
Abstract
Methylene blue (MB) has a long story of use and has been employed for various diseases, however, most of its current rekindled research is related to its function in the mitochondria. MB is gaining interest as a possible treatment because mitochondrial dysfunction is an apparent unifying pathogenic characteristic of a variety of neurodegenerative conditions. Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by impairment of cognitive functions. This study aims to investigate whether MB treatment improves impaired cognitive functions and reduces hippocampal amyloid-β levels and oxidative stress in a D-galactose-induced AD mouse model. Twenty-four wild-type Balb/c mice were randomly divided into four groups (control, D-galactose-induced AD, MB-treated AD, and only MB-treated mice). Mice in the corresponding groups were injected with D-galactose (50 mg/kg, s.c.) for 60 days. In the MB treatment groups, the mice were treated with MB (2 mg/kg, orally) for the last 14 days of treatments. The Morris water maze test was used to assess spatial learning and memory functions. Amyloid β-42, malondialdehyde, superoxide dismutase, and nitric oxide levels in the hippocampi of mice were measured using ELISA and spectrophotometry. MB treatment improved impaired learning and memory functions induced by D-galactose administration and decreased amyloid β-42 concentration in the hippocampi of mice. Malondialdehyde level was found to decrease in MB-treated mice compared to D-galactose-induced AD mice in the hippocampus and plasma. The hippocampus of MB-treated mice displayed increased superoxide dismutase activity while decreased nitric oxide concentration compared to the ones of D-galactose-induced AD mice. MB has been shown to improve learning and memory impairments, as well as reduce Aβ-42 concentration and oxidative stress in the hippocampus of D-galactose-induced AD mice. The findings of this study demonstrate that MB may offer potential benefits as a repurposed agent for AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by impairments of cognitive function especially learning memory and the most common cause of dementia. According to the World Alzheimer Report 2021, there are currently 55 million people suffering from dementia worldwide (International, 2021). The percentage of people aged 60 or over is projected to rise to 21.5% of the global population by 2050 (Shwe et al., 2018). Unfortunately, there is no radical cure for AD yet. For many years, donepezil, galantamine, rivastigmine, and memantine have been the main agents used to avoid worsening the symptoms of AD. Even though promising new drugs such as aducanumab and lecanemab have been approved by the FDA in recent years (Yadollahikhales and Rojas, 2023), there is still a need to develop new and more effective treatment strategies.
There are many hypotheses about the pathogenesis of AD, including the accumulation of amyloid β (Aβ) and tau proteins, cholinergic neuron damage, neuroinflammation, and oxidative stress (Du et al., 2018) that has been considered a key mediator of both neurodegenerative diseases and the normal process of aging (Rehman et al., 2017). Studies have revealed that oxidative stress has an important role in the pathogenesis of AD sustained by the decreased levels of reduced glutathione (GSH), elevated levels of oxidized glutathione (GSSG), and increased production of reactive oxygen species (ROS) detected in AD patients and animal models (Wei et al., 2013; Ali et al., 2015). The brain has been suggested more susceptible to oxidative stress-mediated damage due to the presence of high lipid content and higher oxygen consumption than other organs (Atamna and Kumar, 2010; Garg et al., 2017). Excessive ROS and oxidative stress trigger lipid, protein, or DNA damage which disturbs the normal cellular activity of neurons, ultimately leading to cell death (Ali et al., 2015). Mitochondrial dysfunction plays an initial role in the increased production of ROS. Following mitochondrial dysfunction, brain oxidative stress via a vicious cycle of ROS-induced ROS release in mitochondria eventually leads to brain aging and cognitive impairment, which is the primary symptom of several neurodegenerative diseases including AD (Shwe et al., 2018). Mitochondrial dysfunction in AD can in turn result in synaptic dysfunction and neurodegeneration of the hippocampus and cortical regions of the brain (Atamna and Kumar, 2010).
D-Galactose is a monosaccharide that has a similar configuration to glucose. D-galactose is the main component of the glycolipids in the brain and the myelin sheath of peripheral nerve cells which leads to D-galactose being called the brain sugar. Even though a normal concentration of D-galactose can be metabolized, high concentration is reported to form advanced glycation end products, which induce ROS production (Wei et al., 2013; Cardoso et al., 2015). Chronic systemic exposure of rodents to D-galactose causes the acceleration of aging, age-related cognitive impairments, and the development of neurodegenerative diseases such as AD (Wei et al., 2013; Cardoso et al., 2015; Shwe et al., 2018). Therefore, chronic D‑galactose administration is a widely used model of AD. It has been suggested that the main underlying mechanism of the D-galactose-induced AD model is oxidative stress which also causes mitochondrial dysfunction, inflammation, and apoptosis in neurons (Shwe et al., 2018). Oxidative stress induced by D‑galactose has been shown to increase malondialdehyde (MDA) levels and decrease superoxide dismutase (SOD) and GSH activities (Kumar et al., 2020).
Methylene blue (MB) as the first synthetic drug had already a 120-year history (Schirmer et al., 2011). It is a safe agent that has been used clinically to treat various diseases especially methemoglobinemia (Atamna et al., 2008; Oz et al., 2009; Atamna and Kumar, 2010). MB has been known as an antioxidant and is protective of mitochondria (Zakaria et al., 2016), being effective in counteracting some key basic mechanisms of mitochondrial dysfunction (Atamna and Kumar, 2010). It has been suggested MB may postpone the onset of AD by delaying the decline in mitochondrial enzyme complex IV (cytochrome c oxidase) and increasing heme synthesis which results in improving mitochondrial function (Atamna and Kumar, 2010). MB was also found to increase mitochondrial complex I–III activity and to enhance oxygen consumption and glucose uptake in a mouse hippocampal neuronal cell line (HT-22 cells) (Lin et al., 2012). Since mitochondria are the powerhouse of the cell, their inadequate energy supply to the neurons in AD can be responsible for cell death and synaptic loss (Zakaria et al., 2016). Furthermore, it has been shown that MB can inhibit the aggregation of tau protein (Wischik et al., 1996) and Aβ peptides (Taniguchi et al., 2005; Necula et al., 2007). With its high bioavailability, rapid distribution, and high absorption MB has well-known pharmacokinetic properties and can cross the blood-brain barrier (Peter et al., 2000; Eckert et al., 2012). In addition, MB concentration was found higher in the brain than in plasma, indicating its accumulation in the nervous system (Oz et al., 2009, 2011).
Despite many studies on AD, the inability to find a new drug for many years has brought some of the drugs used for other indications to be tried in the treatment of the pathology. Therefore, MB has been suggested as a potential drug candidate repurposed for AD indication. However, there is a lack of knowledge of the effectiveness and action mechanism of MB in AD. In the present study, we investigated whether MB treatment improves impaired learning and memory functions and ameliorates hippocampal oxidative stress and Aβ accumulation status in a D-galactose-induced AD mouse model.
MATERIALS AND METHODS
Animals
Twenty-four Balb/c mice (16–20 weeks old) were kept in an animal facility under a 12 h light/12 h dark cycle at 23 ± 1°C and provided with food and water ad libitum and acclimatized for 1 week before the experiments. The experimental procedures were approved by the Animal Experimentations Local Ethical Board of Hacettepe University (Decision no. 2021/06-13).
D-Galactose Model and MB Treatment
Mice were divided randomly into four experimental groups each receiving one of the following treatments: control (no treatment), D-galactose, D-galactose + MB, and MB.
In the D-galactose and D-galactose + MB treatment groups, mice were injected with D-galactose (50 mg/kg) subcutaneously for 60 days consecutively. In the control group, mice were injected with saline subcutaneously (same volume as the D-galactose group). In the MB treatment groups, mice were treated with MB (2 mg/kg, oral) for the last 14 days of experiments (Fig. 1). D-galactose and MB were purchased from Sigma Aldrich, Germany.

Schematic representation of experimental groups and treatment schedule (s.c.: subcutaneous; MWM: Morris water maze).
Morris Water Maze Test
The Morris water maze (MWM) test was used to assess the spatial learning and memory of mice at the end of the treatment as explained before (Kazkayasi et al., 2022). Briefly, a circular pool (120 cm diameter, 90 cm high) was filled with water (22 ± 1°C) in which a circular platform (14 cm diameter) was submerged 1 cm below the water’s surface. The test was run in a dim room with different visual clues on the walls. Mice were released from different quadrants of the pool for each trial. Escape latency was measured for each training session by using a video tracking system (Videomot2, TSE systems, Germany). For each session, the mouse was allowed to search the platform for 90 s. They were trained four times a day for 4 consecutive days. On the fifth day of the test, mice were subjected to a probe trial where the platform was removed from the pool. For the probe trial, the percentage time spent in the target quadrant was calculated.
Plasma and Tissue Collection
Blood samples were collected in heparinized tubes and centrifuged at 2500 g for 15 min at 4°C. After centrifugations, plasmas were stored at –80°C until analysis. After blood sampling and removal of the brain, hippocampi were isolated, frozen, and stored at ‒80°C. Our study focused on the hippocampus, as it is the region where the AD pathology typically first develops in patients and D-galactose-induced AD mice (Chadwick et al., 2023).
Aβ-42 Determination Assay
Aβ-42 was detected and quantified using The Invitrogen Mouse β42 ELISA kit according to the manufacturer’s instructions. Briefly, hippocampi were homogenized in 5 M guanidine HCl/50 mM Tris (pH 8.0) buffer containing protease inhibitors. The supernatants were measured for Aβ-42 using a chromogen at 450 nm. The amount of Aβ-42 was determined using a standard curve.
Measurement of Malondialdehyde Levels
The levels of MDA were measured in the plasma and hippocampus. Hippocampi were weighed and then homogenized in a ratio of 1 : 10 (g/mL) potassium chloride (1.5%) using an ultrasonic homogenizer (Bandelin Sonopuls, Germany) for 15 s. Homogenates were centrifuged at 1500 g for 10 min and supernatants were collected. MDA was assessed by a commercial thiobarbituric acid reactive substance assay kit (Cayman, USA). The color intensity of the MDA-TBA complex was measured at 532 nm spectrophotometrically. The amount of MDA was calculated by using tetramethoxypropane as the MDA standard.
Measurement of Superoxide Dismutase Activity
The activity of SOD was measured in the plasma and hippocampi. Hippocampi were homogenized in a ratio of 1 : 10 (g/mL) phosphate-buffered saline (PBS) using an ultrasonic homogenizer (Bandelin Sonopuls, Germany) for 15 s. Homogenates were centrifuged at 1500 g for 10 min and supernatants were collected. SOD was assessed using the Invitrogen EIASODC kit according to the manufacturer’s instructions. The color intensity was measured at 450 nm spectrophotometrically. The results were expressed as units/mL.
Measurement of Nitric Oxide Levels
Hippocampi were homogenized in PBS and centrifuged at 10 000 g for 10 min, then the supernatants were collected. NO, was assessed by a commercial NO colorimetric assay kit (Elabscience, E-BC-K035-M, USA) at 550 nm.
Statistical Analysis
Results are expressed as mean ± SEM. Statistical analyses were performed using Graph Pad Prism software version 6 (Graph Pad Software, San Diego, CA, USA). The difference among trials and groups was assessed by one-way or two-way ANOVA (for swim speed test) tests followed by the Newman-Keuls post hoc test which has more power. p < 0.05 was considered statistically significant.
RESULTS
MB Treatment Improved D-Galactose-Induced Impairment of Learning and Memory in Mice
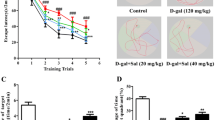
The results of MWM demonstrated that D-galactose administration impaired the learning skills of mice (Fig. 2a). Mice in control and MB-per se treated groups showed improved escape latency time from the first day to the last day of the training sessions where the escape latency time in sessions 3 and 4 were significantly lower than that in session 1 (F (3, 20) = 6.126, p < 0.01 and F (3, 20) = 8.121, p < 0.001 respectively). In the D-galactose-induced AD group, there was no significant change in escape latency times in sessions 2, 3, or 4 compared to session 1 (F (3, 20) = 2.661, p > 0.05), indicating impaired learning (Fig. 2a). MB treatment significantly decreased the escape latency time in all sessions (F (3, 20) = 20.82, p < 0.001) which indicates the beneficial effects of MB on the impaired learning function induced by D‑galactose injection (Fig. 2a). On the fifth day of training sessions, the platform was removed and a probe trial was carried out to evaluate the memory skills of D‑galactose-induced AD and MB-treated mice (Fig. 2b). D-galactose-induced AD mice spent significantly less time in the target quadrant in comparison to control mice (29.8 ± 2.5 s % vs. 43.6 ± 2 s %, p < 0.05). MB treatment increased the time spent in the target quadrant compared to the D-galactose-induced AD group (42.6 ± 4.4 s % vs. 29.8 ± 2.5 s %, p < 0.05, Fig. 2b). MB treatment in control mice did not change the time spent in the target quadrant compared to the control group. The lack of difference in swimming speed between groups showed that there were no motor deficits after D-galactose or MB administrations (Fig. 2c). Representative swimming tracks of mice in all experimental groups during the probe trial are given in Fig. 2d.

The effect of MB treatment on learning and memory functions in D-galactose (Gal) induced AD mice assessed by Morris water maze test. The values are expressed as mean ± SEM (n = 6 in each group). The difference among trials and groups was assessed by one-way or two-way ANOVA (for swim speed test) tests followed by the Newman-Keuls post hoc test. p < 0.05 was considered statistically significant. * p < 0.05, ** p < 0.01, and *** p < 0.001 are statistically significant compared to the related group indicated in the graphs.
MB Treatment Decreased Aβ-42 Concentration Which Increased by D-Galactose in Hippocampi of Mice
D-galactose administration significantly increased Aβ-42 concentration in hippocampi of D-galactose-induced AD mice compared to the control group (45.8 ± 2.5 vs. 28.5 ± 1.1 pg/mL, p < 0.01, Fig. 3a). MB treatment significantly decreased Aβ-42 concentration compared to the D-galactose group (33.02 ± 3.5 vs. 45.8 ± 2.5 pg/mL, p < 0.01, Fig. 3a). MB per se treatment did not change Aβ-42 concentrations compared to the control group (Fig. 3a). The body weight of mice was measured at the beginning and the end of the experiments. Neither D-galactose injection nor MB treatment changed the body weight of mice significantly (Fig. 3b).

The effects of D-galactose injection and MB treatment on Aβ-42 concentrations in hippocampi (a) and body weight (b) of mice. The values are expressed as mean ± SEM. For (a) n = 5 mice per group. One-way ANOVA and post hoc Newman-Keuls test was performed as statistical analysis. p < 0.05 was considered statistically significant. ** p < 0.01 is statistically significant compared to the related group indicated in the graphs.
MB Treatment Decreased MDA Concentration Which Increased by D-Galactose in Hippocampi and Plasma of Mice
MDA concentration in hippocampi and plasma of D-galactose-induced AD mice was significantly increased in comparison with the control group (174.3 ± 33.2 vs. 100 ± 10.8, p < 0.05 and 79.2 ± 11.5 μM vs. 12.2 ± 2.8 μM, p < 0.001, respectively, Figs. 4a and 4b). MB treatment decreased MDA concentration compared to the D-galactose group both in hippocampi and plasma of mice (78.8 ± 17.6 vs. 174.3 ± 33.2, p < 0.05 and 35.8 ± 8.3 μM vs. 79.2 ± 11.5 μM, p < 0.001, respectively, Figs. 4a and 4b). MB per se treatment did not significantly affect MDA concentrations in the hippocampi or plasma of mice compared to the control group.

The effects of D-galactose injection and MB treatment on MDA concentrations in hippocampi (a) and plasma (b) of mice. The values are expressed as mean ± SEM. N = 6 mice per group. One-way ANOVA and post hoc Newman-Keuls test was performed as statistical analysis. p < 0.05 was considered statistically significant. * p < 0.05, *** p < 0.01 is statistically significant compared to the related group indicated in the graphs.
MB Treatment Increased SOD Activity Which Decreased by D-Galactose in the Hippocampi of Mice
SOD activity in hippocampi of D-galactose-induced AD mice was significantly decreased in comparison with the control group (0.73 ± 0.18 vs. 1.25 ± 0.10 U/mg tissue, p < 0.05, Fig. 5a). MB treatment significantly increased SOD activity compared to the D-galactose-induced AD group (1.14 ± 0.18 vs. 0.73 ± 0.18 U/mg tissue, p < 0.05, Fig. 5a). SOD activity in the plasma of D-galactose-induced AD mice was decreased in comparison with the control group (5.79 ± 1.02 vs. 7.32 ± 1.55 U/mL, Fig. 5b). MB treatment increased SOD activity in plasma compared to the D-galactose-induced AD group (8.627 ± 1.95 vs. 5.79 ± 1.02, Fig. 5b). MB per se treatment did not significantly affect SOD activity in the hippocampi or plasma of mice compared to the control group (Figs. 5a and 5b).

The effects of D-galactose (Gal) and MB treatment on SOD activity in (a) hippocampi, (b) in plasma, and (c) on NO concentration in hippocampi. The values are expressed as mean ± SEM. N = 6 mice per group. One-way ANOVA and post hoc Newman-Keuls test was performed as statistical analysis. p < 0.05 was considered statistically significant. * p < 0.05 is statistically significant compared to the related group indicated in the graphs.
MB Treatment Decreased NO Concentration Which Increased by D-Galactose in the Hippocampi of Mice
NO concentration in hippocampi of D-galactose-induced AD mice was significantly increased in comparison with the control group (1176 ± 78.42 vs. 851.9 ± 106.2 μM/mg tissue, p < 0.05, Fig. 5c). MB treatment decreased NO concentration in hippocampi compared to the D-galactose-induced AD group (896.6 ± 98.01 vs. 1176 ± 78.42 μM/mg tissue, p < 0.05, Fig. 5c). MB per se treatment did not significantly affect NO concentrations compared to the control group (Fig. 5c).
DISCUSSION
AD is a progressive neurodegenerative disease characterized by impairments in learning and memory. Severe neurodegenerative alterations occur in the brain including atrophy, loss of neurons and synapses, as well as the selective depletion of certain neurotransmitter systems (Eckert et al., 2012) besides the known classical pathological hallmarks of the disease, i.e. the accumulation of Aβ and hyperphosphorylated tau tangles. Energy hypometabolism, mitochondrial dysfunction, oxidative stress, decline in cytochrome c oxidase, and abnormal iron homeostasis are the other key cytopathologies of AD (Oz et al., 2009; Atamna and Kumar, 2010). The progressive deterioration in cognitive functions has been previously attributed to energy hypometabolism and oxidative damage leading to neurodegeneration based on their earlier onset than Aβ and phosphorylated tau accumulations (Atamna and Kumar, 2010). The brain is particularly more susceptible to impaired energy metabolism and oxidative stress-mediated damage due to the presence of high lipid content and higher oxygen consumption (Atamna and Kumar, 2010). The D-galactose-induced aging model in rodents is a well-known model that is associated with cognitive impairments e.g. hippocampal-dependent spatial memory deficits (Parameshwaran et al., 2010; Wei et al., 2013; Cardoso et al., 2015; Shwe et al., 2018). The underlying mechanism of the model is inflammation, apoptosis in neurons, and most importantly oxidative stress that is caused by mitochondrial dysfunction (Parameshwaran et al., 2010; Shwe et al., 2018). Furthermore, it serves as an established model for AD due to its ability to induce the accumulation of Aβ and phosphorylated tau (Chadwick et al., 2023; Wang et al., 2023). This model was selected based on MB’s promising antioxidant properties and the intention to assess its efficacy in an oxidative stress-inducing AD model.
MB is one of the mitochondrial function enhancer agents that has recently gained some attention as a putative anti-AD drug. In the present study, MB was given at 2 mg/kg as it is the most used dose in clinical applications according to previous reports and is the highest dose without any side effects (Atamna et al., 2008; Oz et al., 2009; Oz et al., 2011; Paban et al., 2014). Even though MB is known as a quite nontoxic medicine, locomotor adverse effects have been reported at higher doses of over 10 mg/kg (Schirmer et al., 2011; Paban et al., 2014). In addition, MB has a hormetic dose response in terms of various behavioral and biochemical responses which high doses may cause ineffectiveness (Bruchey and Gonzalez-Lima, 2008). We found 2 mg/kg MB improved cognitive functions in accordance with previous studies using the same doses (Deiana et al., 2009; Medina et al., 2011; Paban et al., 2014).
Here, D-galactose-induced AD mice showed a prolonged escape latency in the MWM test that was indicative of impaired spatial learning, while the control and MB per se treated mice showed significantly decreased escape latency in consecutive sessions. MB treatment significantly decreased the escape latency time of D-galactose-induced mice in consecutive sessions indicating that MB ameliorated the impaired learning. On the probe trial day of the MWM test, the memory deficit induced by D-galactose seen as less time in the target quadrant was restored by MB treatment. Hereby, MB treatment improved the learning and memory functions that were impaired in D-galactose-induced AD mice, supporting the previous studies showing that MB attenuates cognitive functions in different types of AD mice models (Deiana et al., 2009; Medina et al., 2011; Paban et al., 2014).
MB was shown to be effective against Aβ aggregation and oligomerization (Medina et al., 2011; Zakaria et al., 2016). In vitro studies showed that MB reduces Aβ aggregation and oligomerization (Taniguchi et al., 2005; Necula et al., 2007). In vivo, MB treatment was shown to reduce Aβ levels by increasing chymotrypsin- and trypsin-like activities of the proteasome (Medina et al., 2011) in a study using 3XTg AD mice. In other studies with transgenic AD models such as APP/PS1 or aged PSAPP mice, long-term MB treatment decreased cerebral Aβ deposits, modulated β‑secretase, and improved behavioral impairments (Mori et al., 2014; Paban et al., 2014). A study in a lipopolysaccharide-induced AD mouse model examined MB treatment and showed a significant decrease in Aβ1-42 oligomers (Zakaria et al., 2016). MB was found to interfere with the toxic effects of Aβ and tau by restoring the function of the plasma membrane Ca2+-ATPase pump (Berrocal et al., 2018, 2019). In our study, we investigated the effects of MB on Aβ-42 concentrations in the hippocampi of mice. D-galactose administration increased Aβ-42 concentration compared to the control group. Whereas, MB treatment decreased Aβ-42 concentration which is increased by D-galactose administration. Our results are consistent with the aforementioned studies in which MB treatment was shown to reduce Aβ levels which may be suggested as the reason for the improvement of cognitive functions.
In AD cell lines and transgenic animals, accumulation of APP and Aβ in mitochondria was found to correlate with mitochondrial dysfunction (Eckert et al., 2012). Any disturbances in the electron transport chain lead to elevated ROS levels, ROS, in turn, increases Aβ generation which eventually impairs mitochondrial function. Remarkably, Aβ accumulation and mitochondrial dysfunction create a vicious cycle (Eckert et al., 2012). Even independent of Aβ accumulation, AD is known as a disease in which oxidative stress and mitochondrial dysfunctions are prominent as seen in altered expression of various subunits of the respiration chain complex, declined in complex enzyme activities, reduced ATP levels and superoxide dismutase deficiencies (Eckert et al., 2012). MB has been proposed as a new class of antioxidants that competitively inhibits the reduction of molecular oxygen to superoxide by acting as an alternative electron acceptor in the treatment of neurodegeneration (Oz et al., 2009; Atamna and Kumar, 2010). There is evidence that MB enhances brain metabolism and hemodynamics by enhancing global glucose uptake, cerebral blood flow, and cerebral metabolic rate of oxygen (Lin et al., 2012). MB is also known to avoid the overproduction of free radicals (Atamna and Kumar, 2010; Zakaria et al., 2016), increase complex I–III (Lin et al., 2012) and complex IV activities (Callaway et al., 2004), and enhance oxygen consumption (Atamna et al., 2008) and cell viability (Zakaria et al., 2016). We investigated the effects of MB on different oxidative stress markers in D-galactose-induced mice. MDA concentrations were found significantly increased in the hippocampi and plasma of D-galactose-induced AD mice. This observation supports previous studies claiming that D-galactose increases ROS and thus enhances oxidative stress (Wei et al., 2013; Cardoso et al., 2015). On the other hand, MB treatment decreased MDA concentrations which supports previous studies claiming that the drug reduces ROS production. The fact that aging markers have been associated with cognitive functions (Shwe et al., 2018), the decrease in MDA levels observed as a result of MB treatment provides a plausible explanation for the positive impact on learning and memory in the MWM test. Another important marker in oxidative stress and antioxidant balance is SOD which is the first line of defense against ROS damage. In our study, SOD activity was found significantly decreased in hippocampi of D-galactose-induced AD mice which indicates that the balance has shifted in favor of oxidative stress. A similar pattern of SOD activity in the plasma of D-galactose-induced AD was observed without reaching statistical significance. The reason for the insignificance of SOD activity difference in plasma may be attributed to the weak correlation between the activity of SOD in serum and brain tissues (P. Boriskin, 2019). MB treatment attenuated the reduction in SOD activity provoked by D-galactose treatment in hippocampi. The attenuation of SOD activity may have contributed to the improvement of cognitive functions by restoring the antioxidant capacity of the hippocampus. Nitric oxide (NO) is an endogenously synthesized free radical. NO, and its reactive secondary metabolites oxidize/nitrosate various molecular targets such as proteins, lipids, and nucleic acids during oxidative/nitrosative stress which damages cells. In our study, NO concentration in hippocampi of D-galactose-induced AD mice was found increased which indicates elevated oxidative stress. MB treatment of D-galactose-induced mice decreased NO concentration in the hippocampus which is consistent with a previous study showing that MB decreases NO levels (Wen et al., 2011).
On the other hand, MB was also suggested as a potential drug for AD because of its in vitro tau protein aggregation and tau filament formation-preventing properties (Wischik et al., 1996; Taniguchi et al., 2005). There are some studies using the tau transgenic mouse models showed that MB could reduce soluble tau levels and improve cognition (Wischik et al., 1996; Taniguchi et al., 2005; O’Leary et al., 2010; Spires-Jones et al., 2014; Chalmers and Love, 2007; O’Leary et al., 2010; Congdon et al., 2012; Melis et al., 2015). Furthermore, MB treatment has been tested on tau deposition in phase II and III clinical trials in mild to moderate AD patients which resulted in conflicting outcomes with different doses (Wischik et al., 2015; Gauthier et al., 2016; Wilcock et al., 2018; Schelter et al., 2019; Soeda et al., 2019). Due to the importance of tau accumulation and hyperphosphorylation as a pathological component in AD, these studies support our view that methylene blue may be beneficial in the treatment of AD.
CONCLUSIONS
In conclusion, MB as a safe and almost 100 years-used drug holds promise as a treatment for AD. Many studies have been done to support this issue and some are still ongoing. This is the first study showing that MB improves learning-memory impairments and reduces Aβ-42 concentration and oxidative stress in the hippocampus in such an AD model. Even though our study was focused on the mechanism of action through Aβ accumulation and oxidative stress; anti-inflammatory, anti-apoptotic, and tau aggregation-preventing properties of MB have been reported previously. Since AD is a complex disease in which many genetic and environmental factors have a role, multi-targeted drugs such as MB may be more effective than drugs that act through a single mechanism of action. Considering the positive results obtained from preclinical and clinical studies and our findings, MB deserves to be included in further studies for its use in AD.
REFERENCES
Ali, T., Badshah, H., Kim, T.H., and Kim, M.O., Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model, J. Pineal Res., 2015, vol. 58, pp. 71–85. https://doi.org/10.1111/jpi.12194
Atamna, H. and Kumar, R., Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase, J. Alzheimers Dis., 2010, vol. 20, suppl. 2, pp. S439–S452. https://doi.org/10.3233/jad-2010-100414
Atamna, H., Nguyen, A., Schultz, C., Boyle, K., Newberry, J., Kato, H., and Ames, B.N., Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways, FASEB J., 2008, vol. 22, pp. 703–712. https://doi.org/10.1096/fj.07-9610com
Berrocal, M., Corbacho, I., Gutierrez-Merino, C., and Mata, A.M., Methylene blue activates the PMCA activity and cross-interacts with amyloid beta-peptide, blocking Abeta-mediated PMCA inhibition, Neuropharmacology, 2018, vol. 139, pp. 163–172. https://doi.org/10.1016/j.neuropharm.2018.07.012
Berrocal, M., Caballero-Bermejo, M., Gutierrez-Merino, C., and Mata, A.M., Methylene blue blocks and reverses the inhibitory effect of Tau on PMCA function, Int. J. Mol. Sci., 2019, vol. 20. https://doi.org/10.3390/ijms20143521
Bruchey, A.K. and Gonzalez-Lima, F., Behavioral, physiological and biochemical hormetic responses to the autoxidizable dye methylene blue, Am. J. Pharmacol. Toxicol., 2008, vol. 3, pp. 72–79. https://doi.org/10.3844/ajptsp.2008.72.79
Boriskin, P., Gulenko, O.V., Deviatkin, A., Pavlova, O., and Toropovskiy, A., Correlation of superoxide dismutase activity distribution in serum and tissues of small experimental animals, IOP Conf. Ser.: Earth Environ. Sci., 2019, vol. 403. https://doi.org/10.1088/1755-1315/403/1/012112
Callaway, N.L., Riha, P.D., Bruchey, A.K., Munshi, Z., and Gonzalez-Lima, F., Methylene blue improves brain oxidative metabolism and memory retention in rats, Pharmacol. Biochem. Behav., 2004, vol. 77, pp. 175–181. https://doi.org/10.1016/j.pbb.2003.10.007
Cardoso, A., Magano, S., Marrana, F., and Andrade, J.P., D-galactose high-dose administration failed to induce accelerated aging changes in neurogenesis, anxiety, and spatial memory on young male Wistar rats, Rejuvenation Res., 2015, vol. 18, pp. 497–507. https://doi.org/10.1089/rej.2015.1684
Chadwick, W., Maudsley, S., Hull, W., Havolli, E., Boshoff, E., Hill, M.D.W., Goetghebeur, P.J.D., Harrison, D.C., Nizami, S., Bedford, D.C., Coope, G., Real, K., Thiemermann, C., Maycox, P., Carlton, M., and Cole, S.L., The oDGal mouse: a novel, physiologically relevant rodent model of sporadic Alzheimer’s disease, Int. J. Mol. Sci., 2023, vol. 24. https://doi.org/10.3390/ijms24086953
Chalmers, K.A. and Love, S., Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons, J. Neuropathol. Exp. Neurol., 2007, vol. 66, pp. 158–167. https://doi.org/10.1097/nen.0b013e3180303b93
Congdon, E.E., Wu, J.W., Myeku, N., Figueroa, Y.H., Herman, M., Marinec, P.S., Gestwicki, J.E., Dickey, C.A., Yu, W.H., and Duff, K.E., Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo, Autophagy, 2012, vol. 8, pp. 609–622. https://doi.org/10.4161/auto.19048
Deiana, S., Harrington, C.R., Wischik, C.M., and Riedel, G., Methylthioninium chloride reverses cognitive deficits induced by scopolamine: comparison with rivastigmine, Psychopharmacology (Berl.), 2009, vol. 202, pp. 53–65. https://doi.org/10.1007/s00213-008-1394-2
Du, X., Wang, X., and Geng, M., Alzheimer’s disease hypothesis and related therapies, Transl. Neurodegener., 2018, vol. 7, p. 2. https://doi.org/10.1186/s40035-018-0107-y
Eckert, G.P., Renner, K., Eckert, S.H., Eckmann, J., Hagl, S., Abdel-Kader, R.M., Kurz, C., Leuner, K., and Muller, W.E., Mitochondrial dysfunction—a pharmacological target in Alzheimer’s disease, Mol. Neurobiol., 2012, vol. 46, pp. 136–150. https://doi.org/10.1007/s12035-012-8271-z
Garg, G., Singh, S., Singh, A.K., and Rizvi, S.I., Antiaging effect of metformin on brain in naturally aged and accelerated senescence model of rat, Rejuvenation Res., 2017, vol. 20, pp. 173–182. https://doi.org/10.1089/rej.2016.1883
Gauthier, S., Feldman, H.H., Schneider, L.S., Wilcock, G.K., Frisoni, G.B., Hardlund, J.H., Moebius, H.J., Bentham, P., Kook, K.A., Wischik, D.J., Schelter, B.O., Davis, C.S., Staff, R.T., Bracoud, L., Shamsi, K., Storey, J.M., Harrington, C.R., and Wischik, C.M., Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial, Lancet, 2016, vol. 388, pp. 2873–2884. https://doi.org/10.1016/S0140-6736(16)31275-2
International A.S.D., World Alzheimer Report 2021, 2021.
Kazkayasi, I., Telli, G., Nemutlu, E., and Uma, S., Intranasal metformin treatment ameliorates cognitive functions via insulin signaling pathway in ICV-STZ-induced mice model of Alzheimer’s disease, Life Sci., 2022, vol. 299, p. 120538. https://doi.org/10.1016/j.lfs.2022.120538
Kumar, R., Saraswat, K., and Rizvi, S.I., 2-Deoxy-D-glucose at chronic low dose acts as a caloric restriction mimetic through a mitohormetic induction of ROS in the brain of accelerated senescence model of rat, Arch. Gerontol. Geriatr., 2020, vol. 90, p. 104133. https://doi.org/10.1016/j.archger.2020.104133
Lin, A.L., Poteet, E., Du, F., Gourav, R.C., Liu, R., Wen, Y., Bresnen, A., Huang, S., Fox, P.T., Yang, S.H., and Duong, T.Q., Methylene blue as a cerebral metabolic and hemodynamic enhancer, PLoS One, 2012, vol. 7, p. e46585. https://doi.org/10.1371/journal.pone.0046585
Medina, D.X., Caccamo, A., and Oddo, S., Methylene blue reduces aβ levels and rescues early cognitive deficit by increasing proteasome activity, Brain Pathol., 2011, vol. 21, pp. 140–149. https://doi.org/10.1111/j.1750-3639.2010.00430.x
Melis, V., Magbagbeolu, M., Rickard, J.E., Horsley, D., Davidson, K., Harrington, K.A., Goatman, K., Goatman, E.A., Deiana, S., Close, S.P., Zabke, C., Stamer, K., Dietze, S., Schwab, K., Storey, J.M., Harrington, C.R., Wischik, C.M., Theuring, F., and Riedel, G., Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models, Behav. Pharmacol., 2015, vol. 26, pp. 353–368. https://doi.org/10.1097/fbp.0000000000000133
Mori, T., Koyama, N., Segawa, T., Maeda, M., Maruyama, N., Kinoshita, N., Hou, H., Tan, J., and Town, T., Methylene blue modulates β-secretase, reverses cerebral amyloidosis, and improves cognition in transgenic mice, J. Biol. Chem., 2014, vol. 289, pp. 30303–30317. https://doi.org/10.1074/jbc.M114.568212
Necula, M., Breydo, L., Milton, S., Kayed, R., van der Veer, W.E., Tone, P., and Glabe, C.G., Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization, Biochemistry, 2007, vol. 46, pp. 8850–8860. https://doi.org/10.1021/bi700411k
O’Leary, J.C., 3rd, Li, Q., Marinec, P., Blair, L.J., Congdon, E.E., Johnson, A.G., Jinwal, U.K., Koren, J., 3rd, Jones, J.R., Kraft, C., Peters, M., Abisambra, J.F., Duff, K.E., Weeber, E.J., Gestwicki, J.E., and Dickey, C.A., Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden, Mol. Neurodegener., 2010, vol. 5, p. 45. https://doi.org/10.1186/1750-1326-5-45
Oz, M., Lorke, D.E., Hasan, M., and Petroianu, G.A., Cellular and molecular actions of Methylene Blue in the nervous system, Med. Res. Rev., 2011, vol. 31, pp. 93–117. https://doi.org/10.1002/med.20177
Oz, M., Lorke, D.E., and Petroianu, G.A., Methylene blue and Alzheimer’s disease, Biochem. Pharmacol., 2009, vol. 78, pp. 927–932. https://doi.org/10.1016/j.bcp.2009.04.034
Paban, V., Manrique, C., Filali, M., Maunoir-Regimbal, S., Fauvelle, F., and Alescio-Lautier, B., Therapeutic and preventive effects of methylene blue on Alzheimer’s disease pathology in a transgenic mouse model, Neuropharmacology, Part A, 2014, vol. 76, pp. 68–79. https://doi.org/10.1016/j.neuropharm.2013.06.033
Parameshwaran, K., Irwin, M.H., Steliou, K., and Pinkert, C.A., D-galactose effectiveness in modeling aging and therapeutic antioxidant treatment in mice, Rejuvenation Res., 2010, vol. 13, pp. 729–735. https://doi.org/10.1089/rej.2010.1020
Peter, C., Hongwan, D., Kupfer, A., and Lauterburg, B.H., Pharmacokinetics and organ distribution of intravenous and oral methylene blue, Eur. J. Clin. Pharmacol., 2000, vol. 56, pp. 247–250. https://doi.org/10.1007/s002280000124
Rehman, S.U., Shah, S.A., Ali, T., Chung, J.I., and Kim, M.O., Anthocyanins reversed D-galactose-induced oxidative stress and neuroinflammation mediated cognitive impairment in adult rats, Mol. Neurobiol., 2017, vol. 54, pp. 255–271. https://doi.org/10.1007/s12035-015-9604-5
Schelter, B.O., Shiells, H., Baddeley, T.C., Rubino, C.M., Ganesan, H., Hammel, J., Vuksanovic, V., Staff, R.T., Murray, A.D., Bracoud, L., Riedel, G., Gauthier, S., Jia, J., Bentham, P., Kook, K., Storey, J.M.D., Harrington, C.R., and Wischik, C.M., Concentration-dependent activity of hydromethylthionine on cognitive decline and brain atrophy in mild to moderate alzheimer’s disease, J. Alzheimers Dis., 2019, vol. 72, pp. 931–946. https://doi.org/10.3233/JAD-190772
Schirmer, R.H., Adler, H., Pickhardt, M., and Mandelkow, E., “Lest we forget you—methylene blue...,” Neurobiol. Aging, 2011, vol. 32, pp. 2325.e2327–2316. https://doi.org/10.1016/j.neurobiolaging.2010.12.012
Shwe, T., Pratchayasakul, W., Chattipakorn, N., and Chattipakorn, S.C., Role of D-galactose-induced brain aging and its potential used for therapeutic interventions, Exp. Gerontol., 2018, vol. 101, pp. 13–36. https://doi.org/10.1016/j.exger.2017.10.029
Soeda, Y., Saito, M., Maeda, S., Ishida, K., Nakamura, A., Kojima, S., and Takashima, A., Methylene blue inhibits formation of tau fibrils but not of granular tau oligomers: a plausible key to understanding failure of a clinical trial for Alzheimer’s disease, J. Alzheimers Dis., 2019, vol. 68, pp. 1677–1686. https://doi.org/10.3233/jad-181001
Spires-Jones, T.L., Friedman, T., Pitstick, R., Polydoro, M., Roe, A., Carlson, G.A., and Hyman, B.T., Methylene blue does not reverse existing neurofibrillary tangle pathology in the rTg4510 mouse model of tauopathy, Neurosci. Lett., 2014, vol. 562, pp. 63–68. https://doi.org/10.1016/j.neulet.2014.01.013
Taniguchi, S., Suzuki, N., Masuda, M., Hisanaga, S., Iwatsubo, T., Goedert, M., and Hasegawa, M., Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins, J. Biol. Chem., 2005, vol. 280, pp. 7614–7623. https://doi.org/10.1074/jbc.M408714200
Wang, J., Fasina, O.B., Manzoor, M., Wang, Y., Liu, Q., Mo, J., Ohno, H., Osada, H., Xiang, L., and Qi, J., A new gentiopicroside derivative improves cognitive deficits of AD mice via activation of Wnt signaling pathway and regulation of gut microbiota homeostasis, Phytomedicine, 2023, vol. 113, p. 154730. https://doi.org/10.1016/j.phymed.2023.154730
Wei, H., Wu, G., Chen, J., Zhang, X., Xiong, C., Lei, Y., Chen, W., and Ruan, J., (2S)-5,2',5'-trihydroxy-7-methoxyflavanone, a natural product from Abacopteris penangiana, presents neuroprotective effects in vitro and in vivo, Neurochem. Res., 2013, vol. 38, pp. 1686–1694. https://doi.org/10.1007/s11064-013-1070-8
Wen, Y., Li, W., Poteet, E.C., Xie, L., Tan, C., Yan, L.J., Ju, X., Liu, R., Qian, H., Marvin, M.A., Goldberg, M.S., She, H., Mao, Z., Simpkins, J.W., and Yang, S.H., Alternative mitochondrial electron transfer as a novel strategy for neuroprotection, J. Biol. Chem., 2011, vol. 286, pp. 16504–16515. https://doi.org/10.1074/jbc.M110.208447
Wilcock, G.K., Gauthier, S., Frisoni, G.B., Jia, J., Hardlund, J.H., Moebius, H.J., Bentham, P., Kook, K.A., Schelter, B.O., Wischik, D.J., Davis, C.S., Staff, R.T., Vuksanovic, V., Ahearn, T., Bracoud, L., Shamsi, K., Marek, K., Seibyl, J., Riedel, G., Storey, J.M.D., Harrington, C.R., and Wischik, C.M., Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: cohort analysis as modified primary outcome in a phase III clinical trial, J. Alzheimers Dis., 2018, vol. 61, pp. 435–457. https://doi.org/10.3233/jad-170560
Wischik, C.M., Edwards, P.C., Lai, R.Y., Roth, M., and Harrington, C.R., Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines, Proc. Natl. Acad. Sci. U. S. A., 1996, vol. 93, pp. 11213–11218. https://doi.org/10.1073/pnas.93.20.11213
Wischik, C.M., Staff, R.T., Wischik, D.J., Bentham, P., Murray, A.D., Storey, J.M., Kook, K.A., and Harrington, C.R., Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease, J. Alzheimers Dis., 2015, vol. 44, pp. 705–720. https://doi.org/10.3233/jad-142874
Yadollahikhales, G. and Rojas, J.C., Anti-Amyloid Immunotherapies for Alzheimer’s disease: a 2023 clinical update, Neurotherapeutics, 2023. https://doi.org/10.1007/s13311-023-01405-0
Zakaria, A., Hamdi, N., and Abdel-Kader, R.M., Methylene blue improves brain mitochondrial ABAD functions and decreases Aβ in a neuroinflammatory Alzheimer’s disease mouse model, Mol. Neurobiol., 2016, vol. 53, pp. 1220–1228. https://doi.org/10.1007/s12035-014-9088-8
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The experimental procedures were approved by the Animal Experimentations Local Ethical Board of Hacettepe University (Decision no. 2021/06-13, date of ethical decision is August 24, 2021).
Additional information
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kazkayasi, I., Telli, G. Methylene Blue Attenuates Impaired Cognitive Functions and Reduces Hippocampal Aβ Levels and Oxidative Stress in D-Galactose-Induced Alzheimer’s Disease Mouse Model. Biol Bull Russ Acad Sci 51, 700–710 (2024). https://doi.org/10.1134/S106235902360455X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S106235902360455X