
Abstract
The influence of the accuracy of mass measurements on the number of possible structural compositions and the computation time of computer-aided interpretation of mass spectrometric data has been evaluated. Experimental measurements have been performed for two model objects in the range of small and medium masses using high, ultrahigh, and extreme high resolution electrospray ionization mass spectrometers. The number of possible solutions have been examined and prospects of using machine learning in combination with mass spectrometry for predicting new data on reaction mechanisms and searching for hidden relationships in the chemical space have been demonstrated. It has been shown that there are two types of relationships between the molecular formula and the mass determination error depending on the ion mass: a nonlinear curve is observed for small molecules and a linear relationship is observed for large molecules.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Today mass spectrometry is one of the most important analytical methods for studying the composition and structure of chemical compounds [1–4]. Mass spectrometry is distinguished by high sensitivity up to 10–18 M for routine measurements [5]. The combination of the versatility of the method and its high sensitivity has made it possible to identify complex mixtures. The most important advantage of mass spectrometric analysis is the possibility to accumulate a large body of data on the object under study at a high rate: from thousands to tens of thousands of individual signals in the spectrum are recorded in a short period of time [6, 7]. Thus, the three key factors—versatility, high sensitivity, and high-throughput acquisition of large amounts of data—make mass spectrometry one of the most approachable experimental methods for the development of machine learning and artificial intelligence algorithms.
Machine learning and artificial intelligence technologies attract more and more attention of researchers worldwide due to the advent of affordable computing cost. It turned out that to solve a complex problem, one can construct a set of initial data–correct solution and then try to select an algorithm that describes the relationship between them as accurately as possible. Sometimes, the number of parameters for these algorithms reaches enormous values: tens of millions of parameters for neural networks for image classification, such as ResNet, Inception, and EfficientNet [8]. Since the rules for making a decision are not directly specified, the algorithms find hidden dependencies in the data that are necessary to solve the problem. Currently, these technologies are becoming more integrated into our lives: face recognition [9], self-driving cars [10], natural language processing (for example, translation of texts) [11].
In recent years, machine learning has been increasingly used by researchers to gain new information on chemical processes and to plan experiments. The development of structural solutions based on machine algorithms for mass spectrometric studies is of undoubted importance. Three important directions can be distinguished: the use of mass spectrometric data as vector representations without intermediate identification of compounds, the determination of the structure of compounds represented in the mass spectra, and the use of mass spectrometry as a method for detecting individual compounds. The first direction involves a number of different algorithms and includes the following examples: classification of mass spectra of a biomaterial by the presence of a specific disease [12, 13] or clustering of mass spectra [14]. Two other directions cause difficulties at present, especially for multicomponent samples of complex composition [15]. Solving the problem of identifying compounds in such samples will significantly speed up chemical research by reducing the time spent on interpreting spectra, and will also open up opportunities for identifying complex and implicit dependencies in large datasets collected for chemical processes, which is a target for machine learning and artificial intelligence [16–18].
RESULTS AND DISCUSSION
For interpretation of the mass spectrum, three tasks should be solved: it is necessary to (1) identify groups of peaks related to one compound; (2) determine the bruto formula of a substance by its exact mass, with taking into account the isotope distribution; and (3), if possible, based on the fragmentation spectra by various activation methods, suggest possible structures or structural fragments.
The quality of the spectral data is of key importance for the unambiguous determination of the molecular formula of a substance, especially in complex spectra with overlapping signals. In particular, if a sample contains a large amount of impurities, then the overlapping of signals may distort the determined masses, as well as the peak intensities, which makes the comparison with the theoretical isotope distribution of a possible candidate much more difficult or impossible in principle. Herein, we carried out a comparative study of model samples of small molecules (palladium chloride (PdCl2), MW = 177.33, a catalyst precursor in fine organic synthesis) and large molecules (lysozyme, MW = 14.3 kDa, antibacterial agent, hydrolase enzyme, which destroys bacterial cell walls by hydrolysis of peptidoglycan) on high, ultrahigh, and extreme high resolution mass spectrometers, and the possibility of obtaining high-quality mass spectrometric data for the development of machine learning and artificial intelligence algorithms was tested.
The first task is solved by combining peaks whose masses differ by a given amount. Figure 1 shows a high-resolution electrospray ionization mass spectrum (ESI-MS) obtained for a palladium chloride solution with a concentration of 1 × 10–6 mol/L in acetonitrile in the negative ion mode, in which most of the peak groups were identified. Despite the simplicity of the object under study (PdCl2, CH3CN), the spectrum contains more than 500 individual lines [19]. At present, there are practically no published works in the literature on the interpretation of the observed signals for many simple salts of this type.

ESI-(–)MS spectra of PdCl2 in CH3CN after signal isotopic deconvolution.
Figure 2 shows a fragment of the mass spectrum of the protonated protein lysozyme with a concentration of 1 × 10–6 mol/L in the mass range m/z 1000–2200. In the course of the work, a comparative analysis of the mass spectra recorded using various mass analyzers was carried out: time-of-flight (resolving power up to ~23 000 for m/z 922 during calibration) and a Fourier transform ion cyclotron resonance (FT-ICR) cell (resolution >100 000) [20]. The effect of resolution on the appearance of the spectrum of the analyzed sample is clearly shown. At high resolution (Fig. 2c; resolving power ~10 000 for m/z 922 during calibration), isotopes are not resolved and a broad peak is observed, while at ultra-high resolution (Fig. 2b; resolving power up to ~26 000 for m/z 922 during calibration), the full width at half maximum (FWHM) is 0.0565, which is a necessary but not sufficient condition for unambiguous interpretation of the mass spectrum and protein identification. At the same time, the FT-ICR spectrum (ultrahigh resolution) allows one to reliably identify the isotopic structure and make a conclusion about the composition of the analyzed protein (Fig. 2a).

ESI-(+)MS spectra of lysozyme (1 × 10–6 mol/L) in the positive ion mode: (a) extreme high resolution, (b) ultrahigh resolution, and (c) high resolution. Calculated m/z values are 1788.986469 (z = 8+), 1590.322115 (z = 9+), 1431.390631 (z = 10+), 1301.355780 (z = 11+), and 1192.993405 (z = 12+). The RMS values are given in ppm.
Table 1 presents the comparison of the resolution, FWHM, accuracy of determining the mass number (mass-to-charge ratio), and isotope distribution in the spectra recorded on different mass spectrometers [21, 22].
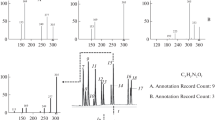
To assess the possible number of options at a given accuracy of mass determination, we carried out exhaustive search for possible values. A clear demonstration of this approach was tested on the spectrum of palladium chloride recorded in the negative ion mode (Fig. 3).

(a) Histogram of relative errors Δ in ppm for generated formulas for Pd2Cl5 (charge is –1, mass number is 386.65230) and (b) the dependence of the number of formulas on the maximum relative error (for each error, the number of formulas is given for which the error does not exceed a specified value).
On the histogram, the number of formulas first increases, and then, on the contrary, decreases (Fig. 3a). This is due to the fact that the difference in mass is getting closer and closer to the mass of one of the elements that was taken into account in the enumeration, namely, of hydrogen. The manifestation of this dependence is observed for the second curve: its right half demonstrates a nonlinear growth, and with a further increase in the error, the growth rate of the number of formulas gradually decreases (Fig. 3b). It can be seen from the performed modeling that, with increasing accuracy (decreasing error ∆, ppm), the number of predicted molecular solutions sharply decreases, tending in the limit to one solution.
In the case of lysozyme, the mass of the protein is large enough; therefore, for a difference of 1 Da, there are a huge number of options for constructing a formula. With an average verification speed of 221 thousand formulas per second (the script is written in the Python, it was run in one thread on an Intel Xeon W-2145 with a frequency of 3.7 GHz), an exhaustive search for lysozyme would take about a thousand years. It is evident that the search optimization can significantly improve the situation, but the search will still take a long time. To obtain data for comparison, the simulation was carried out using the Monte Carlo method. It turned out that the distribution of masses in the interval ±1 Da becomes uniform; i.e., in each sufficiently large section of this interval there is approximately the same number of formulas. As a result, all the columns of the histogram have approximately the same height (Fig. 4a), and the plot for the number of formulas with an accuracy not less than the specified one is just a straight line (Fig. 4b). It can be seen from the performed simulation that, with an increase in accuracy, convergence to one solution is observed, but the plot in Fig. 4b loses its nonlinearity, and the number of variants increases by about six orders of magnitude.

(a) Histogram of relative errors Δ in ppm for generated formulas for lysozyme (charge is +9, mass number is 1589.431144) and (b) the dependence of the number of formulas on the maximum relative error (for each error, the number of formulas is given for which the error does not exceed a specified value).
Comparison of the obtained mass spectra of lysozyme and palladium chloride for different resolutions of the mass spectrometers and the assessment of the use of these data for machine learning have shown that high-resolution mass spectra can provide the correct interpretation of the results on the basis of a precisely determined mass-to-charge ratio by searching for masses less than 1 kDa. Taking into account the isotope distribution of the analyte makes it possible to further improve the quality of ranking of possible formulas by solving the problem of systematic errors. The minimum resolution required to completely separate two neighboring isotope peaks is equivalent to the mass of the detected ion. For example, to resolve the signal of the lysozyme ion (charge is +9, the most abundant mass is 1590.322115), the minimum resolving power of the spectrometer is >15 900 [4]. Further verification of this approach will be carried out using a wide set of mass spectra for the studied class of compounds.
The use of machine learning methods to predict new data on the reaction mechanism and search for hidden dependencies in the chemical space is an important task for a modern scientist. It is proposed to improve the accuracy of the interpretation result by exhaustive search, and, in the case of large masses, to reduce the number of variants by replacing atoms with specific groups (for example, amino acids for proteins). Machine learning algorithms should be used for complex predictive problems that cannot be solved using rules or simple, hand-built models. Experimental estimates and computer simulations carried out in this work have shown that, depending on the ion mass, a different relationship can be observed between the accuracy of determining m/z and the number of possible formulas: a nonlinear S-curve for medium masses and linear dependences for large masses. At the same time, the number of possible formulas begins to grow very quickly in both cases, making the accuracy of measuring the mass-to-charge ratio a key parameter that determines the further use of the spectrum.
EXPERIMENTAL
The following reagents were used: lysozyme enzyme (EC 3.2.1.17, 51 100 units/mg, 14 295.8 Da) purchased from Sigma (Merck-Sigma), palladium(II) chloride (99.9%, ca. 60% Pd) manufactured by ABCR (ABCR GmbH & Co. KG), acetonitrile (LC-MS Grade, J.T. Baker), methanol (LC/MS, LiChrosolv, Merck, Praha), formic acid (LC-MS, Sigma-Aldrich, Germany), and deionized water (milliQ, Merck).
Instrumentation. High-resolution mass spectra were recorded on Bruker micrOTOF (time-of-flight mass analyzer), Bruker maXis QTOF (tandem quarrupole/time-of-flight mass analyzer), and Bruker solariX XR (ICR mass analyzer, a 15-T superconducting magnet) mass spectrometers equipped with an ESI source. The m/z scanning range was 50–3000 for lysozyme and 100–1400 for palladium(II) chloride. The recording time of the mass spectrum on microTOF, maXis, and solariX XR spectrometers was 45 s; in the case of solariX XR, the number of scans was 32 with 4 M data points. External calibration of the mass scale was carried out using a low-concentration calibration solution “Tuning mix” (Agilent Technologies) for microTOF and maXis and a sodium trifluoroacetate solution (0.1 mg/mL in a 1 : 1 acetonitrile/water mixture) for solariX XR. Samples were injected using a 500 μL Hamilton RN 1750 syringe (Switzerland). The measurements were carried out in the positive ion mode (+) for lysozyme (grounded spray needle, 4500-V high-voltage capillary; HV End Plate Offset: –500 V and in the negative ion mode (–) for palladium(II) chloride (grounded spray needle, +4000-V high-voltage capillary; HV End Plate Offset: –500 V). The flow rate during injection was controlled with a syringe pump (3 μL/min). Nitrogen was used as a nebulizer gas (1.0 bar) and dry gas (4.0 L/min, 200°C). To detect a low-concentration lysozyme sample (1 × 10–8 M) on a SolariX XR mass spectrometer, the spectrum was acquired in an ICR cell using 1024 scans and 8 M data points. The data were processed using the Bruker Data Analysis 5.0 software.
Lysozyme samples were prepared by dissolving a weighed portion (1 mg) in an acetonitrile/water (50/50) mixture with the addition of 1 μL of formic acid. The concentration range was from 1 × 10–8 to 1 × 10–4 M. Mass spectra of palladium(II) chloride samples for mass spectrometry were prepared by dissolving a 1-mg portion in 1 mL of acetonitrile followed by dilution to 1 × 10–6 M.
Automatic analysis of mass spectra. All software was written in Python using the NumPy and Pandas data analysis libraries. The plots were built using the Matplotlib library. The spectra were converted to mzXML format and then processed using the pyOpenMS library.
Determination of the number of formulas for masses less than 1000 Da was carried out using an exhaustive search for possible structures. For large masses, the number of possible formulas was estimated using the Monte Carlo method due to the large number of calculations and the impossibility of exhaustive search in a reasonable time. In calculations by the Monte Carlo method, 5 × 109 formulas were randomly selected, for which a comparison with the experimental mass and the corresponding scaling were performed.
For a metal salt, the presence of the following elements (components of the salt itself, possible ligands, and solvent molecules) was assumed: palladium, chlorine, hydrogen, carbon, nitrogen, and oxygen. The elemental composition of lysozyme included hydrogen, carbon, oxygen, nitrogen, and sulfur.
REFERENCES
Dass, C., Fundamentals of Contemporary Mass Spectrometry, New Jersey: Wiley, 2007. https://doi.org/10.1002/0470118490
Kanawati, B. and Schmitt-Kopplin, P., Fundamentals and Applications of Fourier Transform Mass Spectrometry, Amsterdam: Elsevier, 2019. https://doi.org/10.1016/C2016-0-04234-1
Lebedev, A.T., Mass-spektrometriya v organicheskoi khimii (Mass Specrometry in Organic Chemistry), Moscow: Technosphere, 2015.
de Hoffmann, E. and Stroobant, V., Mass Spectrometry: Principles and Applications, 3rd ed., Chichester: Wiley, 2007.
Tsedilin, A.M., Fakhrutdinov, A.N., Eremin, D.B., et al., Mendeleev Commun. 2015, vol. 25, no. 6, pp. 454–456. https://doi.org/10.1016/j.mencom.2015.11.019
Marshall, A.G. and Rodgers, R.P., Acc. Chem. Res., 2004, vol. 37, pp. 53–59. https://doi.org/10.1021/ar020177t
Rodgers, R.P. and McKenna, A.M., Anal. Chem., 2011, vol. 83, no. 12, pp. 4665–4687. https://doi.org/10.1021/ac201080e
Tan, M. and Le, Q.V., Abstracts of Papers, 36th Int. Conf. Mach. Learn. ICML 2019, June 10–15,2019, Long Beach, California, pp. 10691–10700.
Schroff, F., Kalenichenko, D., and Philbin, J., Proc. IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recognit., June 7–12,2015, Boston, pp. 815–823. https://doi.org/10.1109/CVPR.2015.7298682
Bojarski, M., Del Testa, D., Dworakowski, D., et al., arXiv: 1604.07316, 2016, pp. 1–9. http://arxiv.org/abs/1604.07316
Lample, G. and Conneau, A.,arXiv: 1901.07291, 2019. http://arxiv.org/abs/1901.07291
Behrmann, J., Etmann, C., Boskamp, T., et al., Bioinformatics, 2018, vol. 34, no. 7, pp. 1215–1223. https://doi.org/10.1093/bioinformatics/btx724
Huang, Y.-C., Chung, H.-H., Dutkiewicz, P.E., et al., Anal Chem., 2020, vol. 92, no. 2, pp. 1653–1657. https://doi.org/10.1021/acs.analchem.9b03966
Palmer, D.A., Bunch, J., and Styles, I.B., J. Am. Soc. Mass. Spectrom. 2015, vol. 26, no. 2, pp. 315–322. https://doi.org/10.1021/jasms.8b04957
Marshall, A.G., Blakney, G.T., Chen, T., et al., Mass Spectrom. 2013, vol. 2 (Special_Issue), no. S0009. https://doi.org/10.5702/massspectrometry.s0009
Saeys, Y., Inza, I., and Larrañaga, P., Bioinformatics, 2007, vol. 23, no. 19, pp. 2507–2517. https://doi.org/10.1093/bioinformatics/btm344
Granda, J.M., Donina, L., Dragone, V., Long, D.-L., and Cronin, L., Nature, 2018, vol. 559, no. 7714, pp. 377–381. https://doi.org/10.1038/s41586-018-0307-8
Ahneman, D.T., Estrada, J.G., Lin, S., Dreher, S.D., and Doyle, A.G., Science, 2018, vol. 360, no. 6385, pp. 186–190. https://doi.org/10.1126/science.aar5169
Šádek, V., Schröder, D., and Tsierkezos, N.G., Int. J. Mass. Spectrom., 2011, vol. 304, no. 1, pp. 9–14. https://doi.org/10.1016/J.IJMS.2011.02.014
Marshall, A.G., Hendrickson, C.L., and Jackson, G.S., Mass. Spectrom. Rev., 1998, vol. 17, no. 1, pp. 1–35. https://doi.org/10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K
Brenton, A.G. and Godfrey, A.R., J. Am. Soc. Mass. Spectrom., 2010, vol. 21, no. 11, pp. 1821–1835. https://doi.org/10.1016/j.jasms.2010.06.006
Wells, G., Prest, H., and Russ, C.W. IV, Curr. Top.Mass.Spectrom., 2011, pp. 28–33. http://spectroscopyonline.findanalytichem.com/spectroscopy/Articles/Why-Use-Signal-To-Noise-As-a-Measure-of-MS-Perform/ArticleStandard/Article/detail/725238.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by G. Kirakosyan
Rights and permissions
About this article

Cite this article
Burykina, J.V., Boiko, D.A., Ilyushenkova, V.V. et al. Comprehensive Mass Spectrometric Mapping of Chemical Compounds for the Development of Algorithms for Machine Learning and Artificial Intelligence. Dokl Phys Chem 492, 51–56 (2020). https://doi.org/10.1134/S0012501620050024
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0012501620050024