
Abstract
Purpose
Although recessive mutations in GJB2 are the common genetic etiology of sensorineural hearing impairment (SNHI), variants in LRTOMT gene were also identified, mostly in Middle East and North African populations.
Methods
Using Sanger sequencing we screened the exon 7 of LRTOMT in a cohort of 128 unrelated Mauritanian children with congenital deafness.
Results
Only one biallelic missense mutation, predicted as pathogenic (c.179 T > C;p.Leu60Pro) was found at homozygous state in four families. This variant, not reported before, showed a deleterious effect by SIFT (score: 0.01) and a disease-causing effect by Mutation Taster (prob: 1). Exploration of the encoded protein 3D structure revealed a disruption from an organized α helix (in the normal protein structure) into a random conformation. Early fitting of a cochlear implant seemed to improve the audition ability of the mutation carrier.
Conclusion
Further screening using a panel of deafness genes may expose other variants underlying hearing impairment in our population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Deafness is the most common sensorineural disability in developed countries as it affects approximately 5% of the population [1, 2]. In children, 1 to 6 per 1000 newborns are suffering from a form of hearing impairment [3]. Despite the lack of reliable epidemiological data on the affected populations in developing countries, the proportion of hearing loss was estimated to be greater than in states with higher income [4, 5]. In a random WHO survey, the prevalence of deafness was indeed found to be closely affected by widespread issues such as deprived life environment, lack of proper care services but also genetic background including the effect of consanguinity [6, 7]. SNHI was related to about 124 identified causal genes with most cases passed down through variations in GJB2 gene with autosomal recessive inheritance (Hereditary Hearing loss Home page: Last update August 2021). Several variations of LRTOMT gene, mostly in exon 7, have also been associated with deafness in different populations including cohorts from North African populations making this gene the second most frequent deafness causal gene in congenital severe to profound hearing impairment after GJB2 [8,9,10,11,12,13]. In a previous work using Sanger sequencing of GJB2 gene, we detected two pathogenic variants c.35delG (p.Gly12Valfs*2) and c.94C > T (p.Arg32Cys) in only 5 out of the 53 families investigated with sporadic or hereditary deafness [8]. This finding left thus about 95% of the families with other putative genetic cause to be assigned. The aim of this study was to identify the molecular etiology of non-syndromic hearing loss not linked to GJB2 mutations in a large cohort of Mauritanian patients using a targeted mutation screening of exon 7 in LRTOMT gene.
Materials and methods
Patients
The cohort was composed of 128 unrelated Mauritanian children (1 child per family) respectively from 83 families with multiple affected siblings and 45 families with no history of deafness (sporadic cases), recruited from two deaf schools in Nouakchott, Mauritania. Forty-one children of the cohort were identified as negative for GJB2 pathogenic variants in a previous study we carried out on non-syndromic hearing impairment associated GJB2 variants in Mauritania [8] and 87 children were newly recruited. All patients were diagnosed with severe-to-profound non-syndromic congenital hearing impairment following medical examination. This study was approved by the ethics committee of the University of Nouakchott, in Mauritania as stated by the ethics clearance letter No002/2020/CE/UNA. Written informed consent was obtained from all parents or legal guardians of the children. A questionnaire was used to collect demographic data and medical history of the subjects.
Molecular analysis
Blood samples were collected in EDTA tubes and used to extract genomic DNA by Qiagen DNA Blood Minikit procedure (QIAGEN Genomic DNA Handbook.2015). PCR reactions (20 µl) contained 1 µl (20–30 ng) of genomic DNA, 1 µl (10 µM) of each specific primer (5′-AGGATAATAATTGCTACTGGCAAAA-3′ and5′-ATCCCAAATATTCCTTCACTGTCTT-3′),10 µl of AmpliTaqGold 360 Master Mix (Life Technologies) and 7 µl of distilled water. The PCR program presented a DNA denaturation step at 95 °C for 10 min followed by 35 amplification cycles (denaturation at 94 °C for 30 s, annealing at 53 °C for 35 s and extension at 72 °C for 40 s) tailed by a 5 min final extension at 72 °C. PCR products were sequenced on capillary ABI3730 Genetic Analyzer (Applied Biosystems, California, USA). Data obtained were then matched with reference sequences of the LRTOMT gene (NM_001145309.4) using Seqscape3.0 software program package (Gene Codes, MI, and USA). Amino acid change was considered as potentially deleterious if predicted by (PolyPhen2, http://genetics.bwh.harvard.edu/pph2), Sorting Intolerant from Tolerant (SIFT, http://sift.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org/). In families with multiple cases of deafness, DNA of one proband was sequenced. Sanger sequencing was then extended to available parents and all affected siblings carrying the mutation in homozygous state.
LRTOMT protein 3-D structure modeling
The predicted tertiary structure of LRTOMT protein (AF-Q8WZ04-F1) was obtained using AlphaFold protein structure database (https://alphafold.ebi.ac.uk/). Similarity between wild-type and mutated 3D structures of LRTOMT protein was explored by using UCSF Chimera version 1.15.
Results
Screening of exon7 in LRTOMT
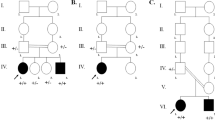
Analysis of sequencing data from exon 7 of LRTOMT gene of the 128 selected patients showed only one biallelic missense variant (c.179 T > C;p.(Leu60Pro) in four (3.1%) unrelated patients. The genomic coordinates of the newly identified variant were Chr11(GRCh37):g.71817077 T > C; NM_001145309.4:c.179 T > C. Sequence alignment revealed that this variant was not reported before. Notably, the arginine residue was highly conserved through evolution among multiple-species (from Human to opossum considering 6 species) (Fig. 1). The variant was predicted to have deleterious effect on gene function by SIFT (score: 0.01) and a disease-causing effect by Mutation Taster (prob: 1). Exploration of effect of p.Leu60Pro on the predicted three-dimensional (3D) structure of the encoded protein showed a disruption from an organized α helix in the wild protein, to a random conformation in the mutant form (Fig. 1). The healthy parents of the four patients in families (F7-DFN, F28-DFN, F31-DFN, F81-DFN) carried the mutation in heterozygous state and all affected children were homozygous for the variant. Three of the four patients had other affected relatives one presented no family history of HI. The four affected families originated from the Trarza region in southern Mauritania (Figs. 2 and 3).

Sequencing data, alignment and prediction structural change in the p.Leu60pro of variant LRTOMT. A Electropherogram plots showing the reference sequence and the mutation in heterozygous and homozygous states. B Sequence alignment presenting conservation of the region of LRTOMT protein containing (Leu60) among various species. C LRTOMT protein tertiary structure modeling performed with Chimera, based on the AlphaFold prediction of the transmembrane O-methyltransferase (AF-Q8WZ0F1). (D) Localized structural change in the wild type alpha helix (Phe57, Leu59, Leu60, Val61, Thr63, Val61, Val64 and Leu65) (left) showing the breaking of the alpha helix induced by substitution of Leu60 by Prolin (right)

Pedigrees of affected children with family history. Segregation of the mutation pattern was compatible with autosomal recessive inheritance. All cases originated from southern Mauritania (map top right)

Pure-tone audiogram of a patient from family F28-DFN who received cochlear implant. Proband, indicated by an arrow, had profound hearing loss in both ears
Cochlear implant outcome
Based on clinical assessment and audiological data, the proband of family F28-DFN, a 7-year-old boy, diagnosed with profound bilateral congenital deafness, received a cochlear implant fitted in the right ear. Now, aged 9 years, signs of auditory recovery are largely perceptible (Fig. 3) on the basis of his score on APCEI scale used to assess his communication skill progress.
Discussion
In this study, we identified a novel biallelic missense pathogenic variant in exon 7 of LRTOMT gene in three Mauritanian children with family history of congenital deafness and one child with no other relative being affected. The variant was found in both girls and boys and only carriers at homozygous state showed the phenotype which supported an autosomal recessive segregation of the variant. Because of their higher number of inter-residue contacts, helices are known to tolerate sequence amino-acid changes without affecting their secondary structure [14]. As a result of this relative robustness, compared to strands and coils conformations, any variation in the helix organization was implied to be enough significant to cause alteration of the protein structure and therefore likely to affect its function. For instance, the wild residue Arg81 formed a salt bridge and hydrogen bonds between the helix and following loop, central for the protein stability. Its substitution with glutamine c.242G > A in exon 7: (p.Arg81Gln) was found to disrupt the alpha helix resulting in local destabilization of the protein tertiary structure [12]. Although involving a radical structurally distinct, our variant (p.Leu60Pro), located in the same exon 7, appeared to be inducing a comparable disorganizing effect on the protein alpha helix which likely resulted in the hearing impairment observed in the four patients of our cohort and also predicted for the (p.Arg81Gln) mutation [11]. Since the first reported case of direct involvement of proline in human diseases [15], mutant proteins for this amino acid have been implicated in the etiology of number of genetic disorders including non-syndromic recessive hearing impairment [16]. For instance, different studies showed that p.Ser50Pro and p.Thr56Pro variants, both consisting of replacement of wild amino acid by proline residue in connexin 50 (Cx50, GJA8), affected the protein contribution to the lens normal survival through angular restriction of α helix or β sheet secondary structure [17, 18]. Proline is indeed a distinctive amino acid with a nitrogen atom covalently locked within a ring inducing a unique constrained φ angle of approximately − 65° [19]. This irregular geometry disrupts protein secondary structure by steric hindrance i.e. in inhibiting the backbone to conform into a normal alpha-helix turn conformation. In addition, the lack of hydrogen on proline's nitrogen (proline cannot donate protons) prevents it from participating in hydrogen bonding essential for the helix structure. The apparent deleterious consequence we observed (SIFT score (0.01) and disease-causing effect (prob: 1) by Mutation Taster) for this novel mutation was therefore likely due to a change of the protein secondary structure induced by the substitution to proline residue consisting in an alpha helix breaking. The wild type leucine residue was also located in an evolutionary highly conserved region [12] suggesting an important functional position of the residue (Fig. 1B). Modeling, using WHATIF server (http://swift.cmbi.ru.nl) on rat COMT crystal structure as template for catechol-O-methyltransferase domain of the human LRTOMT2 (NM_001145309.4) showed that common LRTOMT variants associated with non-syndromic recessive hearing impairment led to hair cell degeneration process [9, 11, 20]. Close to p.Leu60Pro variant, various changes in the same exon 7 were also associated with cases of moderate to profound congenital deafness making of this segment a mutation hot spot in the LRTOMT gene [10]. Besides the above homozygous c.242G > A (p.Arg81Gln) variant found in Tunisian, Libyan and Egyptian families [9, 20] we may also cite missense variants c.122G > A (p.Arg41Gln), c.193 T > C (p.Trp65Arg) in Morocco and Tunisia [11, 12] and a nonsense mutation c.208C > T (p.Arg70*), in Tunisia [11,12,13] (Table 1). In this ethnic context, we noticed that all four affected families carrying the LRTOMT mutation (c.179 T > C;p.(Leu60Pro) identified in this report belonged to the Moors (Maures), a predominant racial group of the Mauritanian population. This group, of Berber-Arab origin, ethnically and culturally self identifies with the neighboring North Africa populations [21].
In a previous study, we detected GJB2 (MIM 121 011) variants in only 5 out of the 53 (9.4%) investigated families with congenital hearing impairment [8]. The present work showed that LRTOMT was also involved in hearing impairment in our population (3.1%; 4/128). However, a limitation of this study was that we have only screened exon 7 of LRTOMT. This screening only in exon 7 was mostly guided by very scarce findings of deafness associated variants in other exons reported by various studies in the region [9,10,11,12,13]. Indeed, a total of 14 pathogenic variants in exon 7 out of 18 causal variants have presently been reported. Search in these sequences along multi-gene hear panel is underway to locate other deafness related variants in our families.
Conclusions
This study identified a novel biallelic predicted pathogenic variant (c.179 T > C;p.(Leu60Pro) in exon 7 of LRTOMT gene likely causing a localized alpha helix breaking in the LRTOMT protein. Being the single exon 7 variant identified in our patients underlined the significance of a genetic screening for this variant in the context of an accurate molecular diagnosis of hearing impairment or in assessing the transmission of the disease in our population and, broadly, among Northern African populations.
Further hearing tests are still required to clearly establish the effect of early cochlear implant fitting on audition rehabilitation in patients carrying mutations in the LRTOMT gene. Screening for other parts of this gene or using multigene panel in a larger population study is also recommended.
Availability of data and materials
The datasets on variants generated during the current study are available from the corresponding author on reasonable request.
References
Wilson BS, Tucci DL, Merson MH, O’Donoghue GM (2017) Global hearing health care: New findings and perspectives. Lancet 390:2503–2512. https://doi.org/10.1016/S0140-6736(17)31073-5
Sheffield AM, Smith RJ (2019) The epidemiology of deafness. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a033258
Kimberly A, Gifford MD, Michael G, Holmes HH, Bernstein DO (2009) Hearing loss in children. Pediatr Rev 30(6):207–215. https://doi.org/10.1542/pir.30-6-207
Prasansuk S (2000) Incidence/prevalence of sensorineural hearing impairment in Thailand and Southeast Asia. Audiology 39(4):207–211. https://doi.org/10.3109/00206090009073080
Elahi MM, Elahi F, Elahi A, Elahi SB (1998) Paediatric hearing loss in rural Pakistan. J Otolaryngol 27(6):348–353
Smith AW (2001) WHO activities for prevention of deafness and hearing impairment in children. Scan Audiol Suppl 53:93–100. https://doi.org/10.1080/010503901750166808
Zakzouk S (2002) Consanguinity and hearing impairment in developing countries: a custom to be discouraged. J LaryngolOtol 116:811–816. https://doi.org/10.1258/00222150260293628
Moctar EC, Riahi Z, El Hachmi H, Veten F, Meiloud G, Bonnet C, Abdelhak S, Errami M, Houmeida A (2016) Etiology and associated GJB2 mutations in Mauritanian children with non-syndromic hearing loss. Eur Arch Otorhinolaryngol 273:3693–3698. https://doi.org/10.1007/s00405-016-4036-z
MosratiMA F-Z, Elgaaied AB et al (2021) Deep analysis of the LRTOMT c.242G>A variant in non-syndromic hearing loss North African patients and the Berber population: Implications for genetic diagnosis and genealogical studies. Mol Genet Genomic Med 9:e1810. https://doi.org/10.1002/mgg3.1810
Ichinose A, Moteki H, Hattori M, Nishio SY, Usami SI (2015) Novel mutations in LRTOMT associated with moderate progressive hearing loss in autosomal recessive inheritance. Ann Otol Rhinol Laryngol 124(Suppl 1):142S-S147. https://doi.org/10.1177/0003489415575043
Charif M, Bounaceur S, Abidi O et al (2012) The c.242G>A mutation in LRTOMT gene is responsible for a high prevalence of deafness in the Moroccan population. Mol Biol Rep 39(12):11011–11016. https://doi.org/10.1007/s11033-012-2003-3
Ahmed ZM, Masmoudi S, Kalay E et al (2008) Mutations of LRTOMT a fusion gene with alternative reading frames cause non syndromic deafness in humans. Nat Genet 40:1335–1340. https://doi.org/10.1038/ng.245
Riahi Z, Bonnet C, Zainine R, Louha M, Bouyacoub Y, Laroussi N, Chargui M, Kefi R, Jonard L, Dorboz I, Hardelin JP, Salah SB, Levilliers J, Weil D, McElreavey K, Boespflug OT, Besbes G, Abdelhak S, Petit C (2014) Whole exome sequencing identifies new causative mutations in Tunisian families with non-syndromic deafness. PLoS ONE 9:e99797. https://doi.org/10.1371/journal.pone.0099797
Abrusán G, Joseph A (2016) Marsh Alpha Helices Are More Robust to Mutations than Beta Strands. PLoS Comput Biol 12(12):e1005242
Schafer IA, Scriver CR, Efron ML (1962) Familial hyperprolinemia, cerebral dysfunction and renal anomalies occuring in a family with hereditary nephropathy and deafness. N Engl J Med 267:51–60. https://doi.org/10.1056/NEJM196207122670201
Batissoco AC, Auricchio MTBM, Kimura L, Tabith-Junior A, Mingroni-Netto RC (2009) A novel missense mutation p.L76P in the GJB2 gene causing non-syndromic recessive deafness in a Brazilian family Brazilian. J Med Biol Res 42:168–171. https://doi.org/10.1590/S0100-879X2009000200004
Adam M, De RA, Meşe G, Li L, Sellitto C, Brink PR, Gong X, White TW (2009) The cataract causing Cx50-S50P mutant inhibits Cx43 and intercellular communication in the lens epithelium. Exp Cell Res 315(6):1063–1075. https://doi.org/10.1016/j.yexcr.2009.01.017
Hadrami M, Bonnet C, Veten F, Zeitz C et al (2018) A novel missense mutation of GJA8 causes congenital cataract in a large Mauritanian family. Eur J Ophthalmol 29(6):621–628. https://doi.org/10.1177/1120672118804757
Alexander AM, Edward R, Dulce EC (2013) Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 8(1):e53785. https://doi.org/10.1371/journal.pone.0053785
Gibriel AA, Abou-Elew MH, Masmoudi S (2019) Analysis of p.Gly12Valfs*2, p.Trp24* and p.Trp77Arg mutations in GJB2and p.Arg81Gln variant in LRTOMT among non-syndromic hearing loss Egyptian patients: implications for genetic diagnosis. Mol Biol Rep 46:2139–2145. https://doi.org/10.1007/s11033-019-04667-0
Vernet R (1986) La Mauritanie, des Origines au Début de l'Histoire. Centre Cultural Francais Nouakchott
Crowgey EL, Washburn MC, Kolb EA, Puffenberger EG (2019) Development of a novel next-generation sequencing assay for carrier screening in old order amish and mennonite populations of Pennsylvania. J Mol Diagnost 21(4):687–694. https://doi.org/10.1016/j.jmoldx.2019.03.004
Babanejad M, Fattahi Z, Bazazzadegan N et al (2012) A comprehensive study to determine heterogeneity of autosomal recessive non-syndromic hearing loss in Iran. Med Genet 10:2485–2492. https://doi.org/10.1002/ajmg.a.35572
Vanwesemael M, Schrauwen I, Ceuppens R et al (2011) A 1 bp deletion in the dual reading frame deafness gene LRTOMT causes a frameshift from the first In to the second reading frame. Med Genet. https://doi.org/10.1002/ajmg.a.34096
Wang R, Han S, Khan A, Zhang X (2017) Molecular analysis of twelve Pakistani families with non-syndromic or syndromic hearing loss. Genet Test Mol Biomark 21(5):316–321. https://doi.org/10.1089/gtmb.2016.0328
Marková S, Brožková DS, Mészárosová A et al (2016) Mutations in eight small DFNB genes are not a frequent cause of non-syndromic hereditary hearing loss in Czech patients. Int J Pediatr Otorhinolaryngol 86:27–33. https://doi.org/10.1016/j.ijporl.2016.04.005
Du X, Schwander M, Marie YE et al (2008) A catechol-O-methyltransferase that is essential for auditory function in mice and humans. PNAS 105(38):14609–14614. https://doi.org/10.1073/pnas.0807219105
Sarmadi A, Nasrniya S, Soleimani Farsani M et al (2020) A novel pathogenic variant in the LRTOMT gene causes autosomal recessive non-syndromic hearing loss in an Iranian family. BMC Med Genet 21:127. https://doi.org/10.1186/s12881-020-01061-7
Wu CC, Lin YH, Liu TC, Lin KN, Yang WS, Hsu CJ et al (2015) Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine 94:e1073. https://doi.org/10.1097/MD.0000000000001073
Acknowledgements
We thank all the families for their participation to the study. This work was supported by Fondation pour l’Audition (FPA-IDA05) and the ANRSI-Mauritania.
Funding
The authors declare that no funds or grants were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
MS, ECM, SMB, CTH and VT collected and organized patients’ files. AD and LAV examined patients and analyzed clinical data. CB, CP and AH contributed in paper conception and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Research in this study has been performed in accordance with the Declaration of Helsinki and was approved by an appropriate ethics committee: the Ethics Committee of the University of Nouakchott, Mauritania (ref. no002/2020/CE/UNA). We confirm that all methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
Informed consent was obtained from all study participants. The informed consent of all patients was also obtained for the data publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Salame, M., Bonnet, C., Moctar, E.C.M. et al. Identification a novel pathogenic LRTOMT mutation in Mauritanian families with nonsyndromic deafness. Eur Arch Otorhinolaryngol 280, 4057–4063 (2023). https://doi.org/10.1007/s00405-023-07907-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-023-07907-z