
Abstract
The chronic lead intoxication was modelled by injecting outbred rats intraperitoneally with lead acetate 3 times a week for 5 weeks. Using an in vitro motility assay, it was shown that lead intoxication causes changes in in vitro actin–myosin interaction, specifically, a decrease in the maximum sliding velocity of native thin filaments over myosin isolated from the left ventricular myocardium of the rat heart. No statistically significant changes were found in the calcium sensitivity and cooperativity of the “pCa–velocity” curve, characteristics of the motile filament fraction, and isometric force. Using electrophoretic separation of proteins, a shift in the ratio of myosin heavy chains toward β-chains with a lower ATPase activity was found.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cardiovascular diseases are the main cause of death in the world [1], and the impact of heavy metals contributes to the incidence and exacerbation of these pathologies [2]. Vast human subpopulations, including the workers of industrial enterprises and people living in the zone of their technogenic influence, are exposed to heavy metals that pollute the surrounding atmosphere [2, 3]. Lead is the most abundant xenobiotic in the human habitat. It is present in air, house dust, soil, water, food, and varied consumption items. Environmental lead accumulation is characterized by stability, creating conditions for toxic exposures many years after the cessation of industrial emissions.
Lead accumulates in human and animal bodies, especially in bones, where its half-life averages decades. It has been shown that there is no safe level of lead in the blood, and its toxic effects are present at levels much lower than supposed previously [2, 4–8].
The literature abounds in data on a toxic effect of lead on all systems of the body, including the cardiovascular [5, 9–17]. Meanwhile, the known published data on the impact of lead on mechanical activity of the myocardium are scarce, do not cover all its aspects, and are, in part, contradictory. The cardiotoxic effect of lead has not been studied at the level of isolated proteins, despite the suggestion that heavy metals affect directly the proteins involved in muscle contraction.
In the ventricular myocardium, there are three myosin isoforms: V1, V2, and V3 [18]. The amino acid sequence of isoforms V1 and V3 of mammalian cardiac myosin is ≥ 93% identical [19]. Isoforms V1 (αα) and V3 (ββ) represent homodimers containing two α- or β-myosin heavy chains (MHC), respectively, while V2 (αβ) is a heterodimer composed of one α- and one β-MHC [20].
In the ventricle of small mammals (e.g., mice and rats), α-MHC is dominant [21–23]. However, gene expression of MHC isoforms in the heart can change in response to the effect of thyroid hormones, hemodynamic load, or varied pathological stimuli, including congestive heart failure; in the myocardium of rodents, these processes can proceed faster and to a larger extent than in humans [22].
Mechanical characteristics of the actomyosin complex depend on the isoforms of cardiac myosin [20]. Using an in vitro motility assay, it was found that the V1 isoform moves actin 2–3 times faster than the V3 isoform [23–25] because V1 has a higher cross-bridge cycling rate [23]. The isometric force developed by the V1 and V3 isomyosin heads is species-specific: in larger animals (rabbits and pigs) the force developed by the V3 myosin head is higher than in V1, and in small mammals (rats and mice), the forces are indistinguishable [23, 24, 26].
To study the cardiotoxic effect of lead salts, we studied the functional characteristics and isoform composition of myosin isolated from the left ventricle of rats. The sliding velocity of thin filaments over myosin was determined by an in vitro motility assay; the ratio of MHCs in the myocardium of the control rat group and the group of rats with chronic lead intoxication was investigated using the electrophoretic separation of proteins.
MATERIALS AND METHODS
Intoxication model
The experiment was carried out on outbred white male rats aged 4 months and weighing 300 g at the beginning of the experiment. All rats were kept under standard conditions, breathed unfiltered air, and were provided with a standard balanced diet. Chronic lead intoxication (“Pb” group) was simulated by intraperitoneal injection of a lead acetate solution, 3 times a week for 5 weeks (up to 15 injections). A single dose was 12.5 mg of lead per 1 kg of body weight. The control group of rats (“C” group) received the same volume of sterile distilled water according to a similar scheme [27, 28].
Experiments were planned and carried out in accordance with the principles of the Basel Declaration and were approved by the Ethics Committee of the Institute of Immunology and Physiology of the Ural Branch of the Russian Academy of Sciences.
Protein extraction
Myosin was isolated from the left ventricle of the rat heart using a standard method [29]. Actin was obtained from rabbit skeletal muscles according to the standard procedure [30], and after polymerization was labeled with TRITC-phalloidin (Sigma-Aldrich, USA) by an addition of 2 mM ATP, 4 mM MgCl2, and 100 mM KCl. Cardiac troponin was isolated from the pig hearts as described elsewhere [31]. Recombinant human tropomyosin was obtained as described in [32]. The use of contractile and regulatory proteins isolated from different species in an in vitro motility assay is a common practice [26, 33]. Regulated thin filaments were reconstituted from actin, troponin and tropomyosin by mixing these proteins at the following concentrations: 400 nM F-actin labeled with rhodamine-phalloidin, 100 nM troponin and 100 nM tropomyosin at 4°C in a buffer containing (in mM) 25 KCl, 25 imidazole, 4 MgCl2, 1 EGTA, and 10 DTT (pH 7.5). Native thin filaments were extracted from the rat left ventricle according to the Spiess protocol [34]. The protein composition of the reconstituted and native thin filaments was checked by electrophoresis in polyacrylamide gel with sodium dodecyl sulfate [35].
In vitro motility assay
An in vitro motility assay was used to determine the sliding velocity of native and reconstituted thin filaments, consisting of actin, troponin and tropomyosin, over myosins extracted from the left ventricle of rats of the “C” and “Pb” groups at various calcium concentrations in solution. Calcium ions interact with a regulated thin filament in a flow cell solution. Calcium binds to troponin C and leads thereby to conformational changes in the thin filament, due to which myosin-binding sites on actin globules open. A mechanical actin–myosin interaction can be evaluated by the characteristics of how the filaments move and stop. Changes in these characteristics, depending on the calcium concentration in a solution with the regulated thin filaments, make it possible to study the mechanisms of calcium regulation of the actin–myosin interaction [33]. Experiments in an in vitro motility assay were carried out at 30°C as described previously [26]. Fluorescently labeled actin filaments in an experimental flow cell were visualized using the Axiovert 200 inverted epifluorescence microscope (Carl Zeiss, Germany) equipped with a 100×/1.45 Plan-Fluar oil alpha-objective. The movement of thin filaments over the myosin-coated surface was recorded using the EMCCD iXon-897BV camera (Andor Technology, UK), while their velocity was analyzed using the GMim-Pro software [36]. The dependence of the sliding velocity of thin filaments on the calcium concentration was analyzed using the Hill equation:
V = V max (1 + 10h(pCa – pCa50))–1, (1)
where V and V max are the velocity and maximum velocity at a saturating calcium concentration, respectively, pCa50 (calcium sensitivity) is the pCa value, at which a half of the maximum velocity is achieved, and h is the Hill cooperativity coefficient.
Determining the relative isometric force developed by myosin in an in vitro motility assay
To determine the isometric force in an in vitro motility assay, we used the “mixture protocol”, in which the myosin under study was mixed with noncycling NEM-modified myosin [37]. The latter, when attached to actin, prevents thin filament sliding and serves as a load. The fraction of NEM-modified myosin, needed to stop filament sliding, is a measure of the relative force that the myosin under study can develop.
Electrophoresis
Isoforms of rat myocardial MHC were determined by electrophoresis in a polyacrylamide gel with sodium dodecyl sulfate [38]. After the completion of electrophoresis, the gel was stained with Coomassie Brilliant Blue R250, then washed from the stain and scanned with a densitometer (BioRad, USA) to determine the percentage of α- and β-MHCs.
Statistical analysis
The statistical significance of the intergroup differences between the mean values of all the obtained parameters was assessed using the nonparametric Mann–Whitney U test; the differences were considered statistically significant at p < 0.05. Data are expressed as mean ± SD.
RESULTS
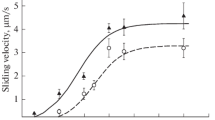
Under the influence of chronic lead intoxication, the maximum sliding velocity of native thin filaments over the left-ventricular myosin decreased compared to the control group from 4.07 ± 0.13 to 3.27 ± 0.12 μm/s (Table 1), i.e. by 20% (Fig. 1). The calcium sensitivity and Hill cooperativity coefficient were not significantly distinguishable between groups “C” and “Pb”.
The characteristics of the dependence of the fraction of motile filaments on the calcium concentration (the maximum value of the fraction, pCa50, Hill cooperativity coefficient) were not significantly different (Table 1, Fig. 2).
Lead intoxication reduces the maximum sliding velocity of native thin filaments over myosin, but does not affect calcium sensitivity and the Hill cooperativity coefficient. The curve of the dependence of the fraction of motile filaments on the calcium concentration shows no significant differences in all parameters, but there is a clear tendency toward a decrease in the fraction under the influence of lead.
Under the influence of chronic lead intoxication, there was a significant shift in the ratio of MHCs toward an increase in the content of β-chains with a lower ATPase activity (55 ± 10% α-MHC and 45 ± 10% β-MHC in group “Pb”, and 86 ± 4% α-MHC and 14 ± 4% β-MHC in group “C”).
To stop the motility of reconstituted thin filaments over the left-ventricular myosin in groups “C” and “Pb”, 34 ± 4% and 42 ± 6% of NEM-modified myosin were required, respectively. This difference was statistically nonsignificant (Fig. 3).

Dependence of the sliding velocity of native thin filaments (a) and the fraction of motile filaments (b) over left-ventricular myosin in rats of groups “C” and “Pb” on calcium concentration in solution. pCa is a negative decimal logarithm of calcium concentration. The regression curve fits the Hill equation. Velocity is represented as mean ± SD for 4 experiments.

Representative electrophoregram to demonstrate a ratio of α- and β-MHCs in the left ventricle of rats in groups “C” (left) and “Pb” (right). Group “C” (left) – 86 ± 4% α-MHC and 14 ± 4% β-MHC. Group “Pb” (right) – 55 ± 10% α-MHC and 45 ± 10 % β-MHC.

Dependence of the sliding velocity of reconstituted thin filaments over left-ventricular myosin in rat groups “C” and “Pb” on NEM-modified myosin concentration. Velocity is represented as mean ± SD for 4 experiments. Differences in the values of NEM-modified myosin amount needed to stop motility in rat groups “C” (34 ± 4%) and “Pb” (42 ± 6%) were statistically insignificant.
DISCUSSION
Studies of the influence of chronic lead intoxication on isolated multicellular preparations of the left ventricle showed that the force of isometric contraction of papillary muscles did not change when changing calcium cycle parameters in cardiomyocytes [39, 40].
Previously, we studied the characteristics of the actin–myosin interaction and contraction of multicellular preparations (trabeculae and papillary muscles) of the right ventricle when exposed to lead ions. It was shown that the maximum sliding velocity of reconstituted thin filaments over right-ventricular myosin decreased under the influence of lead intoxication [27], and the ratio of MHC isoforms shifted toward the slower β-chains [28]. In multicellular preparations of the right ventricle of the same rats that were used in our study, we found an increase in the cross-sectional area of trabeculae and papillary muscles, a decrease in passive (diastolic) mechanical tension, a decrease in the maximum rate of isometric force development and the rate of isotonic shortening of papillary muscles [27].
In our experiments using native thin filaments and myosin from the left ventricle, a similar tendency was observed, namely the maximum velocity of actin–myosin interaction dropped under the influence of chronic lead intoxication by 20% compared to the control group. This was explained by a shift in the α-MHC/β-MHC ratio: an 31% increase in the content of β-chains with a lower ATPase activity and a lower actin-myosin cross-bridge cycling rate. Such a change can be an adaptive energy-saving molecular mechanism in cardiac pathologies [25, 41]. The characteristics of the fraction of motile filaments were statistically indistinguishable; however, there was a tendency toward a decrease in this parameter during lead intoxication.
The differences between groups “C” and “Pb” in the NEM-modified myosin amount required to completely stop the motility of thin reconstituted filaments were statistically insignificant, which can be explained by the absence of differences in the force developed by myosin isoforms in small mammals (rats and mice) [23, 24, 26], although there was a small tendency toward an increase in the isometric force in the left-ventricular myosin of group “Pb”. This effect may be worth a deeper investigation.
Thus, when studying the functional characteristics and myosin isoform composition in the left ventricle of rats with chronic lead intoxication, we revealed a decrease in the rate of actin-myosin interaction and a shift in the ratio of MHCs toward β-chain. These results, along with the aforementioned changes in the contractile characteristics of multicellular right-ventricular preparations of the same rats [27, 28], may result from the development of myocardial hypertrophy caused by pressure overload [28, 42, 43]. The assumption on possible myocardial hypertrophy due to lead intoxication indirectly confirms the increase in the relative heart weight in these rats, as calculated per 100 g of body weight, and in the amplitude of the QRS complex on the electrocardiogram [28], as well as an increase in pressure and the thickness of cardiomyocytes of the left ventricle in rats intoxicated with lower lead doses [44]. Thus, with a professional and environmentally conditioned lead load on the organism, there arises an increased risk of cardiovascular pathology, which may be associated, among other things, with myocardial contractile dysfunction at the molecular level.
REFERENCES
GBD Compare | IHME Viz Hub. https://vizhub.healthdata.org/gbd-compare/. Accessed 28 Dec 2020.
Landrigan PJ, Fuller R, Acosta NJR, Adeyi O, Arnold R, Basu N (Nil), Baldé AB, Bertollini R, Bose-O’Reilly S, Boufford JI, Breysse PN, Chiles T, Mahidol C, Coll-Seck AM, Cropper ML, Fobil J, Fuster V, Greenstone M, Haines A, Hanrahan D, Hunter D, Khare M, Krupnick A, Lanphear B, Lohani B, Martin K, Mathiasen KV, McTeer MA, Murray CJL, Ndahimananjara JD, Perera F, Potočnik J, Preker AS, Ramesh J, Rockström J, Salinas C, Samson LD, Sandilya K, Sly PD, Smith KR, Steiner A, Stewart RB, Suk WA, van Schayck OCP, Yadama GN, Yumkella K, Zhong M (2018) The Lancet Commission on pollution and health. Lancet 391:462–512. https://doi.org/10.1016/S0140-6736(17)32345-0
Kim YD, Eom SY, Yim DH, Kim IS, Won HK, Park CH, Kim GB, Yu SD, Choi BS, Park JD, Kim H (2016) Environmental Exposure to Arsenic, Lead, and Cadmium in People Living near Janghang Copper Smelter in Korea. J Korean Med Sci 31:489. https://doi.org/10.3346/jkms.2016.31.4.489
WHO (2019) Lead poisoning and health. https://www.who.int/news-room/fact-sheets/detail/lead-poisoning-and-health. Accessed 9 Jan 2020.
Afridi HI, Kazi TG, Kazi NG, Jamali MK, Arain MB, Sirajuddin A, Baig JA, Kandhro GA, Wadhwa SK, Shah AQ (2010) Evaluation of cadmium, lead, nickel and zinc status in biological samples of smokers and nonsmokers hypertensive patients. J Hum Hypertens 24:34–43. https://doi.org/10.1038/jhh.2009.39
Vainio H, Heseltine E, Partensky CWJ (1993) Meeting of the IARC working group on beryllium, cadmium, mercury and exposures in the glass manufacturing industry. Scand J Work Environ Health 19:360–363.
Mukhacheva SV (2017) Long-term dynamics of the concentration of heavy metals in the forage and body of the bank vole (Myodes glareolus) during the period of decreasing emissions from the copper smelter. Ecology 6:461–471. (In Russ). https://doi.org/10.7868/s0367059717060087
Mirzaei R (2018) Assessment of Accumulation and Human Health Risk of Trace Elements in the Vicinity of Industrial Estates, Central Iran. Arch Hyg Sci 7:118–125. https://doi.org/10.29252/archhygsci.7.2.118
Nawrot TS, Thijs L, Den Hond EM, Roels HA, Staessen JA (2002) An epidemiological re-appraisal of the association between blood pressure and blood lead: A meta-analysis. J Hum Hypertens 16:123–131. https://doi.org/10.1038/sj.jhh.1001300
Kopp SJ, Bárány M, Erlanger M, Perry EF, Perry HM (1980) The influence of chronic low-level cadmium and/or lead feeding on myocardial contractility related to phosphorylation of cardiac myofibrillar proteins. Toxicol Appl Pharmacol 54:48–56. https://doi.org/10.1016/0041-008X(80)90007-1
Prentice RC, Kopp SJ (1985) Cardiotoxicity of lead at various perfusate calcium concentrations: Functional and metabolic responses of the perfused rat heart. Toxicol Appl Pharmacol 81:491–501. https://doi.org/10.1016/0041-008X(85)90420-X
Alissa EM, Ferns GA (2011) Heavy metal poisoning and cardiovascular disease. J Toxicol 2011:870125. https://doi.org/10.1155/2011/870125
Solenkova NV, Newman JD, Berger JS, Thurston G, Hochman JS, Lamas GA (2014) Metal pollutants and cardiovascular disease: Mechanisms and consequences of exposure. Am Heart J 168:812–822. https://doi.org/10.1016/j.ahj.2014.07.007
Lamas GA, Navas-Acien A, Mark DB, Lee KL (2016) Heavy metals, cardiovascular disease, and the unexpected benefits of chelation therapy. J Am Coll Cardiol 67:2411–2418. https://doi.org/10.1016/j.jacc.2016.02.066
Ruiz-Hernandez A, Navas-Acien A, Pastor-Barriuso R, Crainiceanu CM, Redon J, Guallar E, Tellez-Plaza M (2017) Declining exposures to lead and cadmium contribute to explaining the reduction of cardiovascular mortality in the US population, 1988-2004. Int J Epidemiol 46:1903–1912. https://doi.org/10.1093/ije/dyx176
Chowdhury R, Ramond A, O’Keeffe LM, Shahzad S, Kunutsor SK, Muka T, Gregson J, Willeit P, Warnakula S, Khan H, Chowdhury S, Gobin R, Franco OH, Di Angelantonio E (2018) Environmental toxic metal contaminants and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 362:14–16. https://doi.org/10.1136/bmj.k3310
Sevim Ç, Doğan E, Comakli S (2020) Cardiovascular disease and toxic metals. Curr Opin Toxicol 19:88–92. https://doi.org/10.1016/j.cotox.2020.01.004
Hoh JF, McGrath PA (1978) Electrophoretic analysis of multiple forms of rat cardiac myosin: effects of hypophysectomy and thyroxine replacement. J Mol Cell Cardiol 10:1053–1060.
Alpert NR, Brosseau C, Federico A, Krenz M, Robbins J, Warshaw DM (2002) Molecular mechanics of mouse cardiac myosin isoforms. Am J Physio—Heart Circ Physiol 283:1446–1454. https://doi.org/10.1152/ajpheart.00274.2002
Shchepkin DV, Kopylova GV, Nikitina LV (2011) Study of reciprocal effects of cardiac myosin and tropomyosin isoforms on actin-myosin interaction with in vitro motility assay. Biochem Biophys Res Commun 415:104–108. https://doi.org/10.1016/j.bbrc.2011.10.022
Galler S, Puchert E, Gohlsch B, Schmid D, Pette D (2002) Kinetic properties of cardiac myosin heavy chain isoforms in rat. Pflugers Arch Eur J Physiol 445:218–223. https://doi.org/10.1007/s00424-002-0934-6
Danzi S, Klein S, Klein I (2008) Differential regulation of the myosin heavy chain genes α and β in rat atria and ventricles: Role of antisense RNA. Thyroid 18:761–768. https://doi.org/10.1089/thy.2008.0043
Nikitina LV, Kopylova GV, Shchepkin DV, Katsnelson LB (2008) Study of the interaction between rabbit cardiac contractile and regulatory proteins. An in vitro motility assay. Biochem 73:178–184. https://doi.org/10.1007/s10541-008-2009-6
Malmqvist UP, Aronshtam A (2004) Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry 43:15058–15065.
Hoyer K, Krenz M, Robbins J (2007) Shifts in the myosin heavy chain isozymes in the mouse heart result in increased energy effeciency. J Mol Cell Cardiol 42:214–221. https://doi.org/10.1016/j.physbeh.2017.03.040
Nikitina LV, Kopylova GV, Shchepkin DV, Nabiev SR, Bershitsky SY (2015) Investigations of Molecular Mechanisms of Actin-Myosin Interactions in Cardiac Muscle. Biochem 80:1748–1763. https://doi.org/10.1134/S0006297915130106
Protsenko YL, Katsnelson BA, Klinova SV, Lookin ON, Balakin AA, Nikitina LV, Gerzen OP, Nabiev SR, Minigalieva IA, Privalova LI, Gurvich VB, Sutunkova MP, Katsnelson LB (2019) Further analysis of rat myocardium contractility changes associated with a subchronic lead intoxication. Food Chem Toxicol 125:233–241. https://doi.org/10.1016/j.fct.2018.12.054
Protsenko YL, Katsnelson BA, Klinova SV, Lookin ON, Balakin AA, Nikitina LV, Gerzen OP, Minigalieva IA, Privalova LI, Gurvich VB, Sutunkova MP, Katsnelson LB (2018) Effects of subchronic lead intoxication of rats on the myocardium contractility. Food Chem Toxicol 120:378–389. https://doi.org/10.1016/j.fct.2018.07.034
Margossian SS, Lowey S (1982) Preparation of Myosin and Its Subfragments from Rabbit Skeletal Muscle. Methods Enzymol 85:55–71. https://doi.org/10.1016/0076-6879(82)85009-X
Pardee JD, Spudich JA (1982) Purification of muscle actin. In: Methods in cell biology Methods Cell Biol 24:271–289.
Potter JD (1982) Preparation of troponin and its subnits. Methods Enzymol 85:241–263.
Matyushenko AM, Artemova NV, Shchepkin DV, Kopylova GV, Bershitsky SY, Tsaturyan AK, Sluchanko NN, Levitsky DI (2014) Structural and functional effects of two stabilizing substitutions, D137L and G126R, in the middle part of α-tropomyosin molecule. FEBS J 281:2004–2016. https://doi.org/10.1111/febs.12756
Gordon AM, LaMadrid MA, Chen Y, Luo Z, Chase PB (1997) Calcium regulation of skeletal muscle thin filament motility in vitro. Biophys J 72:1295–1307. https://doi.org/10.1016/S0006-3495(97)78776-9
Spiess M, Steinmetz MO, Mandinova A, Wolpensinger B, Aebi U, Atar D (1999) Isolation, electron microscopic imaging, and 3-D visualization of native cardiac thin myofilaments. J Struct Biol 126:98–104. https://doi.org/10.1006/jsbi.1999.4111
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685.
Mashanov GI, Molloy JE (2007) Automatic detection of single fluorophores in live cells. Biophys J 92:2199–2211. https://doi.org/10.1529/biophysj.106.081117
Haeberle JR, Hemric ME, Chacko S, Pollack J, Reggiani C (1995) Are actin filaments moving under unloaded conditions in the in vitro motility assay? Biophys J 68:306–310.
Reiser PJ, Kline WO (1998) Electrophoretic separation and quantitation of cardiac myosin heavy chain isoforms in eight mammalian species. Am J Physiol Circ Physiol 274:H1048–H1053. https://doi.org/10.1016/S0165-5876(00)00363-3
Silva MA, de Oliveira TF, Almenara CCP, Broseghini-Filho GB, Vassallo DV, Padilha AS, Silveira EA (2015) Exposure to a Low Lead Concentration Impairs Contractile Machinery in Rat Cardiac Muscle. Biol Trace Elem Res 167:280–287. https://doi.org/10.1007/s12011-015-0300-0
Fioresi M, Simões MR, Furieri LB, Broseghini-Filho GB, Vescovi MVA, Stefanon I, Vassallo DV (2014) Chronic lead exposure increases blood pressure and myocardial contractility in rats. PLoS One 2014:1-9. https://doi.org/10.1371/journal.pone.0096900
Reiser PJ, Portman MA, Ning XH, Moravec CS (2001) Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol - Heart Circ Physiol 280:1814–1820. https://doi.org/10.1152/ajpheart.2001.280.4.h1814
Bogaard HJ, Abe K, Noordegmaf AV, Voelkel NF (2009) The right ventricle under pressure. Chest 135:794–804. https://doi.org/10.1378/chest.08-0492
Gupta MP (2007) Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J Mol Cell Cardiol 43:388–403. https://doi.org/10.1038/jid.2014.371
Klinova SV, Minigalieva IA, Privalova LI, Valamina IE, Makeyev OH, Shuman EA, Korotkov AA, Panov VG, Sutunkova MP, Ryabova JV, Bushueva TV, Shtin TN, Gurvich VB, Katsnelson BA (2020) Further verification of some postulates of the combined toxicity theory: New animal experimental data on separate and joint adverse effects of lead and cadmium. Food Chem Toxicol 136:110971. https://doi.org/10.1016/j.fct.2019.110971
Funding
This work was implemented within the Governmental assignment to the Institute of Immunology and Physiology (Ural Branch of the Russian Academy of Sciences, Yekaterinburg); theme reg. Nos. АААА-А19-119070190064-4 and AAAA–A18–118020590135–3 on the institutional equipment of the Center for Collective Use.
Author information
Authors and Affiliations
Contributions
Idea and design of the experiment: L.V.N. and O.P.G.; data collection and processing: O.P.G. and S.R.N.; manuscript writing and editing: O.P.G. and L.V.N.
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have neither evident nor potential conflict of interest associated with the publication of this article.
Additional information
Translated by A. Polyanovsky
Russian Text © The Author(s), 2021, published in Rossiiskii Fiziologicheskii Zhurnal imeni I.M. Sechenova, 2021, Vol. 107, Nos. 6–7, pp. 854–863https://doi.org/10.31857/S0869813921060030.
Rights and permissions
About this article

Cite this article
Gerzen, O.P., Nabiev, S.R. & Nikitina, L.V. Influence of Chronic Lead Intoxication on Functional Characteristics and Isoform Composition of Left Ventricular Myosin in the Rat Heart. J Evol Biochem Phys 57, 896–903 (2021). https://doi.org/10.1134/S002209302104013X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002209302104013X