
Abstract
The affinity between sorafenib and human serum albumin (HSA) was investigated by molecular modeling techniques and spectroscopic methods. The results of fluorescence spectroscopy, three-dimensional (3D) fluorescence, Fourier transform infrared (FTIR) and circular dichroism (CD) spectroscopy suggest that sorafenib has a strong ability to quench the intrinsic fluorescence of HSA. The binding affinity (K A) and the number of binding sites (n) between sorafenib and HSA at 305 K were estimated as 2.64 × 104 L·mol−1 and 1, respectively. The apparent distance between the Trp214 and sorafenib is 4.41 nm. Additionally, the binding of sorafenib induced conformational changes of disulfide bridges of HSA with decrease of the α-helix content. The resonance light scattering (RLS) spectra showed that the dimension of the individual HSA molecules are larger after interaction with sorafenib. These studies provide more information on the potential pharmaceutical effects and toxicological risk assessment of sorafenib.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Proteins exhibit dualism because of the hydrophobic and hydrophilic properties of the amino acids, which causes proteins to interact with amphiphilic molecules. Due to the wide variety of applications like drug delivery, cosmetics and foods, protein–drug interactions have become a topic of considerable interest. Serum albumin is synthesized in the liver and exported as non-glycosylated protein [1, 2]. It binds to a variety of hydrophobic ligands and thus is used as model protein for many biophysical, biochemical and physico-chemical studies. Albumin plays an important role in the transport and deposition of a variety of endogenous and exogenous substances in the blood and is also used in peritoneal dialysis in fighting against the harmful effect of drugs. Moreover, albumin is an important constituent of tissue culture media. Studies have shown that the distribution and metabolism of a large number of biologically active compounds, such as metabolites, drugs, and even some toxins, in blood are dependent to a larger extent on their affinities towards serum albumin [3, 4].
Sorafenib (Fig. 1, formula: C21H16ClF3N4O3) is a type of biological agent called a protein tyrosine kinase inhibitor (TKI). Tyrosine kinase is a protein which acts as a chemical messenger (an enzyme). There are a number of different tyrosine kinases that encourage cancer cells to grow. It works in two ways. It stops signals that tell cancer cells to grow. It also stops cancer cells forming blood vessels, which they need to grow. Treatment that stops blood vessels from forming is called anti angiogenesis treatment [5–7].

Molecular structure of sorafenib
Sorafenib is mainly bound to albumin, whose physiological role is also to serve as a transporter for fatty acids and bilirubin [8]. In several pathological states, these endogenous ligands can accumulate to relatively high concentrations that can displace drugs highly bound to albumin, resulting in a significant increase in these drugs. Patients with advanced solid tumors frequently exhibit denutrition, severe renal or hepatic impairment. In isolated protein solutions, 99.8–79.3 % of sorafenib was bound to HSA. In this context, characterizing the sorafenib binding to albumin is of critical pharmacological and clinical interest. However, the detailed binding process and mechanism are still unknown.
In the present paper, the interaction between sorafenib and HSA was investigated using fluorescence approaches. The binding characteristics between sorafenib and HSA are described and the conformational changes of HSA are explored. The aim is expected to provide important insight into the interaction of the physiologically important biomolecules and sorafenib.
2 Materials and Methods
2.1 Materials
HSA (purity > 98 %) was purchased from Sino-American Biotechnology Company (China) and used without further purification. Sorafenib (purity: 99 %) was purchased from Shandong Xi Bo C., Ltd. (China). NaH2PO4 and Na2HPO4 of guarantee reagent (GR) were purchased from Golden-Shell Pharmaceutical Co., Ltd. (China) and their mixture at pH = 7.4 was used as buffer solution to reach final concentration of 0.02 mol·L−1. Ultrapure water of 18.2 MΩ·cm−1 resistivity from a Milli-Q (Millipore, Bedford, MA) water purification system was used in this study.
2.2 Methods
The fluorescence spectra were recorded on a Cary eclipse spectrofluorimeter (Varian, USA) equipped with a 150 W Xenon lamp. All fluorescence spectra were collected using a 1.0 cm quartz cuvette with both excitation and emission band-width of 5 nm. The intrinsic fluorescence of HSA was measured at an excitation wavelength of 280 nm with both excitation and emission band-width of 5 nm. The spectrum was recorded at three different temperatures: 295, 305, and 315 K, controlled by using a constant-temperature cell holder connected to constant-temperature water circulator (Varian, USA). The synchronous fluorescence characteristics of sorafenib–HSA were measured from the same spectrofluorometer using different values of Δλ = λ ex − λ em. The synchronous fluorescence spectra were measured with Δλ = 15 nm and Δλ = 60 nm at 298 K with the above condition. The RLS spectra were recorded with an F-2500 fluorescence spectrophotometer by simultaneously scanning the excitation and emission spectra from 200 to 600 nm with a slit width of 3.0 nm. Three-dimensional fluorescence spectra were performed on the same instrument under emission and excitation wavelengths set between 200 and 500 nm with increments of 5.0 nm. RLS spectra were obtained by synchronous scanning on the spectrofluorometer with the wavelength range of 200–600 nm at room temperature. The widths of both the excitation slit and emission slit were set at 5 nm.
The time-resolved fluorescence measurements were performed at room temperature, using a single-photon counting spectrometer equipped with pulsed nanosecond LED excitation heads at 280 nm (Horiba, Jobin–Yvon, IBH Ltd, Glasgow, UK). The fluorescence lifetime data were measured to 10,000 counts in the peak, unless otherwise indicated. Data were analyzed using a sum of exponentials, employing a nonlinear least squares reconvolution analysis from Horiba (Jobin–Yvon, IBH Ltd).
Determination of conformational changes of HSA at different concentrations of sorafenib was carried out on Jasco J-810 spectropolarimeter using a quartz cell with a path length of 0.2 cm. Three scans were accumulated at a scan speed of 100 nm·min−1, with data being collected at every nm from 190 to 260 nm. The concentration of HSA was 3 × 10−7 mol·L−1 and the concentrations of sorafenib were 0.001, 0.005, and 0.009 mmol·L−1, respectively. Percentages of secondary structure elements (α-helix, β-pleated sheet, β-turn, and random coils) of HSA were calculated using CDNN 2.1 software.
The infrared spectra of protein–drug solutions were obtained on a Bruker Vertex 70 FTIR spectrometer (Bruker, Germany) via the attenuated total reflection method with resolution of 5 cm−1 and 60 scans, and were recorded at the range of 900–1,800 cm−1. The spectra of protein and buffer solutions were recorded separately under similar conditions. The absorbance of buffer and drug solution was then subtracted from that of the protein–drug solution.
3 Results and Discussion
3.1 Fluorescence Analysis of the HSA–Sorafenib System
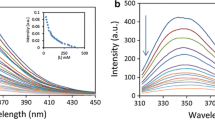
Fluorescence spectroscopy is an important tool for investigating the interaction between small probe molecules and proteins. At the excitation wavelength of 280 nm, HSA displays a strong fluorescence emission peak at 338 nm (Fig. 2). When increasing amounts of sorafenib were titrated into a fixed concentration of HSA, the peak intensity of HSA regularly decreased, suggesting that there were some interactions in the sorafenib–HSA system.

Effect of sorafenib on the fluorescence intensity of HSA. Conditions: T = 305 K, pH = 7.4; c(HSA) = 1.0 × 10−6 mol·L−1; c(sorafenib) a–g: 0, 1.0, 2.0, 3.5, 5.0, 7.0, and 10.0 × 10−5 mol·L−1
The mechanisms of quenching are usually classified as either static or dynamic. Static quenching is due to the formation of a ground-state complex between the fluorophore and the quencher; dynamic quenching results from collision between them. For static quenching, the quenching constants decrease with increasing temperature, while the reverse effect is found for dynamic quenching. For fluorescence quenching, the decrease in intensity is usually described by the well-known Stern–Volmer equation [9]:
where F 0 and F are the fluorescence intensities of the fluorophore in the absence and in the presence of quencher, respectively. [Q] is quencher concentration. The K SV is the Stern–Volmer quenching constant. τ 0 is the average lifetime of the biomolecule in the absence of quencher (τ 0 = 10−8 s), and K q is the quenching rate constant of the bimolecular fluorescence, which equals to K SV/τ 0.
The K sv values at different temperatures were determined from the slopes of F 0/F versus [Q] plots (Fig. 3) and K SV values are given in Table 1. It clearly shows that the values of K SV decrease with increasing temperature, and the corresponding K q values are of the order of 1012 L·mol−1·s−1 at the three different temperatures, which is two orders of magnitude greater than the maximum dynamic quenching constant of various quenchers for biopolymers, 2.0 × 1010 L·mol−1·s−1 [10–13]. These results imply static quenching, caused by complex formation, played a dominant role in this quenching process, rather than collisional quenching [14].

Stern–Volmer plots for the sorafenib–HSA system at 295, 305, and 315 K
To further confirm the quenching mechanism, time resolved decay measurements of HSA under different experimental conditions were carried out at 280 nm. The technique measures the fluorescence life time of the molecule. The decay curves (which show the decay time) of HSA with increasing concentrations of sorafenib are shown in Fig. 4.

Fluorescence life time of HSA in the presence of different concentrations of the drug. Inset is the decay profile of HSA
We have taken the mean fluorescence lifetime as a main parameter to analyze the binding of HSA with sorafenib. The data shows τ 0 /τ equals 1, which means that the quenching occurs through quencher–fluorophore complex formation (static quenching) since the complexed fluorophores are nonfluorescent, and the only observed fluorescence is from the uncomplexed fluorophores, which remain unperturbed and hence maintain the lifetime. The lifetime of HSA remains about 6.4 ns with the addition of sorafenib just as the pure HSA (6.41 ns, shown in Fig. 4 inset). This again confirms a static type quenching process [15].
When small molecules bind independently to a set of equivalent sites on a macromolecule, the equilibrium between free and bound molecules is given by the following equation:
where K A is the binding constant, and n is the number of the binding sites, which can be determined by the ordinate and slope of the double logarithm regression curve (Fig. 5) of log10 (F 0 − F)/F versus log10 [Q] based on the Eq. 4. The values of K a and n are shown in Table 2. The values of n is close to unity indicating that there is only one independent class of binding site on HSA for sorafenib. The decreasing binding constant with increase in temperature resulted possibly due to the reduction of the stability of the sorafenib–HSA complex.

Hill plots for HSA with sorafenib at 295, 305, and 315 K
The interaction forces between a compound and a biomolecule may involve the hydrophobic force, electrostatic interaction, van der Waals interaction and hydrogen bonds. Values of enthalpy change (ΔH°) and entropy change (ΔS°) can be used to determine the model of interaction between a compound and a biomolecule [16, 17]:
-
(1)
ΔH° > 0 and ΔS° > 0, hydrophobic force,
-
(2)
ΔH° < 0 and ΔS° < 0, van der Waals interaction and hydrogen bonds,
-
(3)
ΔH° < 0 and ΔS° > 0, electrostatic interaction.
In order to elucidate the interaction of sorafenib with HSA, the thermodynamic parameters were calculated from Eqs. 5–7. If the temperature does not vary significantly, the standard state enthalpy change (ΔH°) can be regarded as a constant. The standard Gibbs energy change (ΔG°), enthalpy change (ΔH°) and entropy change (ΔS°) can be estimated from the following equation, based on the binding constants at different temperatures:
where R is the gas constant, T is the experimental temperature, and K is the binding constant at the corresponding T. K 1 and K 2 are the binding constant at the experimental temperatures T 1 (295° K) and T 2 (305° K), respectively.
The thermodynamic parameters from the fluorescence quenching experiments are listed in Table 3. The negative value of Gibbs energy (ΔG°) supports the assertion that the binding process is favorable. The negative enthalpy (ΔH° < 0) and entropy (ΔS° < 0) values of the interaction of sorafenib and HSA indicate that van der Waals interaction and hydrogen bonds may play a major role in the binding between sorafenib and HSA.
3.2 Synchronous Fluorescence Spectroscopy Studies
Synchronous fluorescence spectroscopy is a useful method to evaluate the conformational changes of proteins. The shift in the position of the emission maximum reflects the change of the polarity around the chromophore moiety, thus the microenvironment of amino acid residues (such as Trp, Tyr, etc.) can be studied by measuring the shift in the maximum emission wavelength. When the wavelength intervals (Δλ) between the excitation and emission wavelength are 15 and 60 nm, the synchronous fluorescence spectra offer the characteristics of tyrosine and tryptophan residues, respectively [18]. The fluorescence spectra of HSA with various amounts of sorafenib are recorded at Δλ = 15 nm and Δλ = 60 nm and are shown in Fig. 6. It is apparent that the emission intensities of tyrosine residues decrease and the emission maximum wavelength slightly is shifted towards shorter wavelength (from 285 to 286 nm), indicating that the interaction of sorafenib with HSA affected the conformation of the region around the tyrosine residues. In Fig. 6, the emission maximum wavelength of tryptophan residues shows major red shifts (from 278 to 282 nm) with the addition of sorafenib, which suggests that the hydrophilicity around tryptophan residues has increased and the polarity around tryptophan residues has decreased. The above results imply that sorafenib was closer to tryptophan residues than to tyrosine residues, namely the binding sites mainly are focused on the tryptophan moiety.

Synchronous fluorescence spectra of sorafenib with HSA: a at Δλ = 15 nm and b Δλ = 60 nm. Concentrations are consistent with the steady-state fluorescence study
3.3 Three-Dimensional Fluorescence Spectroscopy
Three-dimensional fluorescence spectroscopy has been demonstrated to be a more specific method to comprehensively exhibit fluorescence information of the chromophore and investigate the characteristic conformational changes of the protein. In the present work, it has been utilized to provide additional proofs concerning the conformational changes of HSA in the presence of sorafenib. The three-dimensional fluorescence spectra of the free HSA (A) and sorafenib–HSA (B) complex are shown in Fig. 7 and the corresponding parameters are listed in Table 4. Some useful information about the conformational and micro-environmental changes of HSA can be obtained by comparing the spectral changes of HSA with and without sorafenib. As shown in Fig. 7, peak a is the Rayleigh scattering peak (λ ex = λ em) [19], and the fluorescence intensity increased with the addition of sorafenib. The reasonable explanation should be that an asorafenib–HSA complex formed after the addition of sorafenib, and the diameter of the macromolecule consequently increased, which in turn enhanced the scattering effect. As for peak 1 (λ ex = 280.0 nm, λ em = 349.5 nm), it mainly reveals the spectral behaviors of tryptophan and tyrosine residues, and the maximum emission wave-length and the fluorescence intensity of the residues are sensitively related to the polarity of their microenvironment. What is more, apart from peak 1, there is another strong peak (peak 2, λ ex = 228.0 nm, λ em = 348.5 nm) that mainly displays the fluorescence spectral behavior of the polypeptide backbone structure. As shown in Fig. 7, with the addition of sorafenib, the fluorescence intensities of peak 1 and peak 2 both decrease but to different degrees: the intensity of peak 1 had been quenched by 44.1 %, and by 33.4 % for peak 2. The decrease of fluorescence intensity of the two peaks combining with the molecular modeling and CD spectra results demonstrated that the binding of sorafenib to HSA led to the slight unfolding of the polypeptide of the protein, which led to a conformational change of the serum protein and the increase of the exposure of some hydrophobic regions that had been buried previously [20]. According to all of these phenomena and the overall analysis of peak 1 and peak 2, it can be safely concluded that the binding of sorafenib to HSA induces some microenvironment and conformational changes of HSA.

The three-dimensional fluorescence spectra of the HSA–sorafenib system. Conditions: T = 305 K, pH = 7.4; c(HSA) = 1.0 × 10−6 mol·L−1; c(sorafenib): 0 (A) and 11.0 (B) × 10−5 mol·L−1
3.4 Energy Transfer from HSA to Sorafenib
Fluorescence resonance energy transfer (FRET) has been examined using the absorption spectrum of the protein sorafenib (accepter) and the fluorescence spectrum of HSA (donor) at the same concentration, where there is a significant amount of overlap. The distance between donor and acceptor has been found from the Förster’s theory of non-radiative energy transfer [21, 22]. According to this theory the efficiency of energy transfer (E) has been determined from the given equation:
where R 0 is the Förster distance where the efficiency is just 50 % and r is the actual distance between the donor and acceptor. The value of R 0 can be calculated from the following equation:
The value of J has been calculated from the following formula:
where F(λ) is the fluorescence intensity of HSA (donor) at wavelength l, and ε(λ) is the molar extinction coefficient of sorafenib (acceptor) at wavelength λ. The unit of ε(λ) is normally taken to be L·mol−1·cm−1. The k is the orientation factor of the donor and acceptor and for random orientation of donor and acceptor a value of 2/3 is assumed for k 2. The refractive index of the medium, N, is equal to 1.336. The value of quantum yield Φ of donor is found to be 0.15.
There is a considerable overlap (Fig. S1, Supplementary Data) between the absorption spectrum of sorafenib (acceptor) and the fluorescence spectrum of HSA (donor). So, there is a considerable chance of resonance energy transfer from excited state donor to the acceptor. Moreover the higher value of k q as obtained from fluorescence quenching experiments suggests the possibility of energy transfer rather than a simple diffusion controlled phenomenon. Förster’s theory of non-radiative energy transfer has been used to calculate the distance between HSA and sorafenib. Using Eq. 6 the value of J has been calculated to be 5.14 × 10−13 L·mol−1·cm−1. The efficiency of energy transfer (E) has been found to be 16 % when both the protein and drug concentrations are equal. The values of R 0 and r have been evaluated from Eqs. 3 and 5 as 2.89 and 3.61 nm, respectively. Here the average distance r is less than 8 nm and follows the condition, 0.5R 0 < r < 1.5R 0, which signifies that occurrence of energy transfer from sorafenib to the protein HSA is appreciable.
3.5 UV–Vis Spectra
A static process for the observed quenching can also be supported by the UV–vis spectra of the fluorophore. For a dynamic quenching mechanism, the absorption spectra of the fluorescent substance is not changed, and only the excited state fluorescence molecule is influenced by quenchers, while, for static quenching, a new compound is formed between the ground state of the fluorescent substance and quencher and, therefore, the absorption spectra of fluorescence substance will be considerably influenced [10, 23]. Figure 8 shows the UV absorption of the proteins in the presence and absence of sorafenib. The influences of the absorbance of sorafenib were eliminated by adding in the reference cells solutions of sorafenib of the same concentrations as in the sample solution. As can be seen from Fig. 8, both proteins possess two absorption peaks at around 210 and 280 nm, where that at 210 nm represents the content of α-helix in the protein. A dramatic decrease in the 210 nm absorbance peak of the proteins is observed upon addition of sorafenib to the proteins. This can be attributed to the induced perturbation of the α-helix of the protein by a specific interaction with the ligands. Furthermore, an obvious red shift in the position of the absorbance peak (i.e., from 210.6 to 215.6 nm) can also be observed with the addition of sorafenib. Meanwhile, the absorption intensity of the 280 nm peaks is increased by addition of sorafenib, indicating that the more aromatic acid residues were extended into the aqueous environment. Trp-214 in HSA, which was originally buried in a hydrophobic pocket, was exposed to an aqueous milieu to a certain degree [24]. This result indicates that the microenvironment of the three aromatic acid residues was altered and the tertiary structure of HSA was destroyed. These results show that the interaction between sorafenib and HSA is mainly a static quenching process.

The UV–vis spectra of HSA in the presence of different concentrations of sorafenib; c(HSA) = 5.0 × 10−6 mol·L−1; c(sorafenib): a–g = 0, 1, 2, 4, 6, 8 and 10.0 × 10−6 mol·L−1; conditions: T = 305 K, pH = 7.4
3.6 RLS analysis of the HSA–Sorafenib Complex
RLS is a highly sensitive method for the study of aggregation of chromophores on biological macromolecules. The RLS spectra of the HSA–sorafenib system in phosphate buffer solution (pH = 7.4) are shown in Fig. 9. As is seen in Fig. 9, both free HSA and free sorafenib exhibit very weak RLS signals, but when a fixed concentration of sorafenib was titrated with different amounts of HSA, the intensity of RLS was remarkably enhanced. On the basis of the theory of resonance light scattering, it can be deduced that the RLS intensity is related to the size of the particle formed and directly proportional to the square of molecular volume, and it has been demonstrated that larger particles induce stronger light scattering signals [25]. Hence, the added HSA likely interacted with sorafenib in solution, and a HSA–sorafenib complex formed, resulting in an increase in the RLS signal.

RLS spectra of the HSA–sorafenib system. Conditions: T = 305 K, pH = 7.4; c(HSA) = 1.0 × 10−6 mol·L−1; c(sorafenib) a–d: 0, 2.0, 6.0, and 10.0 × 10−6 mol·L−1. Conditions: T = 305 K, pH = 7.4
3.7 FTIR spectra analysis
FT-IR spectroscopy was used (Fig. 10) to obtain more information about the conformational changes of HSA after binding with sorafenib because the infrared spectra of the proteins exhibit amide bands (I–III), which represent different peptide moiety vibrations. The secondary structure of proteins was determined to occur at 1,650–1,660 cm−1 (amide I, principally due to C=O stretching) and 1,548–1,560 cm−1 (amide II, due to C–N stretching coupled with N–H bending) [26]. Generally, the 1,650–1,660 cm−1 range of the amide I band is attributed to the a-helix, which is used to predict the secondary structure of proteins. In the present study, the characteristic amide I and amide II bands of free HSA were found at 1,655 and 1,549 cm−1, respectively. The peak position of amide I moved gradually from 1,655 to 1,653 cm−1, and for amide II, it moved from 1,549 to 1,545 cm−1, upon binding with sorafenib (Fig. 10). These results show the HSA secondary structure effect in relation to its complexation with sorafenib, resulting in perturbations of the amide I and amide II vibrational frequencies.

FTIR spectra of free HSA (1.0 × 10−4 mol·L−1), and the HSA–sorafenib complex (the molar ratio of HSA/sorafenib was maintained at 1:1, 1:2, and 1:3 for curves b, c, and d, respectively)
3.8 Quantitative Analysis of the Protein Secondary Structure Using CD
CD spectroscopy has been applied to investigate the secondary structure of proteins for many years because of its powerful function in readily estimating of the characteristics of different secondary structures of protein upon interaction with drugs [27]. To get insight into the possible influence of sorafenib binding on the secondary structure of HSA, far-UV CD measurements were performed in the presence of different HSA/sorafenib molar ratios. The CD spectra of HSA exhibited two negative bands at 208 and 222 nm, which are characteristic of the typical α-helix structure of a protein. As shown in Fig. 11, the relative band intensity of curves a–c decrease gradually with increasing addition of sorafenib, which suggests that the changes of protein secondary structure are caused by the formation of the sorafenib–HSA complex. The CD curves of the HSA with and without sorafenib are similar in shape, indicating that α-helix was still the main component of the HSA secondary structure. The algorithm SELCON3 was used to analyze the CD spectra in order to quantify the contents of different secondary structures of HSA. The estimated results for the fraction contents of α-helix, β-turn, β-sheet, and random forms for HSA in the absence and presence of sorafenib are shown in Table 5. A decreasing α-helix content and an increasing β-turn structure content were observed with increasing concentration of sorafenib. The formation of the sorafenib–HSA complex caused the change of α-helical content (the total content of regular and distorted α-helix) from 56 to 51 %. Furthermore, a decrease in the percentage of β-strands and an increase in the unordered structure were observed, which suggested that the binding of sorafenib to HSA induced a little unfolding of the polypeptides of protein. The induction results in the exposure of some hydrophobic regions that were previously buried [28].

CD spectra of HSA in the absence and presence of sorafenib. The concentration of HSA was 3.0 × 10−7 mol·L−1, and the concentrations of sorafenib from a to c were 0.001, 0.005, and 0.009 mmol·L−1
4 Conclusions
A fluorescence method was provided for the rapid and simple determination of the interaction between sorafenib and HSA, as an important carrier protein, under simulated physiological conditions. The method is easy to operate and is reliable, practical and simple. The experimental results indicate that the intrinsic fluorescence of HSA can be quenched by sorafenib through the static quenching mechanism; the binding reaction is spontaneous whereas van der Waals interactions and hydrogen bonds play major roles in the binding process. The number of binding sites (n) for sorafenib was found to be about 1. The binding constants K A, at 295, 305 and 315 K, were determined according to a modified Stern–Volmer equation as (3.85, 2.64 and 1.92) × 104 L·mol−1, respectively. Both theoretical and experimental results suggest a slight conformational change, most probably around the binding pocket of the protein. The distance between the Trp-214 of HSA and sorafenib was evaluated as r = 3.61 nm, in accordance to the fluorescence resonance energy transfer theory. The RLS results elucidated that sorafenib can interact with HSA and cause protein aggregation.
References
Nowak, I., Shaw, L.M.: Mycophenolic acid binding to human serum albumin: characterization and relation to pharmacodynamics. Clin. Chem. 41, 1011–1017 (1995)
Carter, D.C., Chang, B., Ho, J.X., Keeling, K., Krishnasami, Z.: Preliminary crystallographic studies of four crystal forms of serum albumin. Eur. J. Biochem. 226, 1049–1052 (1994)
Zhao, X.C., Liu, R.T.: Recent progress and perspectives on the toxicity of carbon nanotubes at organism, organ, cell, and biomacromolecule levels. Environ. Int. 40, 244–255 (2012)
Wilting, J., van der Giesen, W.F., Janssen, L., Weideman, M., Otagiri, M., Perrin, J.: The effect of albumin conformation on the binding of warfarin to human serum albumin. The dependence of the binding of warfarin to human serum albumin on the hydrogen, calcium, and chloride ion concentrations as studied by circular dichroism, fluorescence, and equilibrium dialysis. J. Biol. Chem 255, 3032–3037 (1980)
Amato, R., Harris, P., Dalton, M., Khan, M., Alter, R., Zhai, Q., Brady, J.R., Jac, J., Hauke, R., Srinivas, S.: A phase II trial of intra-patient dose-escalated sorafenib in patients (pts) with metastatic renal cell cancer (MRCC). J. Clin. Oncol. 25, 5026 (2007)
Takimoto, C.H., Awada, A.: Safety and anti-tumor activity of sorafenib (Nexavar®) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother. Pharmacol. 61, 535–548 (2008)
Choueiri, T.K., Schutz, F.A., Je, Y., Rosenberg, J.E., Bellmunt, J.: Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J. Clin. Oncol. 28, 2280–2285 (2010)
Abou-Alfa, G.K., Schwartz, L., Ricci, S., Amadori, D., Santoro, A., Figer, A., De Greve, J., Douillard, J.-Y., Lathia, C., Schwartz, B.: Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 24, 4293–4300 (2006)
Zhao, X., Liu, R., Teng, Y., Liu, X.: The interaction between Ag(+) and bovine serum albumin: a spectroscopic investigation. Sci. Total Environ. 409, 892–897 (2011)
Hu, Y.-J., Liu, Y., Wang, J.-B., Xiao, X.-H., Qu, S.-S.: Study of the interaction between monoammonium glycyrrhizinate and bovine serum albumin. J. Pharm. Biomed. Anal. 36, 915–919 (2004)
Hu, Y.-J., Yu, H.-G., Dong, J.-X., Yang, X., Liu, Y.: Spectroscopic studies on the interaction between 3,4,5-trimethoxybenzoic acid and bovine serum albumin. Spectrochim. Acta A 65, 988–992 (2006)
Matei, I., Hillebrand, M.: Interaction of kaempferol with human serum albumin: a fluorescence and circular dichroism study. J. Pharmaceut. Biomed. Anal. 51, 768–773 (2010)
Anand, U., Jash, C., Boddepalli, R.K., Shrivastava, A., Mukherjee, S.: Exploring the mechanism of fluorescence quenching in proteins induced by tetracycline. J. Phys. Chem. B 115, 6312–6320 (2011)
Sandhya, B., Hegde, A.H., Kalanur, S.S., Katrahalli, U., Seetharamappa, J.: Interaction of triprolidine hydrochloride with serum albumins: thermodynamic and binding characteristics, and influence of site probes. J. Pharm. Biomed. Anal. 54, 1180–1186 (2011)
Zhao, X.C., Liu, R.T., Chi, Z.X., Teng, Y., Qin, P.F.: New insights into the behavior of bovine serum albumin adsorbed onto carbon nanotubes: comprehensive spectroscopic studies. J. Phys. Chem. B 114, 5625–5631 (2010)
Ross, P.D., Subramanian, S.: Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry 20, 3096–3102 (1981)
Beauchemin, R., N’Soukpoe-Kossi, C.N., Thomas, T.J., Thomas, T., Carpentier, R., Tajmir-Riahi, H.A.: Polyamine analogues bind human serum albumin. Biomacromolecules 8, 3177–3183 (2007)
Gokara, M., Sudhamalla, B., Amooru, D.G., Subramanyam, R.: Molecular interaction studies of trimethoxy flavone with human serum albumin. PLoS ONE 5, e8834 (2010)
Khan, A.Y., Hossain, M., Kumar, G.S.: Investigations on the interaction of the phototoxic alkaloid coralyne with serum albumins. Chemosphere 87, 775–781 (2012)
Kanakis, C., Tarantilis, P., Polissiou, M., Diamantoglou, S., Tajmir-Riahi, H.: Antioxidant flavonoids bind human serum albumin. J. Mol. Struct. 798, 69–74 (2006)
Forster, T.: Delocalized excitation and excitation transfer. In: Sinanoglu, O.E. (ed.) Modern Quantum Chemistry III, pp. 93–137. Academic, New York (1965)
Lu, D.W., Zhao, X.C., Zhao, Y.C., Zhang, B.C., Zhang, B., Geng, M.Y., Liu, R.T.: Binding of Sudan II and Sudan IV to bovine serum albumin: comparison studies. Food Chem. Toxicol. 49, 3158–3164 (2011)
Hu, Y.J., Liu, Y., Xiao, X.H.: Investigation of the interaction between berberine and human serum albumin. Biomacromolecules 10, 517–521 (2009)
Belatik, A., Hotchandani, S., Bariyanga, J., Tajmir-Riahi, H.: Binding sites of retinol and retinoic acid with serum albumins. Eur. J. Med. Chem. 48, 114–123 (2012)
Borissevitch, I.E., Tominaga, T.T., Imasato, H., Tabak, M.: Resonance light scattering study of aggregation of two water soluble porphyrins due to their interaction with bovine serum albumin. Anal. Chim. Acta 343, 281–286 (1997)
Cheng, X.X., Lui, Y., Zhou, B., Xiao, X.H., Liu, Y.: Probing the binding sites and the effect of berbamine on the structure of bovine serum albumin. Spectrochim. Acta A 72, 922–928 (2009)
Yang, Q., Zhou, X.M., Chen, X.G.: Combined molecular docking and multi-spectroscopic investigation on the interaction between Eosin B and human serum albumin. J. Lumin. 131, 581–586 (2011)
Zhao, X., Sheng, F., Zheng, J., Liu, R.: Composition and stability of anthocyanins from purple solanum tuberosum and their protective influence on Cr(VI) targeted to bovine serum albumin. J. Agric. Food Chem. 59, 7902–7909 (2011)
Acknowledgments
This work was supported by Grants from Medical and Technologic Development Project of Shandong province (2011QZ028). The authors gratefully acknowledge Dong-mei Liu and Ji-wen Sheng for their technical guidance and assistance during experiments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lu, Z., Qi, L., Li, Gx. et al. In Vitro Characterization for Human Serum Albumin Binding Sorafenib, A Multi Kinase Inhibitor: Spectroscopic Study. J Solution Chem 43, 2010–2025 (2014). https://doi.org/10.1007/s10953-014-0256-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-014-0256-2