
Abstract
Experimental data for the system GaS–GaSe were subjected to a critical assessment using the differential thermal analysis (DTA), X-ray diffraction and thermodynamic approach. The physicochemical and thermodynamic parameters of the GaS and GaSe compounds were taken from the literature and the authors’ previous assessment, respectively. To reach a self-consistent thermodynamic description for the constituent phases in the system, the experimental on the melting point, the composition of the minimum point and the univariant curves of the GaS–GaSe system were reassessed. An ideal solution model for the liquid, which included non-molecular compounds, was employed to represent phase diagram and Gibbs free energy of mixing data. To make our investigation on invariant points more accurate, a new and complementary experimental DTA determination regarding the compounds was carried out. We clarify the temperature and melting enthalpy of the compounds that are needed to calculate thermodynamic parameters of the system. Our thermodynamic description, compatible with experimental data for the GaS–GaSe system, resulted in an agreement between the calculated and experimental data. The temperature–concentration dependences of the properties (thermodynamic functions, the width of the forbidden band, the heat capacity) of solid solutions of the GaSe–GaS system are established. Optical value was obtained on single-crystal samples. Solid solutions of the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) compounds were grown by the Bridgman method by directional crystallization.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Only a few years ago, low-dimensional structures were an esoteric concept, but now it is apparent they are likely to play a major role in the next generation of electronic devices. The study of layered materials, especially monochalcogenides represented by GaX, is an active and rapidly growing field of research. This field may lead to fabrication of nanodevices in a layer-by-layer fashion using 2D materials. Gallium monochalcogenides (GaX, X = S, Se, Te) are layered compounds like graphite that can be readily reduced to 2D form due to their strong in-plane bonding and weak interlayer van der Waals coupling.
Gallium monoselenide (\(\beta\)-,\(\varepsilon\)-, \(\gamma\)- and \(\delta\)-GaSe) has several polymorphs. Data on the crystal and electron structure of GaX (X = S, Se, Te)-type compounds are shown in [1]. The thermodynamically most stable modification is \(\varepsilon\)-GaSe, which belongs to hexagonal syngony, and is characterized by a layered structure and space group \(D_{3h}^{1} - P\bar{6}m2\) [1]. Layered crystals of GaX type have anisotropic properties, which are caused by the presence of two kinds of interatomic bonding in the crystal [2]. Each layer, e.g., GaSe, contains four atomic planes Se–Ga–Ga–Se perpendicular to the c-axis of hexagonal crystal. Bond is ionic-covalent inside the layers, and neighboring layers are bound by weak forces such as van der Waals forces. GaSe and GaS (space group \(D_{6h}^{4} - P6_{3} /mmc\) for \(\beta\)-type GaS) are wide-gap semiconductors and at room temperature have a band gap at around 1.98 and 2.53 eV, respectively.
Because of its unique properties, GaSe can be used in various fields of science and technology. The wide-gap GaX semiconductor materials have several advantages over other materials: a larger area of operating temperatures, the ability to create on their basis light-emitting devices in the visible spectrum, high values of the critical field of electrical breakdown, radiation resistance [3, 4].
Mamedov et al. by low-temperature calorimetry studied the heat capacity (\(C_{\text{p}}\)) for GaS compounds [5] in the 14–300 K interval and for GaSe [6] in the 60–304 K interval, respectively. The heat capacity anomalies are not found on the \(C_{\text{p}} \left( T \right)\) curve. The thermodynamic parameters of the compounds were calculated. Tyurin et al. [7] by differential scanning calorimetry determined temperature dependence for the heat capacity of GaSe in the 300–700 K interval. Thermodynamic functions were calculated, and it was found that dependence \(C_{\text{p}} /T = f\left( {T^{2} } \right)\) for AIIIBVI compounds is linear in the low-temperature region.
GaSe and GaS compounds are of interest in that they possess semiconducting properties in both the solid and liquid states. GaSe-based materials have low hardness, mechanical strength and susceptible to micro-delamination. To eliminate these drawbacks, it is important to modify the properties of crystals, e.g., by doping or producing GaX-based solid solutions. However, with increasing hardness of crystals it is also necessary to maintain high optical properties and nonlinear susceptibility. Taking into account the practical application of GaSe-based materials makes it relevant to establish the concentration dependences of the properties of the samples of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\). Practical application of these compounds, however, still depends on the ability to prepare them in single-phase form, so phase diagram data for the composition range of the GaS–GaSe system are of key importance.
Rustamov et al. [8] by DTA and XRD, for the first time, built \(T - x\) phase diagram of the GaSe–GaS system with a minimum at 1168 K and 0.68 mol fraction of GaS. The melting point of GaSe and GaS is 1233 and 1238 K, respectively. It was established that continuous rows of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions are forming in the system.
The lack of reliable data in the literature on phase diagram of the quasi-binary GaS–GaSe system necessitated the experimental study of the T-x diagram by differential thermal analysis. Until now, thermodynamic assessment of the GaS–GaSe system has not been performed yet. In order to determine the relationship between the \(T - x\) diagram and thermodynamic functions, we performed thermodynamic analysis of the GaS–GaSe phase diagram. Comparison between deviation values and the additivity of properties (the band gap, heat capacity) of the components of solid solutions of the GaS–GaSe system on the property–composition dependences is carried out.
Aim: modeling and thermodynamic description of the T–x phase diagram of the GaS–GaSe system, determination of temperature–concentration dependences of physicochemical properties of the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions.
Experimental and theoretical technique
Material preparation and experimental details
Ga-5 N gallium, B5 sulfur and OSCh-17-3 selenium with impurity content no higher than 5 × 10−4 mass% were used in the synthesis of GaS and GaSe compounds. Synthesis was performed by melting the initial elements taken in stoichiometric ratios in evacuated (10−3 Pa) and sealed quartz ampules. The ampules were held for 6–8 h at a temperature higher than the liquidus temperature by 25–30 K (melting points of GaS and GaSe are 1288 and 1211 K, respectively) and then left to cool to room temperature. Prepared GaS and GaSe compounds were identified by DTA (a heating/cooling rate of 10 K min−1) and powder X-ray diffraction analysis using the literature data for comparison. DTA of compounds and alloys was carried out using a NETZSCH 404 F1 Pegasus system. The accuracy of measurements was ± 0.5 to 1 K. The XRD analysis was performed on a Bruker D8 ADVANCE diffractometer with the Cu-Kα radiation. For GaSe hexagonal structure, lattice parameters are: a = 3.755 ± 0.002 Å and c = 15.940 ± 0.005 Å at room temperature. The GaSe crystals belong to the \(P\bar{6}m2\) space group and easily cleave along the (0001) plane. Analogous parameters for GaS are: a = 3.583 ± 0.002 Å and c = 15.475 ± 0.005 Å at room temperature.
\({\text{GaSe}}_{{ 1 {\text{ - x}}}} {\text{S}}_{\text{x}}\) solid solutions were prepared analogously by melting stoichiometric weighed portions of preliminarily prepared initial GaS and GaSe components in evacuated quartz ampoules. \({\text{GaSe}}_{{ 1 {\text{ - x}}}} {\text{S}}_{\text{x}}\) elementary cell slightly increases in size when sulfur ion is replaced by selenium ion. \(C_{\text{p}} \left( {T,x} \right)\) temperature–concentration dependences and band gaps (\(E_{\text{g}}\)) of the GaS–GaSe system samples are studied. For \(C_{\text{p}}\) measurements, the vacuum adiabatic calorimeter was used. The error in determining the heat capacity was 0.3% in the investigated temperature interval 10–300 K [9]. \(E_{\text{g}} \left( {T,x} \right)\) of single-crystal samples \({\text{GaSe}}_{{ 1 {\text{ - x}}}} {\text{S}}_{\text{x}}\) was determined by optical method [10].
\({\text{GaSe}}_{{ 1 {\text{ - x}}}} {\text{S}}_{\text{x}}\) single crystals were grown by the Bridgman method [11]. The ampule was moved in the furnace at a rate of 1.1 mm h−1, and the temperature gradient near the crystallization front was 25 ± 3 K.
\(T - x\) phase diagram calculation for GaS–GaSe
Practice shows that it is impossible to uncontrollably raise the temperature in systems containing gallium chalcogenides because at temperatures above melting point of GaX, ampoules may be destroyed due to high vapor pressure of chalcogens. We calculated \(T - x\) dependence of the GaS–GaSe system at constant (atmospheric) pressure, not taking into account the molecular states of chalcogens in gas phase and compared calculation results with experiment.
GaS–GaSe is the example of isomorphous system. The complete solubility in GaS–GaSe occurs because both GaSe and GaS have the same crystal structure, and Se and S have similar radii, electronegativity and valence. In GaSe–GaS system melting occurs over a narrow temperature range, between the solidus and liquidus lines. Solid and liquid phases are at equilibrium in this temperature range.
The equilibrium in the system may be calculated from thermodynamic data using Gibbs energies for all relevant phases. For calculation of the \(T - x\) phase diagram for GaS–GaSe, we used a method including ratios between the concentrations of initial components and their thermodynamic enthalpies of melting [12,13,14]. Thus, the liquidus and solidus lines of the binary system 1–2 with continuous rows of liquid and solid solutions can be analytically described by the formulae
\(x_{\text{i}}^{\text{l}}\) and \(x_{\text{i}}^{\text{s}}\) are the mole fractions of the compound in the equilibrium liquid and solid solutions, respectively, \(T_{\text{i}}^{\text{m}}\) and \(\Delta H_{\text{i}}^{0,\text m}\)—the melting temperature and melting enthalpy of the \(i\)-th component, and \(R\)—universal gas constant. To build \(T - x\) phase diagram for GaS–GaSe, for each particular \(T_{\text{i}}^{\text{m}}\), \({\text{F}}_{ 1}\) and \({\text{F}}_{ 2}\) values were calculated and then via formula (5) \(x_{1}^{\text{l}}\) and \(x_{1}^{\text{s}}\) values were found.
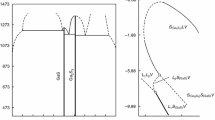
According to DTA data for GaSe and GaS, the following melting parameters of components were chosen: \(\Delta H^{\text{m}}\)(GaSe) = 30300 ± 200 J mol−1, \(T^{\text{m}}\)(GaSe) = 1211 ± 3 К, \(\Delta H^{\text{m}}\)(GaS) = 34800 ± 200 J mol−1, and \(T^{\text{m}}\)(GaS) = 1288 ± 3 К. The calculation results of liquidus (\(T^{\text{l}}\)) and solidus (\(T^{\text{s}}\)) lines of the system GaS–GaSe are shown in Fig. 1.

\(T - x\) phase diagram of the GaS–GaSe system calculated and constructed by us
New experimental measurements of the melting enthalpy for GaSe and GaS compounds along with the data already taken into account in previous thermodynamic assessments have been used in an assessment of the thermodynamic parameters of the GaS–GaSe system. The calculations based on the thermodynamic modeling [12,13,14] are in good agreement with the phase diagram data and experimental thermodynamic values [15,16,17,18,19].
\(\Delta_{\text{f}} G_{\text{T}}^{0} \left( {x,T} \right)\) data are important for modeling the materials preparation process and investigating solid–liquid transitions at varied component concentrations. \(\Delta_{\text{f}} G_{\text{T}}^{0} \left( {x,T} \right)\) composition and temperature dependences for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions were calculated using a modified solution model which included non-molecular compounds [14]. For the Gibbs free energy of formation of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions from their constituent elements at \(p,T = {\text{const}}\), we obtained the equation
where \(x\) is the mole fraction of GaS; 1– \(x\) is the mole fraction of GaSe; \(\Delta_{\text{f}} H_{298.15}^{0}\) and \(\Delta_{\text{f}} S_{298.15}^{0}\) are the enthalpy and entropy of formation of the compounds GaSe and GaS; \(\Delta C_{{{\text{p}},298.15}}^{0}\) is the difference between the heat capacities of the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions and the compounds GaSe and GaS, respectively; and \(\Delta_{\text{f}} G_{\text{T}}^{0,\text ex}\) is the excess molar Gibbs free energy of mixing.
Results and discussion
\(T - x\) phase diagram of GaS–GaSe
Phase diagram for GaS–GaSe system has been studied by various authors before. Version of \(T - x\) phase diagram of GaS–GaSe proposed by Rustamov et al. [8] has been confirmed by our follow-up research. However, our calculation and experimental DTA data on the temperatures of liquidus, solidus and the coordinates of the invariant equilibria in the GaS–GaSe system (Fig. 1) are very different from the data [8] (Table 1). Rustamov [8] shows melting points 1233 and 1238 K for initial GaSe and GaS, respectively. DTA data [8] are also very different from the other literature data [15]. Therefore, a known \(T - x\) phase diagram for GaS–GaSe requires adjustment.
Our calculation results for liquidus and solidus lines of GaS–GaSe system were compared with data from the corresponding literature [8]. For \(T - x\) phase diagram calculation of GaS–GaSe, we used DTA and XRD analysis, taking into account temperatures and enthalpies of melting of initial components [16,17,18,19].
Our studied and calculated phase diagram for GaS–GaSe is shown in Fig. 1. The phase diagram of the GaS–GaSe system is a third type of the Roseboome classification. Calculated solidus and liquidus temperatures differ slightly from the experimental data (solidus difference was ≤ 5 K and liquidus ≤ 15 K). Diagram of the state for GaS–GaSe system is characterized by the minimum (0.7 ± 0.05 mol fraction GaSe and 1200 ± 3 K) and presence of unlimited mutual solubility in the system. This is due to the proximity of the crystal structure source components. It is seen from Fig. 1 that the lines of the liquidus and solidus form a minimum at the point of their tangency. It is obvious that at a given point the compositions of the phases S and L are the same. Therefore, when the compositions of the two equilibrium phases are equal, the GaS–GaSe system degenerates into a one-component system. In this case, the composition corresponding to the minimum point on the equilibrium curves can be considered as a component.
The different phases are not detected in the area of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions. \(T - x\) phase diagram is consistent with thermodynamic calculations of the free energy of mixing (Fig. 2) of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions. Calculation of the temperature dependence of the free energy of mixing of solid solutions [14] was performed in 300–1200 K range.

Concentration–temperature dependence of the Gibbs free energy of mixing calculated by Eq. (4) for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions using data for GaSe and GaS
Thermodynamics of the GaS–GaSe system
The \(\Delta_{\text{f}} G_{\text{T}}^{0} \left( {x,T} \right)\) dependences of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) were calculated by Eq. (2). In Eq. (2), the last two terms are the free energy of mixing in the formation \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) of GaSe and GaS.
To this end, we used the thermodynamic functions of the GaSe and GaS compounds: \(\Delta_{\text{f}} H_{298.15}^{0} \left( {\text{GaS}} \right)\) = − 194650 ± 14650 J mol−1,\(\Delta_{\text{f}} S_{298.15}^{0} \left( {\text{GaS}} \right)\) = − 420 ± 290 J (mol K)−1 [16] \(\Delta_{\text{f}} H_{298.15}^{0} \left( {\text{GaSe}} \right)\) = −159000 J mol−1 [14] and \(\Delta_{\text{f}} S_{298.15}^{0} \left( {\text{GaSe}} \right)\) = − 11 J (mol K)−1. With allowance for the heat capacity of the binary compounds GaS (\(C_{{{\text{p}},298.15}}^{0}\) = 47 ± 1 J mol−1 K) and GaSe (\(C_{{{\text{p}},298.15}}^{0}\) = 48 ± 1 J mol−1 K), the composition dependences of the room temperature heat capacity,\(\Delta C_{{{\text{p}},298.15}}^{0} \left( x \right)\), for the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions were calculated. The partial excess molar Gibbs free energy \(\Delta_{\text{f}} \bar{G}_{\text{T}}^{0, \text ex}\) of the GaSe and GaS compounds was calculated using the phase diagram of the \({\text{GaSe}} - {\text{GaS}}\) system. The values \(\Delta_{\text{f}} \bar{G}_{\text{T}}^{0,{\text{ex}}}\) of the GaX were evaluated as [14]
where T is a temperature on the solidus line; the subscript refers to the GaSe (or GaS) compound. The corresponding parameters were defined by the formula: \(\Delta S^{\text{m}} = \Delta H^{\text{m}} /T^{\text{m}}\), \(\Delta S^{\text{m}} \left( {\text{GaSe}} \right)\) = 25.4 ± 0.1 J (mol K)−1 and \(\Delta S^{\text{m}} \left( {\text{GaS}} \right)\) = 27.3 ± 0.1 J (mol K)−1.
Using the calculated partial values \(\Delta_{\text{f}} \bar{G}_{\text{T}}^{0, \text ex}\) of the GaSe and GaS compounds and taking into account the calculation accuracy, we find the integral excess molar free energy of excess \(\Delta_{\text{f}} \bar{G}_{\text{T}}^{0, \text ex}\) of the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions. Substituting the molar enthalpies and entropies of formation of the GaSe and GaS compounds,\(\Delta C_{{{\text{p}},298.15}}^{0}\), and \(\Delta_{f} G_{T}^{0, \text ex}\) for the energy of mixing \(\Delta_{\text{f}} G_{\text{T}}^{{0,{\text{M}}}}\) of the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions in Eq. (2), we obtain
For thermodynamic description of the formation of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions, it is sufficient to know the composition dependence of the excess molar free energy of mixing under isothermal conditions in the 300–1200 K interval.
Using Eq. (4), we calculated the free energies of mixing for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) formation. The obtained curves are presented in Fig. 2 for the case calculated by Eq. (4) from thermodynamic data for GaSe and GaS. As shown in Fig. 2, the sensitivity of the curve calculated for GaS–GaSe alloys by Eq. (4) is determined primarily by the entropy term. The obtained curves correspond to temperatures where the alloys are in crystalline and liquid states (300–1200 K). The \(\Delta_{\text{f}} G_{\text{T}}^{{0,{\text{M}}}} \left( x \right)\) curve for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) at 800 K (curve 2) is similar to that near the melting point of the alloys, 1200 K (curve 3). Between 800 and 1200 K, there is a well-defined correlation with composition in the range 0 ≤ \(x\) ≤ 1. This suggests the formation of a thermodynamically stable \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) configuration in this composition range.
Thus, it follows from thermodynamic analysis results that \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions have deviations from ideality. This is clearly seen in the composition and temperature dependences of the Gibbs free energy of mixing for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) in differential form, which demonstrates thermodynamic stability of solid solution. According to thermodynamic theory, the formation of solid solutions is energetically favorable if \(\Delta_{\text{f}} G_{\text{T}}^{{0,{\text{M}}}} \le 0\) and ∂2G/∂x2 > 0. The above technique for evaluating the free energy of mixing of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) (Fig. 2) is consistent with this solution theory. As the temperature is varied, the free energy of mixing of alloys of the GaSe–GaS system assumes a negative value, which corresponds to solid solution formation.
The thermodynamic function of Gibbs mixing \(\Delta_{\text{f}} G_{\text{T}}^{{0,{\text{M}}}}\) allows to assess the character and direction of molecular processes accompanying formation of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solutions. The \(\Delta_{\text{f}} G_{\text{T}}^{{0,{\text{M}}}}\) value is defined as the Gibbs mixing energy of ideal solution. In other words, the excess free energy of mixing is non-ideal portion of the isobaric mixing potential. The \(\Delta_{\text{f}} G_{\text{T}}^{{0,{\text{M}}}}\) dependence of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) formation was calculated taking into account the experimental thermodynamic data for GaSe and GaS. For non-ideal \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solutions, the additivity principle of excess free energy of mixing is not observed, and in calculations we took into account the entropy factor. Data on excess free energy of mixing allow to calculate components’ activity coefficients.
\(E_{\text{g}} \left( {T,x} \right)\) dependence of the single-crystal GaS–GaSe system
Solid solutions have an important advantage over compounds, in particular because their band gap \(E_{\text{g}}\) can be varied. The \(E_{\text{g}} \left( x \right)\) data for the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions can be described by empirical equation with allowance for the deviation from additive behavior [14]. The composition dependence of the band gap, \(E_{\text{g}} \left( x \right)\), for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) was represented by the equation
where b is bowing parameter and quantifies the deviation from additivity.
The \(E_{\text{g}} \left( x \right)\) data for the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions are represented by a slightly concave curve, which means a negative deviation. Given that various \(E_{\text{g}}\) values have been reported for GaSe and GaS in the literature [1], we used average of \(b\) = 0.6 values for the deviation coefficient in Eq. (5). Figure 3 shows the 298 K \(E_{\text{g}} \left( x \right)\) curves calculated for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) using Eq. (5) and average \(E_{\text{g}}\) values for GaSe and GaS. The calculated \(E_{\text{g}} \left( x \right)\) curves are in reasonable agreement also with ours experimental data and data for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) at 300 К [20]. The calculation of band gap from spectra of edge absorption assuming indirect transitions has shown that with increasing dopant concentration from 0.1 to 3 mass% the energy gap increases linearly. However, we found that with an increase in the sulfur concentration in alloys and with the formation of solid solutions of GaSe1−xSx in gallium monoselenide, the linearity of the E(x) dependence is violated. As can be seen from the figure at 300 K, the dependence of the band gap width on the triple composition at x ≥ 0.03 is nonlinear and can be expressed by bowing parameter b = 0.6.

Concentration–temperature dependence of the band gap calculated for \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions using Eq. (5)
The \(C_{\text{p}} \left( {T,x} \right)\) dependence of the GaS–GaSe system
We built concentration dependence of heat capacity for samples of the GaS–GaSe system at constant temperature (Fig. 4). \(C_{\text{p}} \left( T \right)\) of pure GaSe and GaS was compared with literature data [1] (for GaS) and [1, 5,6,7, 19] (for GaSe). GaSe and GaS heat capacity demonstrates a reasonable agreement with the measured data available in the literature. As shown in Fig. 4, concentration dependence of heat capacity in the GaS–GaSe system at 10 and 300 K weakly deviated from Neumann–Kopp rule in the negative direction. This can be attributed to the fact that the heat capacity of \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions at low temperatures is weakly related to the parameters of the force interactions of the particles. Relatively minor deviations of the temperature–concentration dependences of the properties from the Vegard’s law indicate an insignificant difference in the short-range order in the initial GaS and GaSe compounds and in the \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions.

Concentration–temperature dependence of the heat capacity of solid solutions of the GaS–GaSe system
Comparison of the \(E_{\text{g}} \left( {T,x} \right)\) dependences of single-crystal samples and \(C_{\text{p}} \left( {T,x} \right)\) (Figs. 3, 4) shows that there is a correlation of these properties not only with each other, but also with the type of phase diagram for the GaS–GaSe system (Fig. 1). These results allow us to conclude that the nature of the interaction of particles in \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) solid solutions undergoes a slight change from the concentration of the components at low and room temperatures.
Conclusions
Much attention has been paid to the consistency between the experimental \(T - x\) phase diagram and thermodynamical calculations. Using current and literature thermodynamic data for GaS and GaSe, the calculation results can reproduce the experimental data \(T - x\) well. We clarify the melting point and melting enthalpy of the GaS and GaSe components that are needed to calculate phase diagram of the GaS–GaSe system. The following values have been determined: \(\Delta H^{\text{m}}\)(GaSe) = 30300 ± 100 J mol−1, \(T^{\text{m}}\)(GaSe) = 1211 К, \(\Delta H^{\text{m}}\) (GaS) = 34800 ± 100 J mol−1 and \(T^{\text{m}}\)(GaS) = 1288 ± 3 К. Calculation curves of liquidus and solidus of \(T - x\) phase diagram for the GaSe–GaS system calculated from the model of ideal solutions vary by 15 and 5 K, respectively, from experimental data. In this case, no fitting parameters are used in the calculations. The satisfactory agreement of the calculated curve with the experimental \(T - x\) phase diagram of GaS–GaSe confirms the validity of applying formula (1) under the conditions for which it is used. Based on the available experimental data within the model of solutions with non-molecular compounds at 300–1200 K, the temperature–concentration dependences of the thermodynamic functions of GaS–GaSe alloys are approximated. Experiments show that in the prepared samples of solid solutions \({\text{GaSe}}_{{ 1- {\text{x}}}} {\text{S}}_{\text{x}}\) the concentration dependences (\(p,T = {\text{const}}\)) of the properties (thermodynamic functions, band gap, heat capacity) are changed regularly. Minor negative deviations from the Vegard’s law on the property–composition relationships of GaS–GaSe alloys indicate the proximity of interaction potentials between particles of binary compounds and solid solutions.
References
Madelung O. Semiconductors data handbook. Berlin: Springer; 2004.
Terhell ICI. Polytypism in the III–VI layer compounds. Prog Cryst Growth Charact Mater. 1983;7:55–110.
Mustafaeva SN, Asadov MM. Currents of isothermal relaxation in GaS <Yb> single crystals. Solid State Commun. 1983;45:491–4.
Mustafaeva SN, Asadov MM. Field kinetics of photocurrent in GaSe amorphous films. Mater Chem Phys. 1986;15:185–9.
Mamedov KK, Kerimov IG, Mekhtiev MI, Masimov EA, Izv Akad Nauk SSSR. Neorg Mater. 1972; 8:2096–98; Bull Acad Sci USSR Inorg Mater (English Transl). 1972; 8:1843–45.
Mamedov KK, Kerimov IG, Kostryukov VN, Mekhtiev MI. Heat capacity of gallium and thallium selenides. Fiz Tekh Poluprovodn (S Peterburg). 1967;1:441–2.
Tyurin AV, Gavrichev KS, Khoroshilov AV, Zlomanov VP. Heat capacity and thermodynamic functions of GaSe from 300 to 700 K. Inorg Mater. 2014;50:233–6.
Rustamov PG. Khal’kogenidy galliya. Baku: Akademiya nauk AzSSR; 1967. s. 130; Rustamov PG. Gallium chalcogenides. Baku: Academy of Sciences of the AzSSR; 1967. p. 130 (in Russian).
Asadov MM, Mustafaeva SN, Mamedov AN, Aljanov MA, Kerimova EM, Nadjafzade MD. Dielectric properties and heat capacity of (TlInSe2)1–x(TlGaTe2)x solid solutions. Inorg Mater. 2015;51:772–8.
Mustafaeva SN, Asadov MM, Kerimova EM, Gasanov NZ. Dielectric and optical properties of TlGa1–xErxS2 (x = 0, 0.001, 0.005, 0.01) single crystals. Inorg Mater. 2013;49:1175–9.
Mustafaeva SN, Asadov MM, Kyazimov SB, Gasanov NZ. T–x phase diagram of the TlGaS2–TlFeS2 system and band gap of TlGa1–xFexS2 (0 = x = 0.01) single crystals. Inorg Mater. 2012;48:984–6.
Asadov MM, Ahmedly KM. Procedure for calculating phase equilibrium in simple binary systems of ideal liquid and solid solution. Inorg Mater. 1996;32:133–4.
Asadov MM, Ahmedly KM. Calculation of some thermodynamic parameters for non-ideal solutions. Solid State Phenom. 2008;138:331–8.
Asadov SM, Mamedov AN, Kulieva SA. Composition- and temperature-dependent thermodynamic properties of the Cd, Ge||Se, Te system, containing solid solutions. Inorg Mater. 2016;52:876–85.
Zheng F, Shen JY, Liu YQ, Kim WK, Chu MY, Ider M, Bao XH, Anderson TJ. Thermodynamic optimization of the Ga–Se system. CALPHAD. 2008;32:432–8.
Abbasov AS. Termodinamicheskiye svoystva nekotorykh poluprovodnikovykh veshchestv (Thermodynamic properties of some semiconductor substances). Baku. Elm; 1981. (in Russian).
Vasilev VP. Correlations between the thermodynamic properties of II–VI and III–VI phases. Inorg Mater. 2007;43:115–24.
Ider M, Pankajavalli R, Zhuang W, Shen JY, Andersone TJ. Thermochemistry of the Ga–Se system. ECS J Solid State Sci Technol. 2015;4:51–60.
Mills KC. Thermodynamic data for inorganic sulphides, selenides and tellurides. London: Butterworth; 1974.
Voevodina OV, Morozov AN, Sarkisov SY, Bereznaya SA, Korotchenko ZV, Dikov DE. Properties of gallium selenide doped with sulfur from melt and from gas phase. KORUS 2005. Sci Technol. In: Proceedings of the 9th Russian-Korean international symposium, 2005; p. 551–5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Asadov, S.M., Mustafaeva, S.N. & Mammadov, A.N. Thermodynamic assessment of phase diagram and concentration–temperature dependences of properties of solid solutions of the GaS–GaSe system. J Therm Anal Calorim 133, 1135–1141 (2018). https://doi.org/10.1007/s10973-018-6967-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-6967-7