
Abstract
Three crystal modifications of phenyl-substituted diphenylphosphoryl thiosemicarbazide (compound (1) and two its solvates with dimethyl sulfoxide (2, 3)) have been obtained and analyzed by the X-ray diffraction (XRD) method. The independent molecules of compounds in crystals 1 and 2 are characterized by the identical (“rolled”) conformation of acyclic framework, stabilized by the intramolecular hydrogen bond N–H⋅⋅⋅O, and their crystals exhibit the formation of an identical chain supramolecular motif, oriented along the axis corresponding to the smallest unit-cell parameter. In crystal 3, the independent molecules are in two different conformations: one is stabilized by an intramolecular hydrogen bond, and the other (“linear”) is due to the molecular adjustment during the formation of a point supramolecular associate with a hydrophilic nucleus and a hydrophobic shell.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Hetero-containing organic compounds are recognized universally as a reliable resource for developing potential and real drugs. For example, organic nitrogen- and sulfur-containing compounds (e.g., sulfonamides and thiazines [1–3]) became an integral part of medical chemistry. Among hetero-acyclic molecules, one may distinguish thiosemicarbazides, the chemistry of which has been actively developing in the last decade in view of the variety of important fields of their application. Free 1,4-disubstituted thiosemicarbazides and their complexes with various metals make up a universal class of compounds exhibiting biological activity (including antibacterial, antifungus, antiinflammatory, antimalarial, viricidal, anticancer) [4–12]. Complexes of thiosemicarbazones with Zn(II) exhibit fluorescent properties and can be used to visualize living cells [13–15]. Thiosemicarbazides may serve as intermediates when synthesizing other important classes of heterocyclic compounds (e.g., 1,2,4-triazoles, 1,3,4-tidiazoles, 1,3,4-oxadiazoles) [16–18]. In addition, thiosemicarbazides are widely used in the production of dyes, photographic films, and plastics, as well as in textile industry [19].
The energies of eight possible conformers of a thiosemicarbazide molecule were estimated within the density functional theory; it was established that the conformation implemented in the crystal is most favorable [20]; i.e., the molecule should not have conformational lability in a crystal. However, functionalization of thiosemicarbazide by various pharmacophoric groups in order to obtain polyfunctional compounds (for aggregation and improvement of pharmacological properties due to the synergetic effect) may increase the acyclic framework of the molecule and its possible conformational mobility in crystals.
Conformation of molecules affects the supramolecular organization in a crystal and its chemical and physicochemical properties (e.g., solubility, stability, complexing, etc.) [21]. From the crystallographic point of view, the compounds whose crystals may exhibit different types of conformations are of interest. It is urgent to reveal the reasons for this molecular behavior of compounds (specifically, phenyl-substituted (diphenylphosphoryl)acetyl thiosemicarbazide), and our study was aimed at solving this problem.
The general scheme of synthesis of (diphenylphosphoryl)acetyl derivatives of thiosemicarbazide and the crystal structure of N-unsubstituted thiosemicarbazide in the form of a solvate crystal with dimethyl sulfoxide (DMSO) were reported previously in [22]. In continuation of the studies of compounds of this series, we will consider the specific features of molecular and crystal structures of the (phosphoryl)acetyl derivative of thiosemicarbazide (N4-substituted (scheme 1)), presented in the form of three crystal modifications, obtained by slow crystallization from (1) ethanol, (2) DMSO, and (3) DMSO/ethanol mixture in a ratio of 1 : 1.

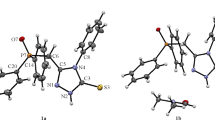
Structural formula of N1-(diphenylphosphoryl)acetyl-N4-phenyl- thiosemicarbazide, with framework atoms enumerated.
EXPERIMENTAL
Single crystal X-ray diffraction (XRD) experiments were performed on a Bruker Kappa Apex II CCD automatic diffractometer (graphite monochromator, МоKα radiation, λ = 0.71073 Å) at temperatures of (1, 3) 150 and (2) 293 K. The data collection, edition and the refinement of unit cell parameters were performed using the APEX2 program [23]. The absorption correction was calculated based on the SADABS program [24]. The crystallographic characteristics and structure refinement parameters are listed in Table 1. The structures were solved by direct methods and refined by the least-squares method, first in the isotropic approximation and then in the anisotropic approximation (for all non-hydrogen atoms) within the SHELXTL software package [25]. The coordinates of the hydrogen atoms of amino groups (except for H2A in compound 2) were determined based on the electron-density difference maps and refined in the isotropic approximation. The coordinates of other hydrogen atoms were determined using stereochemical criteria and refined in the riding models. All calculations were carried out using the WinGX program [26]. The intermolecular interactions were analyzed and images were plotted using the PLATON [27] and Mercury [28] programs. The crystallographic data for compounds 1–3 have been deposited with the Cambridge Crystallographic Data Centre (CCDC) and can be freely accessed at http://www.ccdc.cam.ac.uk/data_request/cif.
Powder XRD patterns were obtained on a D8 Advance automatic X-ray diffractometer (Bruker), equipped with a Vario attachment and a Vantec linear coordinate detector (CuKα1 radiation, λ = 1.54063 Å, a curved Johansson monochromator; the X-ray tube mode was 40 kV and 40 mA). The experiments were performed at room temperature in the Bragg–Brentano geometry. The single crystals of the compounds under study were previously ground, and the thus prepared powders were deposited (without strong pressing) onto silicon plate decreasing the background scattering. The diffraction patterns were recorded in the range of scattering angles 2θ = 3°–95° with a step of 0.008°, the collection time at a point was 4 s. The samples were rotated in their planes at a rate of 15 rpm to eliminate the influence of preferred orientation and average data.
RESULTS AND DISCUSSION
In the solvate-free form, N1-(diphenylphosphoryl)acetyl-N4-phenyl-thiosemicarbazide forms orthorhombic crystals (1) with one molecule in the independent part of unit cell. Two its solvates with DMSO in the ratio of 2 : 1 are (2) monoclinic and (3) triclinic crystals with two independent thiosemicarbazide molecules in their unit cells.
The acyclic-framework conformations of molecules in crystals 1 and 2 are identical; they are formed due to the realization of intramolecular interaction of the N–H⋅⋅⋅O type between hydrogen atom H4 of the terminal amine group and oxygen atom О13 of the phosphoryl group (Fig. 1). The difference between two independent molecules of compound 2 is mainly due to the rotations of phenyl substituents, which is likely caused by the formation of different supramolecular motifs by independent molecules. The structure of molecule А of compound 2 is close to that of a molecule of compound 1 and they form identical supramolecular motifs in the crystal, which will be discussed below.

Conventional superposition of independent molecules of compounds 1 and 2. The intramolecular hydrogen bond N–H⋅⋅⋅O is indicated by a dotted line. The hydrogen atoms of phenyl cycles are omitted.
The О…Н distance in a molecule of compound 1 is 2.54(6) Å, and the corresponding distances in molecules А and B of compound 2 are, respectively, 2.27(4) and 2.14(3) Å. The parameters of strong intra- and intermolecular interactions in the crystals are listed in Table 2 (the geometry of individual molecules in crystals is shown in Figs. S1–S3 in the Supplementary Materials).
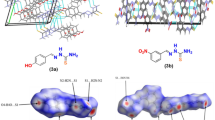
It is likely that, in the case of phosphoryl-acetyl derivatives of thiosemicarbazides, the arrangement of the hydrogen-bond donor and acceptor at different ends of molecule and the length of the spacer between them favors realization of an intramolecular hydrogen bond, which is observed in the crystals of molecules 1 and 2. To establish the generality of this interaction, we searched thiosemicarbazides and their nearest structural analogs (scheme 2) with identical arrangement of donor and acceptor of the N–H⋅⋅⋅O hydrogen bond in the CCDC. The search was restricted by the molecules whose acyclic frameworks contain nine or ten atoms, depending on the acceptor type (phosphoryl, carbonyl, or sulphonyl).

Molecular fragment used to search for thiosemicarbazide derivatives with structures similar to those of compounds 1–3. The covalent bonds of different types are shown by dotted lines.
Based on the search results, 32 compounds were chosen. Some of them, characterized by significant deviations of the interaction parameters from the values corresponding to the formal criteria of hydrogen bond formation, were excluded from consideration (the detailed data are listed in Table S1). Note that molecules of some compounds contained several fragments that were specified for search. Finally, 25 compounds were chosen, 17 of which were in the form of solvate crystals. It is noteworthy that intramolecular hydrogen bond X=O⋅⋅⋅H–N (X = C, S, P) is implemented in the crystals of all these compounds. The О⋅⋅⋅Н distances are in the range of 2.05–2.40 Å; their values are listed in Table 3. Note that the presence of solvate molecules (including strong acceptors. e.g., DMSO and DMF) in the crystals does not hinder realization of intramolecular bonding of this type.
Thus, the following empirical rule is valid for all chosen compounds: if there are no obstacles in a molecule to free rotation along single bonds, its conformation is determined by the above-considered intramolecular interaction, independent of the crystallization type (solvate-free form or a solvate crystal).
One would think that, at the total confirmation of intramolecular interaction by statistical and experimental data, taking into account that the hydrogen-bond acceptor in thiosemicarbazides must occupy a certain position with respect to the hydrogen atom of amine group, the existence of a crystal modification with another molecular geometry is unlikely. However, a radically different conformation of the acyclic molecular framework is implemented specifically in crystal 3 of the other solvate of phosphoryl-acetyl derivative of thiosemicarbazide with DMSO.
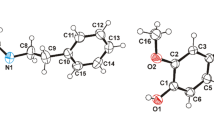
In the crystal of compound 3, two independent molecules are characterized by different acyclic-framework conformations: the previously described “rolled” conformation, stabilized by an intramolecular hydrogen bond, for molecule А with the О⋅⋅⋅Н distance of 2.13(5) Å and a “linear” conformation for molecule B (Fig. 2). Comparison of independent molecules of three investigated compounds indicates the absence of visible reasons for a change in the molecular behavior during crystallization. Most likely, these changes in crystal 3 are due to the influence of the crystalline field and molecular environment rather than incorporation of solvent molecule into the crystal structure.

Conventional superposition of two independent molecules of compound 3. The intramolecular hydrogen bond N–H⋅⋅⋅O is indicated by a dotted line. All hydrogen atoms (except for H4А) are omitted.
It should be noted that the quantum-chemical calculations (B3LYP, 6-31G(d,p)) with optimization of the geometry of independent molecules of crystals 2 and 3 revealed that the “rolled” conformation, stabilized by intramolecular interaction, is less favorable (by 1.7 kcal/mol) than the “linear” one (the calculation results are reported in the Supplementary Materials, Figs. S4–S7). It is not quite clear why the “linear” conformation is somewhat more favorable at the molecular level, and further analysis must be performed to clarify this issue. A possible reason is the influence of molecular environment in the crystal.
One should take into account that the molecule of phosphoryl-acetyl derivative of thiosemicarbazide contains many active functional groups, which can participate in the intermolecular interactions of various nature and call for separate analysis. Nevertheless, let us first consider the realization of classical intermolecular hydrogen bonds in the crystals. Other aspects of the formation of supramolecular structures will be revealed by analyzing the Hirshfeld surface, which represents visually the molecular environment in crystals [29].
As the main forming motif in crystal 1, one can consider the one-dimensional zigzag chain formed due to the N–H⋅⋅⋅O interactions with participation of an oxygen atom of the О13 (N2–H2⋅⋅⋅O13) phosphoryl group and oriented along the axis corresponding to the smallest unit cell parameter a (Fig. 3a). The packing of one-dimensional chains in a crystal is of hexagonal type (Fig. 3b). The N1–H1⋅⋅⋅O11 hydrogen bond also stabilizes these chain supramolecular motifs.

Crystal of compound 1: (a) fragment of a one-dimensional zigzag chain and (b) fragment of a packing of one-dimensional motifs (view along Оа).
A similar supramolecular motif oriented along the c axis (corresponding to the smallest unit-cell parameter) is formed in crystal 2 by molecules А with participation of similar active molecular centers (N1А–H1А⋅⋅⋅O11А and N2А–H2А⋅⋅⋅O13А). One can observe difference in the character of involvement of active centers into the formation of supramolecular motif on the Hirshfeld surfaces for a molecule of compound 1 and molecule А of compound 2 (Fig. S8).
A radically different supramolecular structure is formed by the second independent molecule in crystal 2, despite the fact that the acyclic-framework conformations of molecules A and B are identical. Two molecules B form (with participation of two DMSO molecules) a closed centrosymmetric motif (pseudotetramer) via N–H⋅⋅⋅O hydrogen bonds (N1B–H1B⋅⋅⋅O30 and N2B–H2B⋅⋅⋅O30 with О⋅⋅⋅Н distances of 2.13(5) and 2.13(4) Å, respectively) (Fig. 4).

Pseudotetramer formed by molecules B in crystal 2. The intermolecular interactions are indicated by dotted lines.
On the Hirshfeld surface for molecule B, one can observe also a pair of concentric red regions, corresponding to the centrosymmetric nonclassical С–Н⋅⋅⋅О interaction (С12В–Н12С⋅⋅⋅О11В) with a neighboring molecule B, bound with the initial one by an inversion center (Fig. S9). Due to this interaction, pseudotetramers are bound into a one-dimensional motif (H-chain oriented along the a axis). An interesting aspect of this interaction is that it is involved in the orientation of phenyl rings of the diphenylphosphoryl fragment of molecules B.
Thus, despite the identity of molecular conformations, the morphology of the supramolecular motifs formed by separately independent molecules А and B in crystal 2 differ radically. Taking into account the above-considered structure-forming N–H⋅⋅⋅O and С–Н⋅⋅⋅О interactions in crystal 2, each independent molecule forms its own one-dimensional H-chains of different morphologies, oriented perpendicular to each other. Each independent molecule forms a layer of H-chains, and a crystal can be considered as a stack of layers alternating along the b axis (Fig. 5). In this case, two independent molecules are spatially separated in a crystal, and the packing becomes looser. For example, according to Kitaigorodskii packing coefficient of molecules in the crystals of compounds 1 and 2 are 67.9 and 65.6%, respectively.

Arrangement of independent molecules А (colored dark gray) and B (bright gray) in crystal 2.
Despite the fact that the character of crystallization of crystal 3 is similar to that for compound 2 (with participation of two independent compound molecules and one DMSO molecule), there is no spatial separation of two independent molecules, because their common supramolecular motif is formed. Apparently, one of the reasons is the previously discussed fact that one out of two independent molecules in crystal 3 is in a conformation that is atypical for derivatives of thiosemicarbazides with the acyclic framework. This conformation may be due to the formation of a peculiar supramolecular associate (as the main motif) with participation of two molecules А of the “rolled” conformation; two molecules B of the “linear” conformation; and two DMSO molecules, bound by a set of N–H⋅⋅⋅O interactions (Fig. 6). This motif stabilizes the additional N4B–H4B⋅⋅⋅S3A interactions with participation of thionic fragment. Two pairs of symmetric concentric red regions can be observed on the Hirshfeld surface of molecule B, one of which corresponds to the nonclassical С12B–H12D⋅⋅⋅O11В interaction and the other corresponds to the С25B–H25B⋅⋅⋅O11В interaction (Fig. S10) (the latter orients phenyl cycles of the diphenylphosphoryl fragment). Due to the presence of double hydrogen bonds С12B–H12D⋅⋅⋅O11В, the associates are bound into a one-dimensional cylindrical motif, oriented along the crystallographic a axis, which corresponds to the smallest unit-cell parameter (Fig. S11).

Point associate in crystal 3, formed by four compound molecules and two solvent molecules. The atoms involved in the formation of a chain from H bonds in the supramolecular motif are shown in the sphere–rod representation.
The powder diffraction experiments demonstrated that polycrystalline samples 1 and 3 correspond on the whole to the theoretical data calculated based on the single crystal data (Fig. 7), whereas compound 2, in which two multidirectional one-dimensional motifs are formed, is unstable and passes to the amorphous state.

(1, 3) Theoretical and (2, 4) experimental powder XRD patterns of compounds (1, 2) 1 and (3, 4) 3.
CONCLUSIONS
Based on the results of this study and the data in the literature (using CCDC), it was shown that all derivatives of thiosemicarbazides of considered type in the solid state exhibit the formation of an intramolecular classical hydrogen bond, which leads to the “rolled” conformation of the acyclic molecular framework, independent of the crystallization type (non-solvate form or solvate crystal). The preferred “rolled” type of molecular conformation does not exclude the possibility of forming supramolecular motifs of various dimensionalities and morphologies (including those leading to spatial separation in the crystal regions occupied by independent molecules) in crystals of phosphoryl-acetyl derivatives of thiosemicarbazide due to the intermolecular hydrogen bonds.
The conformational lability, which was observed for independent molecules in crystal 3, is likely due to the induced deviation of molecular geometry from the “rolled” conformation during the formation of a unified supramolecular capsule-type structure by molecules of both conformers.
REFERENCES
S. l. Badshah and A. Naeem, Molecules 21, 1054 (2016). https://doi.org/10.3390/molecules21081054
M. Feng, B. Tang, S. H. Liang, et al., Curr. Top. Med. Chem. 16, 1200 (2016). https://doi.org/10.2174/1568026615666150915111741
F. Carta, A. Scozzafava, and C. T. Supuran, Expert Opin. Ther. Pat. 22, 747 (2012). https://doi.org/10.1517/13543776.2012.698264
G. Küçükgüzel, A. Kocatepe, E. De Clercq, et al., Eur. J. Med. Chem. 41, 353 (2006). https://doi.org/10.1016/j.ejmech.2005.11.005
M. Molnar, M. Tomić, and V. Pavić, Pharm. Chem. J. 51, 1078 (2018). https://doi.org/10.1007/s11094-018-1743-3
T. Plech, M. Wujec, A. Siwek, et al., Eur. J. Med. Chem. 46, 241 (2011). https://doi.org/10.1016/j.ejmech.2010.11.010
M. R. Mlahi, O. A. El-Gammal, M. H. Abdel-Rhman, and I. M. AbdAl-Gader, J. Mol. Struct. 1182, 168 (2019). https://doi.org/10.1016/j.molstruc.2018.12.064
S. K. Kushawaha, R. K. Dani, M. K. Bharty, et al., J. Mol. Struct. 1063, 60 (2014). https://doi.org/10.1016/j.molstruc.2014.01.043
R. Pingaew, S. Prachayasittikul, and S. Ruchirawat, Molecules 15, 988 (2010). https://doi.org/10.3390/molecules15020988
G. Cihan-Üstündag, E. Gürsoy, L. Naesens, et al., Bioorg. Med. Chem. 24, 240 (2016). https://doi.org/10.1016/j.bmc.2015.12.008
S. Güniz Küçükgüzel and G. P. Coşkun, Anti-Cancer Agents Med. Chem. 16, 1288 (2016). https://doi.org/10.2174/1871520616666160219160256
G. P. Coşkun, T. Djikic, T. B. Hayal, et al., Molecules 23, 2 (2018). https://doi.org/10.3390/molecules23081969
A. R. Cowley, J. Davis, J. R. Dilworth, et al., Chem. Commun. 7, 845 (2005). https://doi.org/10.1039/b417206j
D. Dayal, D. Palanimuthu, S. V. Shinde, et al., J. Biol. Inorg. Chem. 16, 621 (2011). https://doi.org/10.1007/s00775-011-0764-0
J. Tang, H.-Y. Yin, and J.-L. Zhang, Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells (Academic, 2017), Ch. 1, p. 1. https://doi.org/10.1016/B978-0-12-803814-7.00001-0
M. R. Ahmed, Eur. J. Med. Chem. 179, 335 (2019). https://doi.org/10.31838/ijpr/2019.11.04.011
T. Li, G. Wen, J. Li, et al., Molecules 24, 1490 (2019). https://doi.org/10.3390/molecules24081490
S. M. Riyadh, S. M. Gomha, and E. A. Mahmmoud, Curr. Org. Synth. 4, 3 (2017). https://doi.org/10.2174/1570179413666151218202004
X. Zhang, P. Lei, T. Sun, et al., Molecules 22, 1 (2017). https://doi.org/10.3390/molecules22122085
C. C. Chambers, E. F. Archibong, S. M. Mazhari, et al., J. Mol. Struct.: THEOCHEM 388, 161 (1996). https://doi.org/10.1016/S0166-1280(96)80029-7
R. R. Saravanan et al., Spectrochim. Acta A 139, 321 (2015). https://doi.org/10.1016/j.saa.2014.12.026
I. A. Krutov, E. L. Gavrilova, R. N. Burangulova, et al., Russ. J. Gen. Chem. 87, 2794 (2017). https://doi.org/10.1134/S1070363218110051
APEX2 (Version 2.1), SAINTPlus. Data Reduction and Correction Program (Version 7.31A), Bruker Advanced X-ray Solutions (BrukerAXS, Madison, WI, 2006).
G. M. Sheldrick, SADABS. Program for Empirical X-Ray Absorption Correction (Bruker-Nonius, Delft, 2004).
G. M. Sheldrick, SHELXTL, Structure Determination Software Suite, v. 6.1 (Bruker AXS, Madison, Wisconsin, USA, 2000).
L. J. Farrugia, J. Appl. Crystallogr. 32, 837 (1999). https://doi.org/10.1107/S0021889899006020
A. L. Spek, J. Appl. Crystallogr. 36, 7 (2003). https://doi.org/10.1107/S0021889802022112
C. F. Macrae, P. R. Edgington, P. McCabe, et al., J. Appl. Crystallogr. 39, 453 (2006). https://doi.org/10.1107/S002188980600731X
J. J. McKinnon, A. S. Mitchell, and M. A. Spackman, Chem. Eur. J. 4, 2136 (1998). https://doi.org/10.1002/(SICI)1521-3765(19981102)4:11<2136::AID-CHEM2136>3.0.CO;2-G
Funding
This study was supported by the Russian Foundation for Basic Research (project no. 19-33-60032) in the part concerning the single crystal crystallographic studies (А.I. Samigullina), a government assignment for FRC Kazan Scientific Center of the Russian Academy of Sciences in the part concerning powder X-ray diffraction experiments (А.T. Gu-baidullin), and the Russian Science Foundation in the part concerning the synthesis of (diphenylphosphoryl)acetyl derivatives of thiosemicarbazide (grant no. 14-23-00073-p).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by Yu. Sin’kov
Supplementary Information
Rights and permissions
About this article

Cite this article
Samigullina, A.I., Krutov, I.A., Gavrilova, E.L. et al. Conformational Behavior of N1-(Diphenylphosphoryl)acetyl-N4-phenyl-thiosemicarbazide in Various Crystal Environments. Crystallogr. Rep. 66, 433–440 (2021). https://doi.org/10.1134/S1063774521030226
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774521030226