
Abstract
The activity ratio of uranium in unknown samples and materials is a crucial signature tool for verification, assay, and tracking of their origin. This paper deals with using γ-ray spectrometric analysis and a new derived formula to calculate the 235U/238U activity ratio. Reference IAEA materials and a highly efficient calibrated HPGe spectrometer were used for the verification and validation of this equation and the results of assay. The calculated specific activities and 235U/238U activity ratios show very good agreement with the declared reference values, 0.046, with differences less than 2%. Natural origin samples were also investigated and assayed with the maximum deviation in the range 1.3–2.9%. Any potential deviation from the reference natural value, 0.046, in uranium samples could be precisely detected, identified, assessed, and quantified using the suggested system and equation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Uranium is a terrestrial naturally occurring radioactive material which always exists in rocks and soils in low quantities [1]. Uranium is found in the earth’s crust with an average content of 3 ppm. Natural uranium (NU) is a mixture of three radioactive isotopes: 238U (99.27% abundance), 235U (0.72% abundance), and 234U (0.0054% abundance) [2]. Enrichment processes are widely practiced to produce enriched uranium (EU) of activity ratio higher than 0.046, used for the production of nuclear energy. On the other hand, depleted uranium (DU) with the activity ratio lower than 0.046 is also produced as a by-product of this enrichment processes. Accordingly, monitoring the isotopic 235U/238U ratio is a very important step associated with any sample that contains uranium [2]. Moreover, the U along with Pb isotope measurements can be used to identify and ensure the geological age of minerals and rocks [3, 4]. The detection of illicit trafficking of nuclear materials, including “nuclear smuggling,” is necessary in order to determine the possible origin of the nuclear material. Investigations are carried out by laboratories capable of handling and analyzing nuclear material of unknown origin [5]. During the past few years, as the international illicit traffic of radioactive/fissionable materials has increased, it has become imperative to have the capability to apply fast and reliable methods for determining the uranium enrichment percentage [6].
This study is mainly focused on verifying and validating the 235U/238U activity ratios for natural origin samples as a signature identification tool using γ-ray spectrometry and derived equation.
THEORETICAL DERIVATION OF THE EQUATION AND CALCULATIONS
The activity of an isotope can be calculated from the following relation [7–9]:
where A is the activity, N is the number of atoms present, and λ is the decay constant. So, the activity ratio can be expressed as follows:
The decay constant is λ = ln2/T1/2, where T1/2 is the half-life of the isotope; thus,
The uranium enrichment percentage (E, %) is defined as follows:
In the case of natural or depleted uranium, the N235 and N234 values are relatively low in comparison to N238 and can be neglected in Eq. (4); thus,
By substituting Eq. (5) in Eq. (3), we obtain
The 235U percentage in natural uranium is 0.72% [10]. The 235U half-life is T1/2(235) = 7.04 × 108 years, and that of 238U is 4.468 × 109 years [10, 11].
By substituting these values into Eq. (6), we obtain
Finally, the 235U/238U activity ratio of can be formulated as
This ratio is almost the same as the certified activity ratio of uranium (0.046) in any natural origin samples with about 0.65% relative difference to this value (0.0457).
EXPERIMENTAL
The radiometric measurement using γ-ray spectrometry is considered to be an essential technique for the analysis of uranium in ores and rock samples. This is attributed to several reasons: (i) it is both simple and cheap for routine analysis, (ii) the samples are measured directly without any chemical or other treatments, (iii) this technique is essential in studying the geochemical prospecting and evaluation of the radioactive mineral deposits, (iv) it has high resolution and progressively improved performance, and (v) it is considered to be the easiest and cleanest method for measuring U concentrations [10, 12].
HPGe Detector Characteristics
A hand-held radioisotope identifier system (TRANS-SPEC) was used in this study for the rapid identification of radioisotopes. The detector consists of a p-type HPGe detector with 40% efficiency. The energy resolution of the detector was found to be ~1.32 and 2.2 at 122 and 1332.5 keV gamma lines, respectively. Control of the acquisition is achieved using a built-in digital electronic system (DigiDart). A Hymatic Stirling cycle cooler system is designed for continuous operation. The count rate and position (geometry) of sample relative to HPGe detectors were fixed during the measurements of the samples.
γ-Ray Spectrometer Energy and Efficiency Calibration
Before the spectrum analysis, both the energy and efficiency calibration of the measuring system were performed using 60Co, 137Cs, 152Eu, and 133Ba point sources. These radioactive isotopes have well-known activities and are routinely used for verification and validation of calibration results. The point sources were positioned on the top at the source-to-detector distance of 25 cm. The counting time was 1800 s (30 min) to obtain high counts with good statistics. In this study, both energy and efficiency calibration of the HPGe system were performed to ensure the accuracy of the detector and, consequently, of the spectrum analysis.
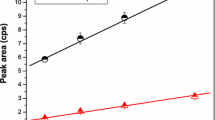
Energy calibration. The energy calibration of the HPGe detector system was performed in the range 53.16–1408.08 keV to obtain a relationship between the channel number of the peak position in the spectrum and the corresponding γ-ray energy. The linear relationship between the channel number and energy is plotted in Fig. 1. This relationship can be described by linear Eq. (9):

Energy calibration curve of HPGe spectrometer.
The radionuclides in reference material and natural samples can then be identified based on their energies.
Efficiency calibration. The absolute photopeak efficiency ξabs (%), which depends on the source–detector geometry and the gamma energy, was measured and calculated using the following equation [13]:
where Ct is the total number of counts recorded per unit time, and Nγ is the number of gamma quanta emitted by the source per unit time; it can be calculated using the equation [14]
where DS is the certified activity or the disintegration rate of the source, and Iγ(Eγ) is the branching ratio or the emission probability per disintegration.
Due to the different geometry and density for the reference material and natural samples, the relative efficiency curves established for different sets of samples were used as reference efficiency curves in Angel-3 software together with the characteristics of the detector crystal, source container, and source–detector geometry [15–18].
The above-mentioned steps are considered to generate different efficiency curves for the same detector but for different containers and geometries.
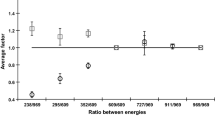
Using the Excel sheet, the efficiency curves of the detector were plotted vs. different photopeak energies for a point source and densities of 1.6 and 2.5 g/cm3 (Fig. 2).

Relative efficiency for different geometries and densities of samples.
Sample Preparation and Readiness for Assay
Two sets of samples were taken. The first set included IAEA-RGU-1 and IAEA-RGTh-1 reference materials (323 g) prepared on behalf of IAEA by the Canada Centre for Mineral and Energy Technology. The samples were prepared by dilution of BL-5 uranium ore (7.09% U) and OKA-2 thorium ore (2.89% Th, 219 μg U/g) with floated silica powder of similar grain size distribution. No evidence for between-bottles inhomogeneity was detected after mixing and bottling. BL-5 has been certified for uranium; the 226Ra and 210Pb measurements confirmed that it was in radioactive equilibrium. The agreement between radiometric and chemical measurements of thorium and uranium in OKA-2 showed that both series were in radioactive equilibrium [19].
The second set included two groups of natural samples. The first group consisted of seven environmental phosphate samples collected from Aswan (coded PS1–PS7). The second group consisted of seven black sand samples collected from the beach of Rosetta–Damietta (coded BS1–BS7). All the natural samples were dried at 100°C and then kept in a plastic cup for a period of more than 28 days. The samples were then counted three times for 24 h using the considered γ-ray spectrometry system.
RESULTS AND DISCUSSION
The γ-transitions emitted from the samples lie in a wide range of energies from approximately 50 keV to 3 MeV, covering the naturally occurring radioactive materials (238U, 235U, 232Th decay series and 40K). The 238U specific activity was determined from γ-rays emitted by its progenies: 234Th (determined from the 63 keV photopeak, Iγ = 0.27) [20] and 234mPa (determined from the 1001 keV photopeak, Iγ = 1.021, which is the most suitable peak for the analytical purposes) [21]. Further decay leads to 226Ra and then to radon. Afterwards, the average of the specific activity of 214Pb and 214Bi, which can be measured using 295.2 (Iγ = 18.414) and 351.9 keV (Iγ = 35.60) lines for 214Pb and the 609.3 keV (Iγ = 45.49) line for 214Bi, can then be taken into consideration [22, 23]. Furthermore, a theoretical calculation of the 226Ra specific activity can be performed by assuming that the entire 186.21 keV (Iγ = 3.555) peak is due to 226Ra. Then, the following correction factor can be used to correct the 226Ra value [14, 24]:
The 235U activity can be estimated at the gamma energy of 185.72 keV [24, 25]:
Also, the γ-energies of 235U (143.7, 163.3 and 205.3 keV) can be used for the activity calculations [22, 26]. Both 40K (1460.83 keV) and 232Th series (583, 911, 968 keV) [27] were observed in the spectra of the analyzed samples before treatment. However, it should be noted that their activities are not considered in this study. The specific activities (A) of the samples are given in Bq/kg for 238U and 235U (A238 and A235). They are estimated by substituting the corresponding count rate of the γ-lines in Eq. (14) [28]:
where M is the mass of the measured sample (kg), ξabs is the absolute detector efficiency at specific energy Eγ with branching ratio Iγ (Eγ), and Cnet is the net count rate in a given photopeak region (ROI) subtracting the background, which is corrected using Eq. (15) [29, 30]:
where C is the measured count rate in the peak of interest in the sample spectrum [s–1], and τ is the analog-to-digital converter (ADC) dead time [11137_2021_8148_Str0_HTML.gif].
For all the measurements, the sample to detector distance for the assayed samples is optimized to keep the dead time correction below 2%.
The Gamma Vision software determines the total uncertainty σt of activity by summing in quadrature the individual uncertainties from various analysis components as in Eq. (16):
where σcount is the counting uncertainty estimate, σnor is the additional normally distributed uncertainty estimate such as balance resolution, σrsum is the random summing uncertainty estimate, σabs is the absorption uncertainty estimate, σnuc is the nuclide uncertainty estimate, σeff is the efficiency uncertainty estimate, σgeo is the geometry uncertainty estimate, and σuni is the uniformly distributed uncertainty estimate such as sample mass determination.
Validation of the Derived Activity Ratio Equation Using IAEA References
For the IAEA-RGU-1 samples, the specific activity of 238U was measured and found to be 4869.6 Bq/kg, and the specific activity of 235U was measured as 229 Bq/kg. The resulting 235U/238U ratios on the average are 0.0470 with the deviation of about 1.7 %. For the IAEA-RGTh-1 samples, the calculated specific activity of 238U was 79 Bq/kg, and the specific activity of 235U was found to be 3.7 Bq/kg. The resulting average 235U/238U ratio is 0.0468 with the deviation of only 1.4%. All the measured and calculated specific activities are in very acceptable agreement with the certified value of 0.46, as shown in Table 1.
Verification of Activities and U Ratio of the Natural Samples
The specific activities of 235U and 238U have been measured in two different groups of the natural samples, and their uranium isotopic contents and ratios were determined.
The average specific activity of phosphate samples, based on 234Th, 234mPa, 226Ra, and average of 214Pb and 214Bi (238U daughters), was found to be 391 ± 22, 387 ± 17, 395 ± 17, and 392 ± 10, respectively; that is, the ratios of 234Th, 234mPa and 226Ra to the average of 214Pb and 214Bi are 1.00 : 1, 0.99 : 1, and 1.01 : 1, respectively. The average specific activity of black sand samples, based on 234Th, 234mPa, 226Ra, and average of 214Pb and 214Bi, was found to be 492 ± 28, 499 ± 21, 472.27 ± 15, 474 ± 8, and 473 ± 19, respectively; that is, the ratios of 234Th, 234mPa, and 226Ra to the average of 214Pb and 214Bi are 0.98 : 1, 1.01 : 1, and 1.06 : 1, respectively (Table 2).
The 235U specific activity in phosphate and black sand samples ranges from 16.8 to 20.6 Bq/kg with the average value of 19.5 Bq/kg and from 20.8 to 22.6 Bq/kg with the average value of 21.6 Bq/kg, respectively (Table 3).
For the phosphate samples, the specific activity of 238U varied from 365 to 467 Bq/kg with an average value of 412 Bq/kg. Thus, the average 235U/238U activity ratio is 0.0460 with a deviation of about 1.30% in maximum.
For black sand samples, the specific activity of 238U varied between 465 and 489 Bq/kg with an average value of 473 Bq/kg. Thus, the average 235U/238U activity ratio is 0.0452 with the deviation of about 2.70% in maximum. The results are given in Table 4.
These values show a slight variation in the natural uranium percentage of the analyzed samples with an average value within the normal signature value, 0.72%. This acceptable variation could be attributed to the change in the detector efficiency at different gamma transitions of 235U (143.7, 163.3, 185.7, and 205.3 keV) and 238U (63.766, 185.99, 295.2, 609.3, and 1001 keV). The calculated 235U/238U activity ratios for the reference IAEA materials and natural samples are depicted against the certified value, 0.046, as shown in Fig. 3. The deviation between the measured and calculated/derived activity ratio values is low (<2%).

235U/238U activity ratios for the verified reference material and natural samples.
Thus, there is good agreement between the theoretical calculation [Eq. (15)] and the certificate values. The average difference does not exceed 1.6% for the reference IAEA materials and about 2.0% for the natural origin samples.
CONCLUSION
The investigation of origin and history of unknown samples by back-tracing approach is an effective tool for identifying undeclared radiological and nuclear activities. This paper introduces a developed mathematical formula for the calculation and verification of 235U/238U activity ratio based on efficient γ-spectrometric analysis measurements. This equation was validated by using reference IAEA materials. The results show deviation of less than 2% from the certified values. The verification of uranium isotopes and activity ratios of natural samples was also carried out with average differences from 1.3 to 2.7%. It may be concluded that the abnormal and deviated activities from the natural origin and signature can be followed and verified by our derived equation and accurate γ-ray spectrometric investigation.
REFERENCES
Sethy, N., Jham, V., Sutar, A., et al., J. Geochem. Explor., 2014, vol. 142, pp. 29–35. https://doi.org/10.1016/j.gexplo.2013.11.009
Ebaid, Y., Rom. J. Phys., 2010, vol. 55, nos. 1–2, pp. 69–74.
Chernyshev, I., Golubev, V., Chugaev, A., and Baranova, A., Geochem. Int., 2014, vol. 52, no. 12, pp. 1013–1029.
Chernyshev, I., Dubinina, E., and Golubev, V., Geol. Ore Depos., 2014, vol. 56, no. 5, pp. 315–321.
Nguyen, C.T. and Zsigrai, J., Nuclear Instrum. Meth. Phys. Res., Sect. B, 2006, vol. 246, no. 2, pp. 417–424.
Van Loat, B., Tam, N.C., Khue, P.D., and Hue, B.M., VNU J. Sci.: Math.-Phys., 2013, vol. 29, no. 2.
Kónya, J. and Nagy, N.M., Nuclear and Radiochemistry, Amsterdam: Elsevier, 2018.
Loveland, W.D., Morrissey, D.J., and Seaborg, G.T., Modern Nuclear Chemistry, Wiley, 2017.
Valkovic, V., Radioactivity in the Environment, Amsterdam: Elsevier, 2019.
Abdel-Rahman, M.A., Abu Shady, H., and El-Mongy, S.A., Z. Anorg. Allg. Chem., 2018, vol. 644, no. 10, pp. 477–482.
Ershov, B.G., Precambrian Res., 2020, p. 105786.
Abdel-Rahman, M.A. and El-Mongy, S.A., Nucl. Eng. Technol., 2017, vol. 49, no. 8, pp. 1752–1757.
Klement, A.W., Handbook of Environmental Radiation, US: CRC, 2019.
Gilmore, G., Practical Gamma-Ray Spectroscopy, New York: Wiley, 2011.
Bell, S., Judge, S., and Regan, P., Appl. Radiat. Isot., 2012, vol. 70, no. 12, pp. 2737–2741.
Mihaljevi, N. and Dlaba, A., Accounting for Detector Crystal Edge Rounding in Gamma-Efficiency Calculations: Theoretical Elaboration and Application in ANGLE Software, 2012.
Khandaker, M.U., Jojo, P.J., and Kassim, H.A., APCBEE Procedia, 2012, vol. 1, pp. 187–192.
Seo, B.K., Lee, K.Y., Yoon, Y.Y., and Lee, D.W., Fresenius’ J. Anal. Chem., 2001, vol. 370, nos. 2–3, pp. 264–269.
Preparation and Certification of IAEA Gamma-Ray Spectrometry Reference Materials RGU-1, RGTh-1 and RGK-1, IAEA, 1987. http://inis.iaea.org/search/search.aspx?orig_q=RN:1808842.
Alharbi, S.H. and Akber, R.A., J. Radioanal. Nucl. Chem., 2017, vol. 311, no. 1, pp. 59–75.
Elzahraa, F., Aly, H.A., and Abdel-Rahman, M.A., Z. Anorg. Allg. Chem., 2012 vol. 31, no. 1, p. 51.
Ibrahim, E.M., El Aassy, I.E., Ghany, H.A.A., and Gamil, S., Environ. Earth Sci., 2018, vol. 77, no. 14, p. 534.
Shazly, A., Kany, A., Osman, A., El Samman, H., and El-Mongy, S., in Proc. 8th Conf. on Nuclear and Particle Physics, Hurghada (Egypt), Nov. 20–24, 2011.
Elsayed, A.F., Hussein, M.T., El-Mongy, S.A., Ibrahim, H.F., and Shazly, A., Phys. Part. Nucl. Lett., 2021, vol. 18, no. 2, pp. 202–209. https://doi.org/10.1134/S1547477121020060
Abdel-Rahman, M.A. and El-Mongy, S.A., Z. Anorg. Allg. Chem., 2018, vol. 644, no. 1, pp. 29–32.
Huynh, N.P.T. and Le, C.H., J. Environ. Manag., 2020, vol. 271, p. 111001.
Khattab, M.R., Ebyan, O.A., and Abdel-Rahman, M.A., Radiat. Prot. Environ., 2017, vol. 40, no. 3, p. 126.
AN63, ORTEC®
Upp, D.L., Keyser, R.M., Gedcke, D.A., Twomey, T.R., and Bingham, R.D., J. Radioanal. Nucl. Chem., 2001, vol. 248, no. 2, pp. 377–383.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Elsayed, A.F., Hussein, M.T., El-Mongy, S.A. et al. Signature Verification of 235U/238U Activity Ratio for Ores and Natural Samples Using γ-Ray Spectrometry and a Derived Equation. Radiochemistry 63, 627–634 (2021). https://doi.org/10.1134/S1066362221050118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1066362221050118