak absorption bands at near 280 nm belonged to the π → π* transition of the aromatic amino acids such as Trp, Tyr and Phe. It was obvious that the intensity of UV-absorption of α-LA increased with the addition of sorafenib. However, the observed red shift of maximum peak of α-LA at 210 nm and blue shift of maximum peak of α-LA at 280 nm were also noticed probably due to the formation of α-LA- sorafenib complex.

UV spectra of α-LA (10 μM) in the presence of sorafenib under physiological condition (pH 7.4) at room temperature.
Molecular docking studies. Molecular docking studies can provide some insights into the interaction between small molecule and bio macromolecule [32, 41]. In this work, to complement our experimental observations and investigate the interactions between sorafenib and various amino acid residues of α-LA in the binding site, molecular docking studies was performed. the results of these studies can show the best probable binding site of sorafenib on α-LA. According to Fig. 7a, sorafenib binds near the flexible loop of alpha domain of α-LA. Sorafenib interacted with Trp104, Thr33, Tyr103, Val99, Trp60, Leu105, Asn56, Lys58, Ile59 and hydrophobic interaction played the major role in the binding. In addition, the Ser47, Ala106 Residues formed two hydrogen bonds with sorafenib (Fig. 7b). The Gibbs free energy (ΔG0) of binding data calculated from docking analyses was equal to –27.11 kJ mol–1 which is near to experimental results in Table 2. The reason for the slight differences between these values is probably due to the differences between the X-ray structure of the protein and the α-LA in the aqueous medium and/or due to the role of water and DMSO molecules in the hydrophobic interaction of sorafenib with α-LA. On the other hand The fluorescence emission experiments have been done in solution environment that is vital condition but the molecular modeling results have been obtained in cyberspace.In addition, experiment conditions in two techniques are different [42, 43].

(a) Stereo image presentation of binding site of α-LA and sorafenib; (b) Lig plot diagram of the interaction between α-LA and sorafenib.
However, the results obtained from the docking studies are in agreement with those from fluorescence spectroscopy measurement in which the binding process is spontaneous.
By comparison between the conformation of sorafenib in complex with free sorafenib (Figs. 8a, 8b), it can be found that the conformation of sorafenib in sorafenib-α-LA complex significantly changes, suggesting that the flexibility of sorafenib plays an important role in increasing the stability of the whole system upon association of sorafenib with α-LA.

The conformation of sorafenib in sorafenib-α-LA complex and free sorafenib. (a) Free sorafenib, (b) complex.
MD simulation analysis. Molecular dynamic simulations were performed on the obtained lowest docking energy’s trajectories and the best conformer structures. Root mean square deviations (RMSD) is one criterion that is used to determine the system’s arrival time to a stable state and to confirm the stability of the simulations [44, 32]. In this work, the RMSD plots of the complexes and α-LA protein alone were obtained from the back bone atoms of protein. The attained RMSD plot of complex and α-LA as a reference (Fig. 9a) shows that the RMSD value was increased sharply until approximately 2 ns, and then was remained constant until the end of the simulation. This phenomenon indicates that the complex was relatively stable after 2 ns, so it is reasonable to investigate the binding site based on the snapshots which have been extracted from 2 to 20 ns. The root mean square fluctuation (RMSF) values is other criteria obtained from MD simulation that is used for investigation of the protein mobility [45]. The RMSF of all residue over all time scale was calculated for α-LA and complex and results was shown in (Fig. 9b). Generally, the fluctuations were found to be very similar to those of pure α-LA and the sorafenib-α-LA complex. However, local changes upon binding of the ligand occurred not only in the binding sites but also in the distant regions of the protein, which may suggest an overall conformational change of the protein. To further analyze the binding mode during the simulation, average conformation of the binding pocket of α-LA complexed with sorafenib was obtained from MD simulation. As can be seen in Fig. 10, the most key residues have been appeared around the sorafenib in the binding pocket on the basis of average MD-simulated complex structure are approximately similar to the results obtained from docking however. Analysis of MD results shows that hydrogen bonds which are formed between NH group of Trp60 and the carbonyl group of sorafenib and NH2 group of the Ala160 were remained stable throughout the simulation, which indicates that these hydrogen bond interactions were strong. In addition, hydrophobic interactions (Val99, Asp44, Tyr103, Asp46, Ile59, Leu105, Trp104, Thr33, Gln54) played an important role in stabilization of sorafenib in the binding pocket (Fig 10).

(a) The root means square deviations (RMSD) versus the MD simulation time for α-LA (orange) and α-LA-sorafenib complex (blue) in the MD simulation system; (b) RMSF value of each residue of α-LA during the entire simulation time, in absence and presence of sorafenib.

Plot of the MD-simulated structures of sorafenib in the binding site of α-LA.
EXPEREMENTAL
Materials and Methods
Chemical and reagents. α-LA from bovine milk-type III-calcium depleted (L 6010) was supplied by Sigma-Aldrich. Sorafenib was procured from Baran Co. (Tehran, Iran). A stock solution of α-LA (10 μM) was prepared in phosphate buffer (0.05 mol L–1, pH 7.4) and were kept in the refrigerator at dark place. Stock solution of sorafenib (4.5 mM) was prepared in DMSO. Experimental solutions of α-LA and sorafenib were prepared by appropriate dilution of their stock solutions.
Spectroscopic experiment. Fluorescence measurements were performed on a PC based spectrofluorometer (JASCO Japan FP-8300) equipped with a Xenon lamp and 1.0 cm quartz cell.
Fluorescence titrations were carried out using a fixed concentration of α-LA (10 × 10–6 mol L–1) in the presence of different amounts of sorafenib (0.0–50.0 × 10–6 mol L–1; interval concentration: 10 × 10–6 mol L–1).The excitation wavelength was set to 280 nm, and the emission spectra were recorded at wavelengths from 300 to 500 nm. Prior to fluorescence measurements, samples were thermally equilibrated at 298, 303, and 308 K for about 10 min. The titrations are done manually using the 10 μL micro syringe. The excitation and emission slit widths were set at 5 nm. In order to decrease the inner filter effect, fluorescence intensity was corrected for the absorption of excited light according to the following equation [24]:
Where \({{F}_{{{\text{cor}}}}}\) and \({{F}_{{{\text{obs}}}}}\) are corrected and observed fluorescence intensity, respectively, \({{A}_{{{\text{ex}}}}}\) and \({{A}_{{{\text{em}}}}}\) are the absorption of the system at excitation and emission wavelength, respectively.
UV spectra of all α-LA solutions in the absence and presence of sorafenib were recorded from 200 to 400 nm at room temperature on a SQ4802 UV-Vis diode-array spectrophotometer equipped with 1.0 cm quartz cells.
Bruker Tensor 27 FT-IR spectrometer coupled with the OPUS software was used to obtained IR spectra (spectral resolution 4 cm−1; 32 scans, from 400 to 4000 cm−1 wavenumber). The IR spectra for α-LA solution in buffer and α-LA–sorafenib solutions were obtained and Then converted to absorbance. The FT-IR results provided evidence of possible conformational changes in the protein molecule.
Molecular docking studies. The chemical structure of sorafenib was sketched in Gauss View 5. Then the structure treated by semi-empirical method (PM3) level and then optimized by density functional theory (DFT) at B3LYP/6-31+g(d,p) level using Gaussian 03W molecular modeling package [25]. The X-ray crystal structure of α-LA was fetched from Protein Data Bank (PDB ID code: 1HFZ) [26]. Then, the polar hydrogen atoms in α-LA molecule were added [27]. In the next step, the partial atomic charges of α-LA and sorafenib were calculated using Gasteiger–Marsili method [28], respectively. Finally, the different conformers of sorafenib were generated by using Lamarckian genetic algorithm (LGA) [29] with the same parameters of each docking. Autodock 4.2 [30] program was employed to generate a docked conformation of sorafenib with α-LA. In order to recognize best binding site of α-LA, grid-size was kept at 40 × 48 × 50 Å with a grid-point spacing of 0.375 Å is investigated to ensure an appropriate docking size of sorafenib accessible. Based on calculated root mean square cluster tolerance between proposed structures, the complexes of sorafenib with α-LA were sorted. Finally, according to the Autodock scoring function, the conformation of α-LA-sorafenib complex with the lowest binding energy was selected as the most stable conformation of α-LA-sorafenib complex.
Molecular dynamic simulations. In order to evaluate the in-vivo changes of the the α-LA and α-LA-sorafenib complex equilibrium and also to investigate the reliability of the docking results, 20 ns the molecular dynamic calculations (simulation and trajectory analysis) was performed in the presence of an explicit solvent by using the GROMACS 5.1.2 program, with the GROMOS 43a7 force field. The topologies parameters using the PRODRG2 Server. The simulation conditions for both protein and complexes were the same. The solvated system was neutralized by adding Na+ counter ions in the simulation box. energy minimization was performed using the the steepest descent algorithm for 50 000 steps. Canonical NVT (constant Number of particles, Volume, and Temperature) and NPT (constant Number of particles, Pressure, and Temperature) ensemble were performed for equilibration. NVT ensemble at 300 K was used with periodic boundary conditions, and the temperature was kept constant by the Berendsen thermostat [31]. NPT ensemble at 300 K was used and pressure was kept constant by the Parrinello–Rahman. Both phase of equilibration was performed by 50 000 steps and 100 ps. Cut-off distances for the calculation of Coulomb and van der Waals interactions were 1.0 nm and time step were set to 2 fs for all phase. Finally, the full system was run at constant temperature (300 K) and pressure (1 bar).
CONCLUSION
In the current study, the interaction of sorafenib with α-LA was investigated by fluorescence, UV-Vis and FT-IR spectroscopies. These procedures were combined with molecular modeling and MD method to further analyze the α-LA-sorafenib system under simulated physiological conditions.
The results of this study indicated that sorafenib can bind to of α-LA and quench the intrinsic fluorescence of α-LA due to the formation of α-LA-sorafenib complex. The number of binding sites in α-LA-sorafenib complex is single and binding interaction with α-LA is relatively strong. The obtained thermodynamic parameters revealed that the main forces in the binding process of sorafenib with α-LA are hydrogen bonding and van der Waals interactions. The UV-Vis and FT-IR results showed that the secondary structure of α-LA slightly changes after binding with sorafenib, while the conformation of sorafenib is obviously change, indicating that the flexibility of sorafenib plays a key role in the binding process of sorafenib with α-LA. MD results showed that the α-LA and α-LA-sorafenib complex achieved stability after about 2 ns. Analysis of sorafenib binding mode after MD showed the existence of different interactions between sorafenib and α-LA.
Finally, the results of this investigations can help us to obtain more information about the influence of sorafenib on the mother’s milk.
REFERENCES
Belatik, A., Kanakis, C., Hotchandani, S., Tarantilis, P., Polissiou, M., Tajmir-Riahi, H., J. Biomol. Struct. Dyn., 2012, vol. 30, pp. 437–447. https://doi.org/10.1080/07391102.2012.682209
Markus, C.R., Olivier, B., Panhuysen, G.E., Van der Gugten, J., Alles, M.S., Tuiten, A., Westenberg, H.G., Fekkes, D., Koppeschaar, H.F., and de Haan, E., Am. J. Clin. Nutr., 2000, vol. 71, pp. 1536–1544. https://doi.org/10.1093/ajcn/71.6.1536
Teschemacher, H., J. Pept. Sci., 1997, vol. 43, pp. 99–117. https://doi.org/10.1002/(SICI)1097-0282(1997)43:2<99:AID-BIP3>3.0.CO;2-V
Kimpel, F., Schmitt, JJ., J. Food Sci, 2015, vol. 80, pp. 2361–2366. https://doi.org/10.1111/1750-3841.13096
Fitz Gerald, R.J., Murray, B.A., Walsh, D.J., J. Nutr., 2004, vol. 134, pp. 980–988. https://doi.org/10.1093/jn/134.4.980S
Chamani, J., J. Mol. Struct., 2010, vol. 979, pp. 227–234. https://doi.org/10.1016/j.molstruc.2010.06.035
Svensson, M., Sabharwal, H., Hkansson, A., Mossberg, A.K., Lipniunas, P., Leffler, H., Svanborg, C., and Linse, S., J. Biol. Chem., 1999, vol. 27, pp. 6388–6396. https://doi.org/10.1074/jbc.274.10.6388
Delavari, B., Saboury, A.A., Atri, M.S., Ghasemi, A., Bigdeli, B., Khammari, A., Maghami, P., Moosavi-Movahed,i, AA., Haertl’e, T., and Goliaei, B., Food Hydrocolloids, 2015, vol. 45, pp. 115–124. https://doi.org/10.1016/j.foodhyd.2014.10.017
Astrup-Jensen, A., Bates, A.C.J., Begg, E.J., Edwards, S., Lazarus, C., Matheson, I., Mountford, P.J., Neville, M.C., Notarianni, L.J., Prentiss Ajay Rane, A., Reinhardt, D., and Walsh, C., Drugs and Human Lactation, Bennett, P.N., Ed., Amsterdam: Elsevier, 1996.
American Academy of Pediatrics, Pediatrics, 2001, vol. 108, pp. 776–789. https://doi.org/10.1542/peds.108.3.776
Amato, R., Harris, P., Dalton, M., Khan, M., Alter, R., Zhai, Q., Brady, J.R., Jac, J., Hauke, R., and Srinivas, S., J. Clin. Oncol., 2007, vol. 10, pp. 153–158. https://doi.org/10.1016/j.clgc.2012.03.001
Takimoto, C.H. and Awada, A., Cancer Chemother. Pharmacol., 2008, vol. 61, pp. 535–548. https://doi.org/10.1007/s00280-007-0639-9
Choueiri, T.K., Schutz, F.A., Je, Y., Rosenberg, J.E., and Bellmunt, J., J. Clin. Oncol., 2010, vol. 28, pp. 2280–2285. https://doi.org/10.1200/JCO.2009.27.2757
Keating, G.M. and Santoro, A., Target. Oncol., 2017, vol. 2, pp. 243–253. https://doi.org/10.1007/s11523-017-0484-7
Waguespack, S.G., Sherman, S.I, Williams., M.D, Clayman., G.L, and Herzog, C.E., Thyroid, 2008, vol. 19, pp. 407–412. https://doi.org/10.1089/thy.2008.0429
Dasari, A., Gore, L., Messersmith, W.A., Diab, S., Jimeno, A., Weekes, C.D., Lewis, K.D., Drabkin, H.A., Flaig, T.W.,Camidge, D.R., Invest. New Drugs, 2013, vol. 31, pp. 115–125. https://doi.org/10.1007/s10637-012-9812-z
Sharifi-Rad, A., Mehrzad, J., Darroudi, M., Saberi, M. R., Chamani, J., J. Biomol. Struct. Dyn., 2020, vol. 39, pp. 1–15. https://doi.org/10.1080/07391102.2020.1724568
Mokaberi, P., Babayan-Mashhadi, F., M.R., Chamani, J., J. Biomol. Struct. Dyn., 2020, https://doi.org/10.1080/07391102.2020.1766570
Iyer, R., Fetterly, G., Lugade, A., Thanavala, Y., Expert Opin. Pharmacother., 2013, vol. 11, pp. 1943–1955. https://doi.org/10.1517/14656566.2010.496453
Kamshad, M., Jahanshah Talab, M., Beigoli, S., Sharifirad, A., and Chamani, J., J. Biomol. Struct. Dyn., 2018, vol. 37, pp. 2030–2040. https://doi.org/10.1080/07391102.2018.1475258
Mokaberi, P., Reyhani, V., Amiri-Tehranizadeh, Z., Saberi, M.R., Beigoli, S., Samandar, F., and Chamani, J., J. Chem., 2019, vol. 43, p. 8132. https://doi.org/10.1039/C9NJ01048C
Shakibapour, N., Dehghani Sani, F., Beigoli, S., Sadeghian, H., and Chamani, J., J. Biomol. Struct. Dyn., 2019, vol. 37, pp. 359–371. https://doi.org/10.1080/07391102.2018.1427629
Sohrabi, T., Hosseinzadeh, M., Beigoli, S., Saberi, M.R., and Chamani, J., J. Mol. Liq., 2018, vol. 256, pp. 127–138. https://doi.org/10.1016/j.molliq.2018.02.031
Rashidipour, S., Naeeminejad, S., and Chamani, J., J. Biomol. Struct. Dyn., 2016, vol. 34, pp. 57–77. https://doi.org/10.1080/07391102.2015.1009946
Frisch, M., Trucks, G., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., and Mennucci, B., Gaussian 09, revision a.02, Gaussian, Inc., Wallingford, CT, 200, 2009.
Ghuman, J., Zunszain, P.A., Petitpas, I., Bhattacharya, A.A., Otagiri, M., and Curry, S., J. Mol. Biol., 2005, vol. 353, pp. 38–52. https://doi.org/10.1016/j.jmb.2005.07.075
Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J., J. Comput. Chem., 2009, vol. 30, pp. 2785–2791. https://doi.org/10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
Gasteiger, J. and Marsili, M., Tetrahedron, 1980, vol. 36, pp. 3219–3228. https://doi.org/10.1016/0040-4020(80)80168-2
Morris, G.M., Huey, R., Lindstrom, W., and Sanner, M.F., J. Comput. Chem., 2009, vol. 30, pp. 785–2791. https://doi.org/10.1002/jcc.21256
Sanner, M.F., J. Mol. Graphics Modell., 1999, vol. 17, pp. 57–61.
Berendsen, H.J., Postma, J.V., van Gunsteren, W.F., DiNola, A.R.H.J., and Haak, J.R., J. Chem. Phys., 1984, vol. 81, pp. 3684–3690. https://doi.org/10.1063/1.448118
Mousavi, S.F. and Fatemi, M.H., Bioorg. Chem., 2019, vol. 90, p. 103037. https://doi.org/10.1016/j.bioorg.2019.103037
Faridbod, F., Ganjali, M.R., Larijani, B., Riahi, S., Saboury, A.A., Hosseini, M., and Pillip, C., Mol. Biomol. Spectrosc., 2011, vol. 78, pp. 96–101. https://doi.org/10.1016/j.saa.2010.09.001
Feng, X.Z., Lin, Z., Yang, L.J., Wang, C., and Bai, C.L., Talanta, 1998, vol. 47, pp. 1223–1229. https://doi.org/10.1016/S0039-9140(98)00198-2
Hou, H.N., Qi, Z.D., OuYang, Y.W., Liao, F.L., Zhang, Y., and Liu, Y., J. Pharm. Biomed. Anal., 2008, vol. 47, pp. 134–139. https://doi.org/10.1016/j.jpba.2007.12.029
Chi, Z., Liu, R., and Zhang, H., Biomacromolecules, 2010, vol. 11, pp. 2454–2459. https://doi.org/10.1021/bm100633h
Ross, P.D. and Subramanian, S., Biochem., 1981, vol. 20, pp. 3096–3102. https://doi.org/10.1021/bi00514a017
Ojha, B. and Das, G., J. Phys. Chem., 2010, vol. 114, pp. 3979–3986. https://doi.org/10.1021/jp907576r
Moosavi-Movahedi, A.A., Chamani, J., Ghourchian, H., Shafiey, H., Sorenson, C.M., and Sheibani, N., J. Protein Chem., 2003. vol. 22, pp. 23–30. https://doi.org/10.1023/A:1023011609931
Moosavi-Movahedi, A.A., Golchin, A.R., Nazari, K., Chamani, J., Saboury, A.A., Bathaie, S.Z., and Tangestani-Nejad, S., Thermochim. Acta, 2004, vol. 414, pp. 233–241. https://doi.org/10.1016/j.tca.2004.01.007
Mousavi, S.F. and Fatemi, M.H., Struct. Chem., 2019, vol. 30, pp. 1–12. https://doi.org/10.1007/s11224-018-1178-1
Dehghani Sani, F., Shakibapour, N., Beigoli, S., Sadeghian, H., Hosainzadeh, M., and Chamani, J., J. Lumin., 2018, vol. 203, pp. 599–608. https://doi.org/10.1016/j.jlumin.2018.06.083
Shakibapour, N., Dehghani Sani, F., Beigoli, S., Sadeghian, H., and Chamani, J., J. Biomol. Struct. Dyn., 2018, vol. 37, pp. 359–371. https://doi.org/10.1080/07391102.2018.1427629
Fani, N., Bordbar, A.K., and Ghayeb, Y., J. Lumin.,2013, vol. 141, pp. 166–172. https://doi.org/10.1016/j.jlumin.2013.03.001
Jana, S., Dalapati, S., Ghosh, S., and Guchhait, N., J. Photochem. Photobiol. A, 2012, vol. 231, pp. 19–27. https://doi.org/10.1016/j.jphotochem.2011.12.002
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
No animals were involved in this work. No human subjects were involved in this work.
Conflict of Interests
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bagheri, F., Fatemi, M.H. Investigation of the Interaction of Sorafenib with Alpha-Lactalbumin: Spectroscopic and Molecular Modeling. Russ J Bioorg Chem 47, 864–873 (2021). https://doi.org/10.1134/S1068162021040038
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162021040038