
Abstract
The paper presents the results of modeling, theoretical and experimental investigations on the physicochemical conditions of the synthesis, calculation of the Т–х phase diagram, thermodynamics and kinetics of crystallization, and correlation of the properties with the composition and grain sizes of semiconducting solid solutions in the two-dimensional (2D) GaS–GaSe system, in which the complete range of GaSxSe1 – x (0 ≤ х ≤ 1) solid solutions is formed. GaSxSe1 – x (0 ≤ х ≤ 1) solid solutions are synthesized and, using the methods of physicochemical analysis, individual thermodynamic phases are identified. In the context of the developed model of solutions close to ideal solutions, the physicochemical and thermodynamic patterns of behavior of the GaS–GaSe Т–х phase diagram are determined. Using a thermodynamic technique, the temperature-concentration dependences of the change in the Gibbs free energy for GaSxSe1 – x (0 ≤ х ≤ 1) solid solutions are determined. The regularities of the kinetics of nucleation and crystallization, as well as the thermodynamics of cluster formation in multicomponent 2D systems are modeled. Electronic band structures of 2D GaS and GaSe are calculated within the framework of density functional theory. From theoretical calculations, the band gaps for GaS and GaSe monolayers are determined. The electrical absorption spectra of amorphous-film and bulk single-crystal GaSe samples are analyzed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 INTRODUCTION
Investigations of low-dimensional, in particular, semiconducting, materials are interdisciplinary and involve physical technology, chemistry, engineering, and biology. The synthesis, characterization, assembling, and processing of such materials and nanoparticles has advanced considerably. In this field, materials based on two-dimensional (2D) systems are still a developing subject. Although synthesis of 2D materials is one of fields of materials science and technology currently being developed, there are no general rules for synthesizing (e.g., nanoparticles), designing micro- and nanostructures, and processing bulk materials based on nanoparticles [1].
As a result, it is important to comprehensively study specific 2D systems and ensure that they are systematically considered for application in semiconductor electronics. Nevertheless, the common patterns of the synthesis and correlations of the properties with the structure, composition, and important physicochemical and technological factors of multicomponent 2D systems are absent.
In relation to this, vital and interesting but intricate systems based on layered chalcogenides of the III group elements, in particular gallium chalcogenides, have been intensively studied in recent years. Investigation of the correlation of the properties with the composition and grain size focused on the thermodynamic and kinetic features of obtaining solid solutions in the GaS–GaSe system is the aim of the present research.
Since the size of an atom is ~1 Å, a cluster of 1 nm in radius includes ~25 atoms and most atoms are situated at the surface. If the size of nanoparticles in such a cluster are smaller than the critical size, then they characterize many physical phenomena or quantum effects in materials, and in particular, the wavelength and the electron mean free path in semiconductors; i.e., when the dimension is smaller than these critical lengths, new physical and physicochemical properties arise. This makes nanoparticles a fascinating and unique class of material compared to large molecules of hundreds or thousands of atoms.
Below, we briefly consider examples of semiconducting systems including 2D materials based on III–VI compounds. The interest in the 2D semiconducting materials based on III–VI compounds, for which quantum effects are typical, is due to the potential of their application in nanoscale devices. In 2D III–VI crystals, the type of the chemical bond within layers is covalent; and between the layers, it is the weak Van der Waals bond. Due to this, for III–VI compounds, anisotropic properties are typical. In terms of the optical and electrical properties, the III–VI crystals, e.g., GaS [2, 3], GaSe [4, 5], InSe [6, 7], and TlS [8, 9], are close to the promising materials of nanotechnology, such as graphene [10] and topological insulators.
The data on the crystal structure and electronic structure of the GaS and GaSe compounds are known [5, 11]. The GaS and GaSe crystals pertain to the hexagonal system and are characterized by a layered structure and the \(D_{{3h}}^{1} - P\bar {6}m2\) space group. They have different polytypes. For instance, GaSe has four polytypes: β-, ε-, γ-, and δ-GaSe [5, 11]. At room temperature, β-GaS and ε-GaSe, respectively are thermodynamically more stable polytypes. GaS and GaSe are wide-gap semiconductors and have band-gap energies of 2.53 and 1.98 eV, respectively, at room temperature. They possess a series of advantages over other 2D materials: a wide range of operating temperatures, the possibility of creating light-emitting devices in the visible region based on them, high values of the critical field of electrical breakdown, and radiational stability [12–16]. GaS and GaSe form the complete range of GaSxSe1 – x (0 ≤ х ≤ 1) solid solutions between each other [5, 17–19].
However, GaS and GaSe polycrystals and single crystals, as well as GaSxSe1 – x solid solutions, formed from a melt often have an inhomogeneous distribution of the density of the dislocations, just as other multicomponent melts [20–25]. These disturbances lead to mechanical stress and the formation of intrinsic point defects in the lattice. As a consequence, the resulting material possesses irreproducible electrical, optical, photoelectric, luminescent, and other physical characteristics. In addition, the synthesis and investigation of the properties of GaS and GaSe are complicated by the formation of different polytypes.
In comparison to their bulk analogues, 2D crystals, including GaS and GaSe, exhibit new physical properties and potential for application in electronic and optoelectronic devices of the new generation. Nevertheless, the controlled synthesis of large homogeneous crystals and 2D layers remains a technological challenge. Let us consider several specific examples.
Two-dimensional nanostructures are attracting attention due to the quantum effects discovered in them. In [26], the results on the synthesis of 2D materials based on GaSe with several nanolayers for high-performance photodetectors are presented. Comparison of the critical parameters of 2D nanostructured photodetectors shows that, e.g., the quantum efficiency of a GaSe monolayer is 85 times greater than that of graphene and 4557 times greater than that of graphene oxide.
The results of the investigation of the controlled vapor phase growth of 2D GaSe single crystals with a high photoresponse are given in [27]. It is shown that, using vapor-phase deposition, homogeneous (up to 60 mm in the cross section) 2D GaSe single crystals were deposited on SiO2/Si substrates. The process was controlled by the duration, region, and temperature of growth, as well as the carrier gas (argon) flow rate. However, obtaining a stoichiometric GaSe composition in this way was problematic.
In [28], a synthesis of multilayer GaS1 – xSex with adjustable compositions in the complete range of х = 0–1 was reported. The synthesis was carried out in a quartz-tube reactor by the vapor-phase chemical transport. As the starting components, Ga2S3 (99.99%, Sigma-Aldrich) and Ga2Se3 (99.99%, Alfa) powders were used. To grow multilayer nanosheets, a Si substrate was employed to deposit layers onto it at 700°C. The composition was controlled by the evaporation temperature and the proportion between the compositions of the two starting powders. For the uniform growth of 300-nm-thick monolayers on the SiOx/Si substrates, a two-zone furnace was used, in which zones I (starting components) and II (for monolayer growth) were heated to 950 and 700°C, respectively. Next, the growth was conducted at these temperatures for 2 to 10 min at a pressure of 20 Torr in an argon atmosphere. It was shown that the obtained multilayer nanoplatelets of alloys in the GaS–GaSe system have a band-gap energy of Eg = 2.0–2.5 eV, and the Eg value can be tuned linearly in the spectral range from the red to the green light. The epitaxial growth of monolayers leads to a drastic increase in Eg up to 3.3 to 3.4 eV, which shifts radiation to the UV region. The ab initio calculations predict that Eg for GaS and GaSe monolayers is 3.325 and 3.001 eV, respectively. When the number of layers is increased to three, the Eg energies of both direct and indirect transitions decrease considerably and Eg for the indirect transition approaches the value for a multilayer platelet. It is worth noting that formation of a stoichiometric composition of multilayer nanoplatelets and obtaining reproducible results using the described technologically challenging technique is problematic.
The reasons for the nonuniform distribution of the structural defects across the volume of the crystals formed in the GaS–GaSe system are determined by several processes. The main causes are crystallization, postcrystallization cooling, and the thermal treatment of the crystals, resulting in gradients of the concentration, chemical potential, and temperature. However, the parameters affecting the formation of the structure and the physical properties of GaSxSe1 – x in the context of the thermodynamic modeling and theory of crystallization of nanophases are still not established.
From this it follows that applying the thermodynamic and kinetic theories of crystallization to describe the formation and evolution of specific structures and polytypes in the form of bulk and thin-film samples based on the GaS–GaSe system is considerably complicated by a series of unsettled issues. In particular, in creating multicomponent materials with a desired set of properties, it is crucial to not only synthesize the substance with a certain chemical composition but also directedly form its structure.
This study aims to carry out a theoretical and experimental investigation on the physicochemical conditions of synthesis, calculation of the T–x phase diagram, thermodynamics and kinetics of crystallization, and correlation of the properties with the composition of phases and grain size of 2D GaSxSe1 – x (x = 0–1.0 mole fraction) solid solutions.
2 EXPERIMENTAL AND THEORY
Materials
GaS and GaSe binary compounds were synthesized from pure elemental gallium (Ga-5 N), sulfur (B5), and selenium (17-3 special-purity grade). The compounds were synthesized by melting the initial elements taken in stoichiometric proportions in the evacuated (10–2 Pa) quartz cells. The cells with the corresponding components for the synthesis of 2D GaS and GaSe were placed in an electric furnace and the temperatures were increased higher than the melting points of these compounds by 10 to 15 K (the melting points of GaS and GaSe are 1288 and 1211 K, respectively). At temperatures higher than the GaS and GaSe melting temperatures, the cells can break because of the high pressure of chalcogen vapors. The cells with molten GaS and GaSe were held for 6 to 8 h, then they were cooled to 500 K and the furnace was switched off. In this way, the cells with GaS and GaSe were spontaneously cooled to room temperature.
The synthesized p-type GaS and GaSe semiconductor compounds were identified by differential thermal analysis (DTA) (NETZSCH 404 F1 Pegasus derivatograph) and powder X-ray diffraction (XRD) (Bruker D8 ADVANCE diffractometer with Cu Kα radiation) [5]. The temperature-phase changes were determined with an accuracy of ±1 K. The error of the determination of the cell parameters did not exceed 0.003 Å.
After that, by melting stoichiometric amounts of the GaS and GaSe compounds obtained earlier in evacuated (10–2 Pa) quartz cells, samples of GaSxSe1–x solid solutions (x = 0–1.0) in the GaS–GaSe system were obtained. GaSxSe1 – x single crystals were grown by directional solidification. The cells with the samples were grown to the melting points of the samples, and then the melts were solidified. The solidification interface velocity was varied in the range of 0.5 to 1.1 mm/h (Bridgman method). The properties were studied with both film [12] and single-crystal samples.
The band structure and electronic properties of GaS and GaSe with a layered structure were investigated in the context of the ab initio method based on the density functional theory (DFT). Geometry optimization and calculation of the total energy was carried out using the package for simulation of the exchange-correlation potential in a version of a generalized gradient approximation (GGA) by Perdew, Burke, and Ernzerhof (PBE) [10]. The total and partial densities of the electronic states in the GaS and GaSe compounds and their monolayers were analyzed.
Factors of Isomorphic Interchangeability of Atoms in Crystals
In the estimation of the interchangeability of the isomorphic GaS and GaSe components, we considered four well-known factors affecting the isomorphic interchangeability of atoms in the solid-solution crystals: the criterion of chemical indifference of the atoms, the factor of atomic size, the factor of the likelihood of the nature of the interatomic bond, and the factor telling us whether the crystal structures are isostructural or not. In particular, according to the first criterion, isomorphic mixtures are formed due to the kinetic energy of the particles’ motion. Such mixtures are close to ideal solutions, in which the forces of interaction between the molecules of different components are equal to the forces at which the molecules of the same component act on each other. In other words, if the GaS particles form an isomorphic mixture with the GaSe particles, then, for an ideal solution, the force of interaction between the GaS and GaSe particles will be the same as that between the GaS and GaS particles and that between GaSe and GaSe. This condition rules out the formation of chemical compounds.
Thermodynamic Properties
The T–x phase diagram for the quasi-binary GaS–GaSe system with a complete range of solid solutions was calculated from the thermodynamic data using the Gibbs energies for the corresponding GaS (1) and GaSe (2) phases. For the thermodynamic calculation of the T–x phase diagram of the binary system 1–2, we used the technique proposed in [5, 29] involving the relationships between the concentrations of the initial components and their thermodynamic functions of melting \((F_{i}^{{\text{m}}})\). Therefore, the liquidus lines (the lines in which the primary solidification begins) and solidus lines (the lines in which the primary solidification ends) in the T–x diagram of system 1–2 with the complete ranges of liquid and solid solutions were described using the formulas
where \(x_{1}^{l}\) and \(x_{1}^{s}\) are the mole fractions of compound (1) in equilibrium liquid and solid solutions, respectively, \(T_{i}^{{\text{m}}}\) and \(\Delta H_{i}^{{0,{\text{m}}}}\) are the melting temperature and melting enthalpy for the ith component, and R is the universal gas constant. The equilibrium T–x phase diagram for the GaS–GaSe system was calculated as follows. Firstly, for each specific liquidus value \(T_{i}^{{\text{m}}}\), the \(F_{1}^{{\text{m}}}\) and \(F_{2}^{{\text{m}}}\) values were calculated, and, then, using Eq. (1), the values of compositions \(x_{1}^{l}\) and \(x_{1}^{s}\) were approximated.
The concentration-temperature dependences of the Gibbs free energy \(\left( {\Delta G_{T}^{0} = f\left( {x,~T} \right)} \right)\) of the formation of the GaSxSe1 – x solid solutions (0 ≤ x ≤ 1) were calculated using the model of a solution including nonmolecular compounds [5, 30]. The equation for \(\Delta G_{T}^{0}\) of the GaSxSe1 – x solid solutions made of the binary components constituting them, at p, T = const, has the form
where x is the GaS mole fraction; (1 – x) is the GaSe mole fraction; \(\Delta H_{{298}}^{0}\) and \(\Delta S_{{298}}^{0}\) are the enthalpy and entropy of formation of the GaSe and GaS compounds; \(\Delta C_{{p,298~}}^{0}\) is the difference in the heat capacities of the GaSxSe1 – x solid solutions and GaSe and GaS compounds, respectively; and \(\Delta G_{T}^{{0,{\text{ex}}}}\) is the excess molar Gibbs free energy of mixing, related to the chemical interaction of different sorts of atoms. From the thermodynamics, \({{\Delta }_{f}}G_{T}^{0} = {{\Delta }_{f}}G_{{298}}^{0} + ~\Delta G_{T}^{{{\text{0}}{\text{,ex}}}}\), where \({{\Delta }_{f}}G_{T}^{{0,{\text{ex}}}}\) is the molar Gibbs free energy of the component in the given phase and \({{\Delta }_{f}}G_{{298}}^{0}\) is the Gibbs free energy in the reference state for this phase. The excess thermodynamic functions of mixing, in particular, \({{\Delta }_{f}}G_{T}^{{0,{\text{ex}}}}\) characterize deviations of the real systems (solutions) from the ideal systems: \({{\Delta }_{f}}\Phi _{{{\text{mix}}}}^{{{\text{gen}}}} = {{\Delta }_{f}}\Phi _{{{\text{mix}}}}^{{{\text{id}}}} + {{\Delta }_{f}}\Phi _{{{\text{mix}}}}^{{{\text{ex}}}}\).
3 RESULTS AND DISCUSSION
Physicochemical Properties
The structure and properties of bulk samples of GaS (light-yellow) and GaSe (dark-red) binary compounds are known to have been studied in the literature in detail. In synthesis and solidification, different polytypes of these semiconductor compounds with a layered structure form. Both of them belong to the GaS structure type, in which each gallium atom is bonded to three chalcogen atoms and another gallium atom.
The XRD and DTA data for the samples of the GaS and GaSe components confirm that the components are completely soluble in each other in both the liquid and solid states. As an example, Fig. 1 shows the X-ray powder pattern for pure β-GaS, which crystallizes in a hexagonal system with the space group \({{P{{6}_{3}}} \mathord{\left/ {\vphantom {{P{{6}_{3}}} {mmc}}} \right. \kern-0em} {mmc}}~\). This crystal polytype has the following unit cell parameters: a = 4.002 ± 0.002 Å and с = 15.44 ± 0.005 Å. For the β-GaSe hexagonal structure, the lattice parameters are 3.755 ± 0.002 Å and с = 15.475 ± 0.005 Å at room temperature. The obtained unit cell parameters for GaS and GaSe agree with the data of [5, 11].

X-ray powder pattern for single crystal of stable β-GaS polytype.
Thermodynamic Calculations
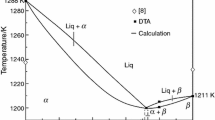
The T–x phase diagram of the GaS–GaSe system studied by us and calculated using Eq. (1) is shown in Fig. 2. From the DTA data, for the GaSe and GaS components, the following melting parameters are obtained [5]: \({\Delta }{{H}^{{\text{m}}}}\)(GaSe) = 30 300 ± 200 J/mol, \({{T}^{{\text{m}}}}\)(GaSe) = 1211 ± 3 K, \({\Delta }{{H}^{{\text{m}}}}\)(GaS) = 34 800 ± 200 J/mol, and \({{T}^{{\text{m}}}}\)(GaS) = 1288 ± 3 K. The GaS–GaSe T–x phase diagram, or, to be more precise, the liquidus (\({{T}^{{\text{l}}}}\)) and solidus (\({{T}^{{\text{s}}}}\)) lines, were calculated taking into account the indicated data for the process of melting the components.

Calculated (curves) and experimental (dots) phase diagram in GaS–GaSe system.
The T–x phase diagram of the GaS–GaSe system is characterized by the complete mutual solubility of the starting components. More specifically, the components form a complete range of solid solutions and the T–x diagram has a minimum in the liquidus curve. The minimum corresponds to the following coordinates: ~0.7 GaSe mole fraction and 1200 ± 1 K. At these coordinates, the compositions of the two equilibrium phases are equal and the GaS–GaSe system degenerates into a quasi-single-component one; i.e., the composition corresponding to the minimum in the GaS–GaSe T–x phase diagram can be regarded as an individual component.
The GaS–GaSe T–x phase diagram with the minimum point belongs to the third type in the Roseboom classification. The analytical solidus and liquidus temperatures are somewhat different from the experimental data (for the solidus, the difference was ~5 K and that for the liquidus was ~10 K). In the minimum point of the T–x phase diagram, the melt with the corresponding composition melts congruently; i.e., the liquid phase passes into the solid phase with the same composition.
The formation of complete GaSxSe1 – x solid solutions in the GaS–GaSe system corresponds to the empirical criteria: the size of atoms Δr/r ≤ 15% (Holdschmidt’s rule), the same crystal structures, and similar electronegativity: |χA – χB| ≤ 0.6. The crystals of the GaSxSe1–x isomorphic mixture are uniform and single-phase, and the species of the isomorphic mixture are distributed in the unit cells of the GaS and GaSe crystals statistically. The interaction between the GaS and GaSe components leads to the formation of phases with variable composition, and the properties change continuously within the homogeneity region. The XRD analysis of GaSxSe1 – x (0 ≤ x ≤ 1) reveals that these solid solutions form substitutional phases without changes to the symmetry of the crystal structure.
At the minimum point of the T–x diagram, the change in the total Gibbs free energy ΔGtot when the melt is cooled with subsequent solidification takes place due to two contributions. The first part of ΔGtot is due to a decrease in the system’s energy during solidification. The system’s energy ΔGtot decreases by the value G1 = VΔGV ~ n3 (V is the volume of the monomer nucleus–germ, ΔGV is the specific difference in the free energies when the melt transits to the crystalline state, and n3 is the number of particles). Conversely, the second part of ΔGtot increases by the value equal to the work of the formation of the interface G2 = sσ ~ n2 (σ is the specific surface tension at the crystal–liquid interface and s is the germ’s surface area). Supposing that each formed nucleus of the thermodynamic phase is a sphere with radius r, we may write ΔGtot = r3ΔGV + 6r2σ.
New clusters with subcritical sizes (r < r*) formed from monomers characterize germs, and clusters with a radius larger than r* characterize nuclei. From here, it follows that the germs larger than the critical size (r > r*) cause a decrease in ΔGtot; i.e., such germs will be stable and capable of further growth. Consequently, up to the first approximation, the process of the formation of an ordered phase agrees with the criteria of the thermodynamic theory of the formation of a new phase.
Let us analyze the solutions in the GaS–GaSe system thermodynamically taking into account the Gibbs free energy G, which, like any other extensive (mass-dependent) thermodynamic function of a solution, is determined by the external parameters (p and T, the pressure and temperature) and by the solution’s composition: \(G = f\left( {p,~T,{{x}_{1}},{{x}_{2}}} \right)\). On condition that \(p = {\text{const}}\), the dependences \({{\Delta }_{f}}G_{T}^{0}\left( {x,~T} \right)\) were calculated for the formation of GaSxSe1 – x solid solutions using Eq. (2). In Eq. (2), the last two terms are the free energy of mixing during the GaSxSe1 – x formation, which involve the partial molar thermodynamic quantities for the GaS and GaSe components.
According to the theory of solutions, the function of mixing \(\bar {\Phi }_{i}^{{{\text{mix}}}}\) (or the relative partial molar quantity) characterizes the difference between the partial molar quantity for the component in the solution and the molar quantity for the pure component; i.e., \(\bar {\Phi }_{i}^{{{\text{mix}}}}\) characterizes the change in the total thermodynamic function when 1 mole of a pure substance is mixed with an infinitely large amount of solution with the given composition at constant T and p. From here, it follows that the partial molar function of mixing \(\overline {~\Phi } _{i}^{{{\text{mix}}}} = {{\Phi }_{i}} - \Phi _{i}^{0}\) is the function of mixing characterizing the individual solution component, where \({{\Phi }_{i}}\) is the total partial molar quantity and \(\Phi _{i}^{0}\) is the molar quantity for the pure component i.
To describe the thermodynamic functions of state, in particular, the Gibbs energy of the pure components in a condensed state using the CALPHAD approach (CALculation of PHAse Diagrams), generally, the Scientific Group Thermodata Europe (SGTE) database (DB) is employed. In the SGTE DB, there are data for calculating the Gibbs energy of the elements in different phase states, including metastable ones, for wide temperature ranges. However, in the approximation of the GaS–GaSe T–x phase diagram, we used the experimental and tabulated reference thermodynamic data. For the calculations, the following averaged molar thermodynamic functions for the GaS and GaSe compounds were taken: \({{\Delta }_{f}}H_{{298}}^{0}\left( {{\text{GaS}}} \right)\) = –194 650 ± 14 650 J/mol, \({{\Delta }_{f}}S_{{298}}^{0}\left( {{\text{GaS}}} \right)\) = 420 ± 290 J/(mol K); \({{\Delta }_{f}}H_{{298}}^{0}\left( {{\text{GaSe}}} \right)\) = –159 000 J/mol, and \({{\Delta }_{f}}S_{{298}}^{0}\left( {{\text{GaSe}}} \right)\) = –11 J/(mol K).
The concentration dependences \(~\Delta C_{{298}}^{0} = f\left( x \right)\) of the heat capacity of the GaSxSe1 – x solid solutions were calculated taking into account the heat capacities \(C_{{p,298}}^{0}\) of the GaS and GaSe binary compounds; at room temperature, \(C_{{p,298}}^{0}\left( {{\text{GaS}}} \right)\) = 47 ± 1 J/(mol K) and \(C_{{p,298}}^{0}\left( {{\text{GaSe}}} \right)\) = 48 ± 1 J/(mol K).
Considering the phase diagram of the GaS–GaSe system, for the GaS and GaSe compounds, the excess partial molar Gibbs free energies \({{\Delta }_{f}}\bar {G}_{T}^{{0,{\text{ex}}}}\) were calculated [5]:
where \({{T}^{s}}\) is the temperature in the solidus line of the T–x phase diagram. The entropy parameters of melting \({{\Delta }_{f}}S_{i}^{m}\) of the corresponding compounds were determined from the formula \({{\Delta }_{f}}S_{i}^{m} = {{{{\Delta }_{f}}H_{i}^{m}} \mathord{\left/ {\vphantom {{{{\Delta }_{f}}H_{i}^{m}} {T_{i}^{m}}}} \right. \kern-0em} {T_{i}^{m}}}\). \({{\Delta }_{f}}S_{i}^{m}\left( {{\text{GaS}}} \right)\) = 27.3 ± 0.1 J/(mol K) and \({{\Delta }_{f}}S_{i}^{m}\left( {{\text{GaSe}}} \right)\) = 25.4 ± 0.1 J/(mol K).
Using the partial quantities \({{\Delta }_{f}}\bar {G}_{T}^{{0,{\text{ex}}}}\) for the GaSe and GaS compounds, the integrated excess molar free energy \({{\Delta }_{f}}G_{T}^{{{\text{ex}}}}\) for GaSxSe1 – x solid solutions (0 ≤ \(x\) ≤ 1) was calculated. Next, taking into account the molar enthalpy and entropy of formation of the GaS and GaSe compounds, as well as \({{\Delta }_{f}}G_{T}^{{{\text{ex}}}}\) and \(\Delta C_{{p,298}}^{0}\), with the use of Eq. (2), we determined the free energy of mixing \({{\Delta }_{f}}G_{T}^{{{\text{mix}}}}\) for the formation of GaSxSe1 – x solid solutions:
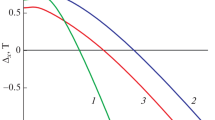
Employing Eq. (4), in the temperature range 300–1200 K, the temperature dependence of the free energy of mixing components in the GaS–GaSe system was approximated. The calculated concentration-temperature dependences, in particular, \(~{{\Delta }_{f}}G_{T}^{{{\text{mix}}}} = f\left( {x,~T} \right)\) of the components, agree with the GaS–GaSe T–x phase diagram (Fig. 3). As is seen from the figure, at different temperatures, the sensitive quantity \({{\Delta }_{f}}G_{T}^{{{\text{mix}}}}\) is correlated to the concentration dependence of the free energy of the distortion of the chemical bonds in the GaSxSe1 – x solid solutions.

Concentration-temperature dependence of Gibbs free energy of mixing calculated for GaSxSe1 – x solid solutions (x = 0–1 mole fraction) using Eq. (4) and data for GaS and GaSe. (1) 300, (2) 800, and (3) 1200 K.
For the mixing reaction, the temperature dependence of the enthalpy of mixing was calculated according to Kirchhoff’s law:
At Т > 300 K, the \({{C}_{p}}\) of the solid solutions obeys the additive rule (Neumann–Kopp law). The \({{\Delta }_{f}}H_{T}^{{{\text{mix}}}}\) quantity does not differ significantly from \({{\Delta }_{f}}G_{T}^{{{\text{mix}}}} = f\left( {x,~T} \right)\) with change in the concentration and temperature of GaSxSe1 – x solid solutions.
The formation of a solid solution (sol. solution) from a mixture of GaS and GaSe compounds can be written in the form of the \(x{\text{GaS}} + \left( {1 - x} \right){\text{GaSe}} \to \) \({\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}\) reaction. For this mixing reaction
For this reaction, \({{\Delta }_{r}}G_{T}^{{{\text{ex}}}} < ~\,\,0\), and it proceeds in the direction from the reactants to the products (sol. solution): \(G\left( {{\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}} \right) < G~\left( {{\text{GaS}}} \right) + G\left( {{\text{GaSe}}} \right)\); i.e., thermodynamically, the solid solution is more stable. The \({{\Delta }_{f}}G_{{298}}^{{{\text{ex}}}}\) value differed by <5% from the total thermodynamic quantity \({{\Delta }_{f}}G_{{298}}^{0}\left( {{\text{GaS}} - {\text{GaSe}}\,{\text{sol}}{\text{.}}\,{\text{solution}}} \right)\). Therefore, in the synthesis, if the effect of the kinetic factors in negligible, solid solutions will form and they will not be decomposed with decreasing temperature.
Thus, the Gibbs free energy of mixing for the systems close to ideal ones, including the complete range of solid solutions, will be the difference between the value of this function for the solid solution and that for the mixture of the initial components. If we take any thermodynamic function, then, for ideal systems, e.g., А–В,
However, there turns out to be a small deviation of the calculated liquidus and solidus from the experimental data in GaSxSe1 – x (0 ≤ \(x\) ≤ 1). This can be related to the aggregate state of the phases which are distinctive in the intermolecular interaction between molecules. In other words, this deviation may be connected with the disturbance of the proportion between the attractive forces and the repulsive forces between the molecules of the starting substances in different phases.
If we consider that, at a distance of r = r0 (where r0 is the equilibrium distance between molecules), the resulting force F = 0, i.e., the attractive and repulsive forces cancel out each other, then, in the presence of thermal motion, \(F \ne ~\,0\). In other words, at r < r0, repulsive forces dominate (F > 0), and, at r > r0, attractive forces dominate (F < 0). At distances r > 10–9 m, there are no forces of intermolecular interaction (F → 0).
When increasing the distance between molecules by dr, the elementary work δA of force F is done through the decrease in the mutual potential energy (Epot) of the molecules; i.e., \(\delta A = Fdr = ~ - d{{E}_{{{\text{pot}}}}}\). From the characteristic dependence \({{E}_{{{\text{pot}}}}} = f~\left( r \right)\) for molecular systems, it follows that, if the molecules are situated at a distance of r → ∞ from each other, then Epot = 0.
When molecules are drawn to each other, attractive forces (F < 0) appear between them and \(\delta A = Fdr > 0\). Then Epot decreases and reaches the minimum at r = r0. With decreasing r, when r < r0, repulsive forces (F > 0) rise and the work done against them will be \(\delta A = Fdr < 0\). In addition, Epot rises and becomes Epot > 0. Therefore, the system of two interacting molecules in the state r = r0 has the minimum of the potential energy \((E_{{{\text{pot}}}}^{{{\text{min}}}}\)).
Kinetics of Nucleation in Multicomponent Systems
In a multicomponent system, determining the conditions of a clustering process is mainly hindered by the kinetic aspects of nucleation indicated below: determining the form of the distribution of the clusters and the contribution of the interface and diffusion fluxes to the nucleation process, and calculating the nucleation rate.
As opposed to the single-component case, during the kinetic development, the composition of a multicomponent cluster differs from that of a cluster in the initial phase. Nucleation and growth of this cluster will include both attachment of molecules at the interface and transfer of the cluster through the initial phase to the interphase region. The link between the indicated processes is complicated compared to growth due to the stochastic character of the cluster’s evolution in the vicinity of the critical size. Under these conditions, the interphase rates are likely to be different for different types of atoms due to long-distance diffusion, and the rate of growth of large clusters will be connected with diffusion-limited growth. The change in the composition in the regions of the initial phase and in the vicinity of each cluster will depend on time. The composition of clusters themselves may depend on the time and size. A technological prehistory is also known to have a noticeable effect on the subsequent character of nucleation.
The composition corresponding to the minimum point in the Т–х diagram of the 2D GaS–GaSe system can be considered as single-component. Let us analyze such a thermodynamically stable composition GaS0.3Se0.7 within the scope of the homogeneous nucleation theory taking into account the energy barrier of the nucleation. Let us assume that the phase transformations in the region of metastability are initiated in the starting phase (GaS0.3Se0.7) by the nucleation of small regions of a new phase, which then grow to a microscopic size. Nucleation is characterized by oscillations of the so-called order parameter for densities, atomic structures, or chemical composition of the starting and new phases. These fluctuations for clusters of several particles (or molecules), in a configuration of a new phase, will be localized and statistically distributed in both space and time. The probability of the onset of oscillations is determined by the thermodynamic conditions, in particular, the minimum work necessary for creating oscillations.
The nucleation energy barrier arises due to the loss of energy for the creation of the interface between the cluster and the initial phase. If the phase transition is thermodynamically favorable, large clusters of a new phase should have a smaller free energy than that of the same particles preserving the configuration of the initial phase. However, the particles in the interface region between the initial and new phases have a higher energy state than that in the two macroscopic phases. For small clusters of the new phase, the majority of particles will be in the interphase region; therefore, creation of such clusters will consume work.
For the critical cluster with radius r* containing n* particles, the work of cluster formation will have maximum W*, which is a nucleation barrier. As the clusters grow beyond the critical size, the proportion of particles in the interphase region decreases, as does the work of cluster formation. That is why the clusters larger than r* (or n*) will grow to complete the phase transformation. The number of clusters crossing this critical size per unit time will be the nucleation rate.
Let us consider the thermodynamics of cluster formation. Based on the statistical grounds, entropy S is determined by the number of available states (w) in a thermodynamic system. Oscillations in a small volume will be accompanied by changes in entropy (ΔS) from the bulk (or average) value (\(\bar {S}\)), which reflects the change in the number of available states from \(\bar {S}\), i.e., from w0 to w: \(\Delta S = S - \bar {S} = \,\,~{{{{k}_{{\text{B}}}}\ln w} \mathord{\left/ {\vphantom {{{{k}_{{\text{B}}}}\ln w} {{{w}_{0}}}}} \right. \kern-0em} {{{w}_{0}}}}\), where \({{k}_{{\text{B}}}}\) is the Boltzmann constant. When a homogeneous medium is balanced out in a time shorter than that necessary for a fluctuation to form, probability P of the emergence of a fluctuation is \(P\,\,~ \propto {\text{exp}}\left( {{{\Delta S} \mathord{\left/ {\vphantom {{\Delta S} {{{k}_{{\text{B}}}}}}} \right. \kern-0em} {{{k}_{{\text{B}}}}}}} \right)\), which is expressed through the minimum work W min required for the formation of a fluctuation: \(~P~ \propto {\text{exp}}\left( {{{ - {{W}^{{{\text{min}}}}}} \mathord{\left/ {\vphantom {{ - {{W}^{{{\text{min}}}}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right)\).
Kinetics of Crystallization
Let us consider the solid–liquid reactions typical for obtaining semiconductor materials. Investigating systems consisting of small molecules, Ostwald (Ostwald, 1897), apparently, for the first time in the literature, described the rule of stages and conditions of the “ripening” of crystals. It is the so-called process of Ostwald ripening. According to the rule of stages of crystallization, the process of the transformation of unstable (or metastable) states into stable ones proceeds step-by-step. Usually, the products formed first are thermodynamically unstable. They are intermediate products possessing the free energies close to those in the initial state.
The second issue dealing with the metastable state is the contribution of the interphase free energy to the total free energy of the system. This energy contribution is small for crystals larger than 1 µm. In the process taking place under equilibrium conditions, i.e., during the Ostwald ripening, large crystals grow through the dissolution of smaller ones. At the beginning of the process, a multitude of small crystals can form in the system; next, most of them disappear, except for several crystals which are enlarged by incorporating other crystals.
Let us consider the physical kinetics of the growth of nanocrystallites in a liquid disperse medium. For such a case, diffusion is the main kinetic factor affecting both the nucleation and growth of crystals. Diffusion is described with the diffusion coefficient
where ED is the diffusion activation energy, which, in the first place, determines the nucleation and growth of a crystal.
According to the classical notions, the growth of crystals from a solution which is a heterogeneous disperse system is related to diffusion effects. These effects can cause the deposition of grains of the second phase in a supersaturated solution containing one or several solid substances distributed in a liquid disperse medium. Such a diffusion process of deposition corresponds to the Ostwald ripening. The kinetic model corresponding to such a diffusion mechanism can be presented as the process in which several grains grow and then dissolve later. At the same time, other grains will be enlarged and incorporate the extra grains which they encounter.
For the mentioned crystallization processes, in the kinetic theory of deposition of nanoparticles, it is assumed that the particle radius changes as the power law \({{\bar {r}}^{n}} - \bar {r}_{0}^{n} \propto t\), where n is the index of the power law. Its value depends on the limitation of the stage of the phase growth and characterizes the solubility of a particle as a function of the particle size.
Under the conditions of diluted solutions, in which the diffusion of ions through a solution is a limiting stage, for the power law, n = 3. Under the conditions of concentrated solutions, e.g., in the case of the formation of germs in alloys, depending on the limiting stage involved in the interphase reactions (dissolution or redeposition), the coefficient n = 2–4.
Growth of a nanoparticle from a liquid disperse medium (from a suspension) can proceed via a mechanism that is different from the Ostwald ripening mechanism. If we represent the growth process as a chemical reaction, then the particles distributed in the liquid disperse medium should contact each other, and their intergrowth becomes possible; i.e., in such an interaction between two particles, depending on their orientation, the third phase should form and a new phase will be deposited. For such a reaction similar to a chemical reaction and the subsequent deposition process, the particle size can be determined by the expression [31]
where \({{r}_{{{\text{equi}}}}}\) is the particle’s equivalent radius at moment t; \({{r}_{i}}\) is the average radius of the initial particle; \(a = ~{{N}_{{\text{A}}}}{{k}_{{\text{B}}}}T{{\left[ {{{A}_{0}}} \right]~} \mathord{\left/ {\vphantom {{\left[ {{{A}_{0}}} \right]~} \eta }} \right. \kern-0em} \eta }\) is the constant determined by temperature T and the viscosity of the medium \(\eta = {{\eta }_{0}}{{e}^{{{{{{E}_{\eta }}} \mathord{\left/ {\vphantom {{{{E}_{\eta }}} {{{N}_{{\text{A}}}}}}} \right. \kern-0em} {{{N}_{{\text{A}}}}}}{{k}_{{\text{B}}}}T}}}\), \(\left[ {{{A}_{0}}} \right]\) is the initial concentration of the crystallites (nanoparticles), \({{N}_{{\text{A}}}}\) is the Avogadro number, \({{\eta }_{0}}\) is the kinetic preexponential factor, which depends on temperature weakly compared to the exponential term in Eq. (5), and \({{E}_{\eta }} = {{E}_{\eta }}\left( T \right)\) is the activation energy of the viscous flow, depending on T on the whole. Having determined all the terms in Eq. (6), we can estimate the average radius of the nanoparticles as a time function at the beginning stages of synthesis.
For different rates of nucleation of the first germ, familiar [32] expressions are listed in Table 1.
In Table 1, N is the number of growth nuclei present at time moment t and \({{K}_{{{\text{nuc}}}}}\) is the nucleation rate constant. If the crystallization process is forced, multistep crystallization takes place and the rate of germ nucleation, in particular, when the solution is heated, is expressed in the form \(J = Zw{{t}^{{\left( {w - 1} \right)}}}\) (differential form) or \(J = Z{{t}^{w}}\) (integrated form), where w is the heating rate and Z is the complex parameter consisting of \({{{{N}_{0}}{{{({{K}_{{{\text{nuc}}}}}t)}}^{{~w}}}} \mathord{\left/ {\vphantom {{{{N}_{0}}{{{({{K}_{{{\text{nuc}}}}}t)}}^{{~w}}}} w}} \right. \kern-0em} w}\). The isothermal crystallization rate for metals and alloys is estimated using the Johnson–Mehl–Avrami–Erofeev–Kolmogorov relationship. It is assumed that crystallization is controlled at the interface, and the formula for the rate of such diffusion-controlled crystallization is expressed by the Avrami–type equation
where \({{\alpha }_{{\text{T}}}}\left( t \right)\) is the dimensionless degree of the transformation in time t or the kinetic degree of the transformation (crystallization) (\(0\,\,~ \leqslant {{\alpha }_{{\text{T}}}}\left( t \right) \leqslant 1)\), KT is the overall rate constant (including both the nucleation rate and the growth rate), and \(n = {{n}_{{{\text{nuc}}}}} - {{n}_{{{\text{grow}}}}}\) is the parameter (Avrami index) depending on the crystallization mechanism (Table 1).
Let us suppose that the formed phase of the end product in the bulk form, e.g., crystals, is internally uniform and has the same structure, composition, and properties. Then, the Gibbs free energy of the formation of a crystalline germ with radius r is
where \(\Delta {{G}_{{{\text{cr}} - {\text{l}}}}} \leqslant 0\) for \(T < {{T}_{m}}\) is the difference between the Gibbs free energies for a crystal and a liquid per crystal unit volume, which is thus the driving force of crystallization, \(\gamma ~\,\, > ~\,\,0\) is the interface energy, i.e., the energy per unit surface area of the crystal-liquid interface (\(\gamma \) does not depend on the curvature of the interface at a radius r), and \(\Delta {{G}_{{{\text{elast}}}}}\) and \(\Delta {{G}_{{{\text{deform}}}}}\) are the elastic deformation energy and the lattice deformation energy, respectively.
Correlations of Properties with Composition, Structure of Phases, and Particle Size
The experimental results on the dependence of the band-gap energy Eg on the composition of the GaSxSe1 – x solid solutions (x = 0–1) confirm the isomorphic mixing between the isostructural GaS and GaSe compounds of the same polytype (ε- and β-GaX polytypes) (Fig. 4). With the changing composition of the complete range of solid solutions, isovalent substitutions of selenium (by convention, Se–2 with an ionic radius in a crystal of rc = 0.198 nm) for sulfur (by convention, S–2 with rc = 0.186 nm) leads to a monotonic change in Eg for stable ε- and β- GaS and GaSe polytypes at different temperatures. At 298 K, for GaS and GaSe, the Eg value is 2.53 and 1.98 eV, respectively. The concentration dependence of the band-gap energy \({{E}_{{\text{g}}}} = f\left( x \right){\text{Ga}}{{{\text{S}}}_{x}}{\text{S}}{{{\text{e}}}_{{1 - x}}}\) (x = 0–1) is approximated by Eq. [5]

Dependence of band-gap energy of solid solutions in GaS–GaSe system on composition and temperature: (1) 77 and (2) 300 K.
where b is the bending parameter, which quantitatively determines the deviation from additivity (Fig. 4). The band-gap energy for GaSxSe1–x solid solutions (0 ≤ х ≤ 1) increases as a consequence of the change in х from 0 to 1 mole fraction. Such a run of Eg = f(x) holds true at least for two stable ε- and β-polytypes.
The effective charges of the atoms, evaluated by the Bader scheme [33], indicate that there is a flow of a small (~0.17 e) electronic charge from one [Ga-X] layer to another [Ga-X] layer. Such charge states of atoms differ from the states obtained using the ion model (Ga3+, Х2–). The chemical bonds in GaX layers are likely to be of a mixed covalent-ionic type, whereas the interlayer interactions are Van der Waals in behavior.
Within the scope of the DFT, the ab initio calculations of the electronic band structures for the GaS and GaSe monolayers were performed. For separately located GaS and GaSe layers, primitive supercells were used. Sampling at the k point was carried out using the DFT method [10]. The primitive supercell consists of two Ga atoms and two S (or Se) atoms in two layers. To preclude interaction between neighboring cells along the c axis, a vacuum was maintained within 18 Å along the direction normal to the layer plane.
The band-gap energies for the optimized monolayer configurations of GaS and GaSe are, respectively, 3.8277 and 3.072 eV for direct (Γ → Γ) electron transition, and 0.571 and 0.087 eV for ΔEg = Eg (direct) – Eg (indirect) electron transition taking into account the energy of the indirect transition (Fig. 5). These values agree with the known data for GaS and GaSe. The electronic band structures for 2D GaS and GaSe and the plots of energy bands for monolayer and bulky GaS and GaSe along the high-symmetry k points are analogous and differ in the maximum values for the valence bands. The band-gap energy is a function of the number of layers.

Theoretical electronic band structures for 2D GaS and GaSe. (a) Schematic of first Brillouin zone for hexagonal monolayer structure, where \({{\vec {a}}_{1}}\) and \({{\vec {a}}_{2}}\) are vectors. Plots of energy bands for (b) GaS and (c) GaSe monolayers along high-symmetry k points. In calculated band structures, Fermi level is set to zero energy.
Semiconducting structures based on thin-film GaSe are highly photosensitive [12]. The GaSe film sample was obtained by explosive evaporation in a vacuum. The thickness of the evaporated GaSe films was on the order of one micrometer. In particular, spectra of electroabsorption in the resulting thin-film sample were taken (curve 1) with an alternating (Fac = 6 × 104 V/cm) and constant (Fdc = 106 V/cm) electric field applied to it (Fig. 6). The electroabsorption in a single-crystal GaSe sample was measured in an electric field of Fdc = 5 × 104 V cm–1 applied parallel to the c axis of the crystal. In both samples, aluminum was used as the contacts.

Electroabsorption spectra for (curve 1) Al/GaSe/SnO2 semiconductor structure with amorphous GaSe film thickness of d ≈ 1000 nm and (curve 2) GaSe single crystal grown by Bridgman method.
A comparison of the results of measurements for the semiconductor samples of gallium monoselenide shows that the electroabsorption spectra for the amorphous GaSe film (curve 1) and those for the GaSe single crystal (curve 2) differ strongly. For the experimental optical spectrum of the amorphous GaSe film, there is no excitonic electroabsorption (curve 1), which was typical for the single-crystal GaSe sample (curve 2). The electroabsorption signal in the GaSe film was observed in an alternating electric field over a long time after the constant electric field was switched off. The presence of the electroabsorption signal when only an alternating electric field is applied to the GaSe sample indicates that there is a chaotic internal field in the GaSe film.
The existence of such an internal field in the GaSe film is likely to be due to the presence of microscopic inhomogeneities in the amorphous structure such as crystallite boundaries, creating potential barriers for the drift and recombination of the charge carriers. In addition, in the near-contact region of the GaSe film, the charge captured by the deep traps in the band gap and injected from the contact accumulates. The emergence of an extra negative peak with an increasing electric field is likely to be due to the change in absorption in this region.
In a macroscopic semiconductor crystal, the energy levels of a GaSe particle form bands. The valence band is filled and the conduction band is empty at 0 K. The bands are separated by an energy gap. When an electron receives the action of, e.g., thermal excitation, an electron–hole pair is generated. The electron in the conduction band and hole in the valence band are bound when they approach each other at a finite distance.
This bound pair, i.e., exciton, is delocalized over the whole crystal. The exciton Bohr radius is given by \({{a}_{{\text{B}}}} = {{{{h}^{2}}\varepsilon } \mathord{\left/ {\vphantom {{{{h}^{2}}\varepsilon } {4{{\pi }^{2}}{{e}^{2}}}}} \right. \kern-0em} {4{{\pi }^{2}}{{e}^{2}}}}\left[ {{1 \mathord{\left/ {\vphantom {1 {{{m}_{{\text{e}}}}}}} \right. \kern-0em} {{{m}_{{\text{e}}}}}} + {1 \mathord{\left/ {\vphantom {1 {{{m}_{{\text{h}}}}}}} \right. \kern-0em} {{{m}_{{\text{h}}}}}}} \right]\), where ε is the material dielectric permittivity, me and mh is the effective mass of an electron and a hole, respectively, and e is the elementary charge.
The quantum size effects become observed at the nanocrystal length of the exciton radius of aB. It is ~25 Å for the GaSe thin film. The unit cell sizes for semiconductor materials are smaller than the characteristic de Broglie wavelength (λ). In such materials, λ = h/mv is in the range of nanometers and strong confinement effects become apparent only when the particle size approaches this value. In this dimension, most materials have structures similar to the structures of their bulk analogues.
The electronic structure of materials is closely linked to their nature. In a three-dimensional object of a large size, the electronic structure is not limited by the size of the material. The electron wavelength is much smaller than the length of the material. When the electronic motion is limited in one dimension and is free in the other two dimensions, it results in the creation of quantum wells and quantum films. In this case, electrons feel a potential well, since they will be captured by the quantum film. In such films, the density of states is stepwise in behavior. In the case of a one-dimensional system, i.e., when electrons can move freely in only one direction, the density of states turns out to show the shape of the Lorentz line. Such a situation is observed with carbon nanotubes. If electrons are restricted to some point, a zero-dimensional system results, in which electrons cannot move freely. Here, states with a molecular character appear.
In bulk materials, the density of states changes smoothly, whereas, in closed quantum systems, discontinuities occur. This leads to steps in the 2D confinement, peculiarities in the one-dimensional confinement, and retention and discrete lines in the zero-dimensional confinement.
Among the properties that change dramatically with size are the optical properties, including both the absorption and emission of light, as seen from Fig. 6. Similarly to nanocrystals, GaSe films have discrete orbitals. As is well known, the block model predicts the energies at the levels \({{{{n}^{2}}{{h}^{2}}} \mathord{\left/ {\vphantom {{{{n}^{2}}{{h}^{2}}} {8{{m}_{{\text{e}}}}{{a}^{2}}}}} \right. \kern-0em} {8{{m}_{{\text{e}}}}{{a}^{2}}}}\left( {n = 1,2, \ldots } \right)\) for particles. Then the energy of the first level of a particle will be shifted from the position typical for the bulk value for these particles by \({{{{h}^{2}}} \mathord{\left/ {\vphantom {{{{h}^{2}}} {8{{m}_{{\text{e}}}}{{a}^{2}}}}} \right. \kern-0em} {8{{m}_{{\text{e}}}}{{a}^{2}}}}\), where a is the particle diameter. The band-gap energy increases with a decrease in the particle size (a). As a consequence, GaSe thin films can be sensitive to light in a broad range; therefore, the color range can be varied versus the particle size.
The band-gap energy increases with a decreasing particle size, which is accounted for by the above-mentioned size effect. The band-gap energy of the GaSe film at room temperature is 2.2 eV, whereas, for the GaSe single crystal this value was 1.98 eV, and, for all compositions of the GaSxSe1–x solid solutions, the measured band-gap energy rose to 2.5 eV as х was increased.
CONCLUSIONS
In the GaS–GaSe system, under the conditions of the experiment carried out, taking the published data into account, the formation of continuous phase regions in which a condensed phase coexists in equilibrium with a liquid phase was found. The results of the calculations show that the curves of equilibrium in the T–x phase diagram of the quasi-binary GaS–GaSe system in the liquid and solid phases are quite well approximated by the thermodynamic parameters of melting (\({\Delta }{{H}^{{\text{m}}}}\)(GaS) = 34 800 ± 200 J/mol, \({{T}^{{\text{m}}}}\)(GaS) = 1288 ± 1 K, \({\Delta }{{H}^{{\text{m}}}}\)(GaSe) = 30 300 ± 200 J/mol). The calculated solidus and liquidus temperatures differ by ≤3 to 5% from the experimental ones within the error limits. Such a result is explained by the smooth strengthening of the intermolecular interaction between the GaS and GaSe binary compounds and between the chalcogen ions, emerging due to the analogous electronic structure in the vicinity of phase transitions (liquid ↔ solid) in the complete GaSxSe1 – x solid solutions (0 ≤ x ≤ 1). This means that the energy of the intermolecular interaction between the initial components in the liquid phase, compared to the covalent bonds, is stronger than that in the solid phase, in which a long-range order also acts between particles, i.e., in liquid–crystalline phases, as early as in the disordered phase, there is a relatively strong anharmonicity of the lattice vibrations, which is strongly enhanced when transiting to the liquid state. Since the intermolecular interactions have an electric nature and are the sum of attractive forces (orientation, induction, and dispersion forces) and repulsive forces, in a liquid state, the latter become higher, and the liquidus line differs from the experimentally obtained values more than the solidus line does. This leads to the origination of extra minima of the potential energy, which facilitates the covalent-ionic transport of particles in a liquid phase. Therefore, when the experimental DTA curves are provided, the proposed model makes it possible to theoretically predict the T–x phase diagram of the system including the initial compounds with the analogous structure in a disordered phase, and enables us to say if its continuous transition to an ordered state is possible. In modeling the T–x phase diagram for GaS–GaSe, we took into account our experimental data and the thermodynamic data for the initial GaS and GaSe components. The approximated concentration-temperature dependence of the Gibbs free energy of mixing for GaSxSe1 – x solid solutions (0 ≤ x ≤ 1) agrees with the T–x phase diagram of the GaS–GaSe system.
Based on the analysis of the thermodynamic and kinetic conditions of the growth and solidification of phases in the 2D GaS–GaSe system, the structural-chemical mechanism of formation of GaSxSe1–x solid solutions (0 ≤ x ≤ 1) does not change with the composition at the temperatures of the synthesis. This is explained by stability of the phases during isovalent substitution, which does not change the mechanism of the formation of phases in the system.
An increase in the concentration of one of the components leads to the formation of phases with a layered structure of the GaS type, in which a portion of the chalcogen ions is substituted for the ions of another chalcogen, with the band-gap energy (Eg) changing monotonically within the homogeneity region.
In the context of the density functional theory, the calculated (GGA approximation) values of the band-gap energy Eg for the samples were, in particular, for GaS and GaSe monolayers, 3.8277 and 3.072 eV, respectively. These Eg values differ strongly from the experimental data for bulk GaS (2.53 eV) and GaSe (1.98 eV) samples at 298 K. The reason for the inconsistencies in the Egcalc and Egexp values, apart from the particle size, may be the problem of the adequacy of the local exchange-correlation potential used in the calculation.
Comparison between the electroabsorption spectra for the amorphous GaSe film samples and bulk GaSe samples reveals that the properties of the investigated amorphous film are close to those of the nanocrystalline material. The excitonic electroabsorption, which was observed with the single-crystal GaSe sample, is not typical for the thin-film GaSe. The electroabsorption signal in the GaSe film was observed in the alternating electric field over a long time after the constant electric field was switched off.
REFERENCES
Lukichev, V.F. and Amirov, I.I., Research and development in the field of micro and nanosystems, Istor. Nauki Tekh., 2018, no. 8, pp. 92–99.
Asadov, S.M., Mustafaeva, S.N., and Lukichev, V.F., Charge transport in layer gallium monosulfide in direct and alternate electric fields, Russ. Microelectron., 2019, vol. 48, no. 6, pp. 422–427. https://doi.org/10.1134/S1063739719
Mustafaeva, S.N. and Asadov, M.M., Currents of isothermal relaxation in GaS〈Yb〉 single crystals, Solid State Commun., 1983, vol. 45, no. 6, pp. 491–494. https://doi.org/10.1016/0038-1098(83)90159-X
Asadov, S.M., Mustafaeva, S.N., Lukichev, V.F., and Guseinov, D.T., Effect of the composition on the dielectric properties and charge transfer in 2D GaS1–xSex materials, Russ. Microelectron., 2019, vol. 48, no. 4, pp. 203–207. https://doi.org/10.1134/S1063739719040024
Asadov, S.M., Mustafaeva, S.N., and Mammadov, A.N., Thermodynamic assessment of phase diagram and concentration-temperature dependences of properties of solid solutions of the GaS–GaSe system, J. Therm. Anal. Calorim., 2018, vol. 133, no. 2, pp. 1135–1141.
Mustafaeva, S.N., Asadov, M.M., and Ismailov, A.A., Charge transfer along localized states in InSe and InSe〈Sn〉 single crystals, Low Temp. Phys., 2010, vol. 36, no. 4, pp. 310–312. https://doi.org/10.1063/1.3388822
Mustafaeva, S.N., Asadov, M.M., and Ismailov, A.A., Effect of γ irradiation on the parameters of localized states in p-InSe and n-InSe〈Sn〉 single crystals, Low Temp. Phys., 2010, vol. 36, no. 7, pp. 642–644. https://doi.org/10.1063/1.3479690
Mustafaeva, S.N., Asadov, M.M., and Ismailov, A.A., Charge transfer over localized states in a TlS single crystal, Phys. Solid State, 2008, vol. 50, no. 11, pp. 2040–2043. https://doi.org/10.1134/S1063783408110073
Mustafaeva, S.N., Asadov, M.M., and Ismailov, A.A., Dielectric and baric characteristics of TlS single crystal, Phys. B (Amsterdam, Neth.), 2014, vol. 453, pp. 158–160. https://doi.org/10.1016/j.physb.2014.03.095
Asadov, M.M., Guseinova, S.S., and Lukichev, V.F., Ab initio modeling of the electronic and energy structure and opening the band gap of a 4p-element doped graphene monolayer, Russ. Microelectron., 2020, vol. 49, no. 5, pp. 314–323.
Medvedeva, Z.S., Khal’kogenidy elementov III B podgruppy periodicheskoi sistemy (Chalcogenides of Elements IIIB Subgroup of the Periodic System), Moscow: Nauka, 1968.
Mustafaeva, S.N. and Asadov, M.M., High field kinetics of photocurrent in GaSe amorphous films, Mater. Chem. Phys., 1986, vol. 15, pp. 185–189. https://doi.org/10.1016/0254-0584(86)90123-9
Jung, C.S., Shojaei, F., Park, K., Oh, J.Y., Im, H.S., Jang, D.M., Park, J., and Kang, H.S., Red-to-ultraviolet emission tuning of two-dimensional gallium sulfide/selenide, ACS Nano, 2015, vol. 9, no. 10, pp. 9585–9593. https://doi.org/10.1021/acsnano.5b04876
Hu, P.A., Wen, Z.Z., Wang, L.F., Tan, P.H., and Xiao, K., Synthesis of few-layer GaSe nanosheets for high performance photodetectors, ACS Nano, 2012, vol. 6, no. 7, pp. 5988–5994. https://doi.org/10.1021/nn300889c
Li, X.F., Lin, M.W., Puretzky, A.A., Idrobo, J.C., Ma, C., Chi, M.F., Yoon, M., Rouleau, C.M., Kravchenko, I.I., Geohegan, D.B., and Xiao, K., Controlled vapor phase growth of single crystalline, two-dimensional GaSe crystals with high photoresponse, Sci. Rep., 2015, vol. 4, pp. 1–9. https://doi.org/10.1038/srep05497
Tan, L., Liu, Q., Ding, Y., Lin, X., Hu, W., Cai, M.-Q., and Zhou, H., Effective shape-controlled synthesis of gallium selenide nanosheets by vapor phase deposition, Nano Res., 2020, CN 11-5974/O4. https://doi.org/10.1007/s12274-020-2653-8
Ho, C.H., Wang, S.T., Huang, Y.S., and Tiong, K.K., Structural and luminescent property of gallium chalcogenides GaSe1 – xSx layer compounds, J. Mater. Sci.: Mater. Electron., 2009, vol. 20, pp. S207–S210. https://doi.org/10.1007/s10854-007-9539-3
Bereznaya, S., Korotchenko, Z., Redkin, R., Sarkisov, S., Tolbanov, O., Trukhin, V., Gorlenko, N., Sarkisov, Y., and Atuchin, V., Broadband and narrowband terahertz generation and detection in GaSe1 – xSx crystals, J. Opt., 2017, vol. 19, no. 11, p. 115503. https://doi.org/10.1088/2040-8986/aa8e5a
Kolesnikov, N.N., Borisenko, E.B., Borisenko, D.N., Tereshchenko, A.N., and Timonina, A.V., Synthesis and growth of GaSe1 – xSx (x = 0–1) crystals from melt. Phase composition and properties, Inorg. Mater.: Appl. Res., 2018, vol. 9, no. 1, pp. 66–69. https://doi.org/10.1134/S2075113318010173
Mullin, J.W., Crystallization, 4th ed., Oxford, UK: Butterworth-Heinemann, 2001.
Phase Transformations in Multicomponent Melts, Herlach, D.M., Ed., Weinheim: Wiley-VCH, 2008.
Aaronson, H.I., Enomoto, M., and Lee, J.K., Mechanisms of Diffusional Phase Transformations in Metals and Alloys, 1st ed. Boca Raton: CRC, Taylor and Francis Group, 2010.
Kukushkin, S.A. and Slezov, V.V., Dispersnye sistemy na poverkhnosti tverdykh tel (evolyutsionnyi podkhod): mekhanizmy obrazovaniya tonkikh plenok (Dispersed Systems of the Solid Surface (Evolutionary Approach): Mechanisms for the Formation of Thin Films), St. Petersburg: Nauka, 1996.
Chernov, A.A., Modern Crystallography III, Berlin etc.: Springer, 1984. https://doi.org/10.1007/978-3-642-81835-6
Nucleation in Condensed Matter: Applications in Materials and Biology, Kelton, K.F. and Greer, A.L., Eds., Vol. 15 of Pergamon Materials Series, Amsterdam: Elsevier, Pergamon, 2010.
Hu, P.A., Wen, Z.Z., Wang, L.F., Tan, P.H., and Xiao, K., Synthesis of few-layer GaSe nanosheets for high performance photodetectors, ACS Nano, 2012, vol. 6, no. 7, pp. 5988–5994. https://doi.org/10.1021/nn300889c
Li, X.F., Lin, M.W., Puretzky, A.A., Idrobo, J.C., Ma, C., Chi, M.F., Yoon, M., Rouleau, C.M., Kravchenko, I.I., Geohegan, D.B., and Xiao, K., Controlled vapor phase growth of single crystalline, two-dimensional gase crystals with high photoresponse, Sci. Rep., 2015, vol. 4, pp. 1–9. https://doi.org/10.1038/srep05497
Jung, C.S., Shojaei, F., Park, K., Oh, J.Y., Im, H.S., Jang, D.M., and Kang, H.S., Red-to-ultraviolet emission tuning of two-dimensional gallium sulfide/selenide, ACS Nano, 2015, vol. 9, no. 10, pp. 9585–9593. https://doi.org/10.1021/acsnano.5b04876
Ahmedly, K.M. and Asadov, M.M., Procedure for calculating phase equilibrium in simple binary systems of ideal liquid and solid solution, Inorg. Mater., 1996, vol. 32, no. 2, pp. 133–134.
Asadov, M.M., Mustafaeva, S.N., Mamedov, A.N., and Tagiyev, D.B., Effect of composition on the properties of (TlInSe2)1 – x(TlGaTe2)x solid solutions, Inorg. Mater., 2015, vol. 51, no. 12, pp. 1232–1236. https://doi.org/10.1134/S0020168515110011
Ribeiro, C., Lee, E.J.H., Longo, E., and Leite, E.R., A kinetic model to describe nanocrystal growth by the oriented attachment mechanism, Chem. Phys. Chem., 2005, vol. 6, pp. 690–696. https://doi.org/10.1002/cphc.200400505
Illeková, E. and Šesták, J., Crystallization of metallic micro-, nano-, and non-crystalline alloys, in Thermal Analysis of Micro, Nano- and Non-Crystalline Materials, Dordrecht: Springer Science, 2013, pp. 257–289.https://doi.org/10.1007/978-90-481-3150-1_13
Bader, R.F.W., Atoms in Molecules. A Quantum Theory, Oxford, UK: Clarendon, 1994.
Funding
This study was supported by the Science Development Foundation under the President of the Republic of Azerbaijan, project no. EİF-BGM-4-RFTF-l/2017-21/05/l-M-07 and the Russian Foundation for Basic Research, project no. 18-57-06001 no. Az_a 2018.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they do not have conflict of interest.
Additional information
Translated by Z. Smirnova
Rights and permissions
About this article

Cite this article
Asadov, S.M., Mustafaeva, S.N. & Lukichev, V.F. Modeling of the Crystallization and Correlation of the Properties with the Composition and Particle Size in Two-Dimensional GaSxSe1 – x (0 ≤ х ≤ 1). Russ Microelectron 49, 452–465 (2020). https://doi.org/10.1134/S1063739721010042
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063739721010042