
Abstract
Potential mechanisms for the realization of the neuroprotective action of nicotinamide during acute lethal short-term hypoxia were evaluated based on current knowledge about its effect on metabolic processes. Attention was drawn to the role of mitochondrial dysfunction and excitotoxicity followed by axonal degeneration and apoptosis of neurocytes and neuroglia during the development of an inflammatory response in trauma and cerebral ischemia. A decrease in the level of ATP in the cells during hypoxia affects the generation of the mitochondrial membrane potential, promotes an increase in membrane permeability, the release of NAD from mitochondria, the entry of sodium into the cell, and the development of intracellular edema. Activation of poly (ADP-ribose)-polymerase 1, induced by DNA damage during reoxygenation, reduces the level of NAD in the cell as a substrate for its reaction and causes dysfunction of the respiratory mitochondrial complex. High doses of nicotinamide have neuroprotective properties in traumatic brain injury, ischemia and stroke, as well as in neurodegenerative Alzheimer’s, Parkinson’s and Huntington’s diseases. It is generally accepted that the neuroprotective effect of nicotinamide, firstly, is because, being a substrate for the synthesis of nicotinamide mononucleotide and NAD+, it can maintain and prevent the NAD+ content decrease under conditions of acute hypoxia. Secondly, nicotinamide, being a blocker of poly (ADP-ribose) polymerase 1, can provide the required level of NAD+ in the cell and reduce its use in the reactions with this polymerase. Thirdly, nicotinamide is a substrate for NAD+ synthesis and maintains the NAD+/NADH complex, which is important for the functioning of the antioxidant system of the mitochondrial respiratory chain under conditions of acute hypoxia. It is doubtful that these mechanisms are sufficient to implement the action of nicotinamide for the reason that there was no decrease in the level of NAD in mitochondria during the death of animals under conditions of short-term lethal hypoxia. A variant of the mechanism of nicotinamide action through GABA/benzodiazepine receptors, which causes inhibition of the activation of neurocyte glutamate receptors during acute hypoxia, was considered. In hypoxia, there is an excessive hyperactivation of glutamate receptors and the development of acute cellular hypoxia, which leads to cell death from overexcitation (excitotoxicity). Nicotinamide reduces the death of neurocytes from excitotoxicity by acting on benzodiazepine receptors. GABA agonists prevent the effect of glutamate during excitotoxicity in this way.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Nicotinamide and nicotinic acid are B vitamins, which are important for the functioning of the central and peripheral nervous system [1]. Nicotinamide at high doses has neuroprotective properties in traumatic brain injury, ischemia, and stroke, as well as in neurodegenerative Alzheimer’s, Parkinson’s, and Huntington’s diseases [2]. Nicotinamide and nicotinic acid are used as vitamin B3 at a dose of 20 mg; for medicinal purposes the vitamin is used at higher doses (0.5–5.0 g) [3]. Possible indirect ways of realizing the action of nicotinamide have been proposed, which reduce the manifestations of the secondary cascade of effects of tissue damage to the brain (inflammation, generation of free radicals, death of neurocytes from excitotoxicity, and others) [4]. However, due to the multi-vector effect of nicotinamide in the body, there is no complete clarity on the mechanism of its neuroprotective properties, considering specific pathology scenarios and pharmacodynamics for various manifestations of the pharmacological action of the drug. This brief review is devoted to the analysis of potential mechanisms for the implementation of the neuroprotective properties of nicotinamide in conditions of acute short-term lethal hypoxia.
PATHOPHYSIOLOGICAL FEATURES OF ACUTE BRAIN HYPOXIA
The general picture of brain tissue damage due to acute hypoxia is characterized by mitochondrial dysfunction, overexcitation of neurocytes (excitotoxicity), generation of reductive and oxidative stress followed by the development of an inflammatory response, axonal degeneration, and cell death [5–7]. A characteristic feature of the brain is its high energy consumption, which is ten times higher than the energy consumption of other tissues. A person loses consciousness within 15–45 s in the absence of oxygen supply to the brain [5, 8]. For the same reason, death of the organism may occur in various diseases due to energetic stoppage of the functioning of the brain.
The first main sign of the manifestation of acute tissue hypoxia is a sharp decrease in the level of ATP in the cells; this is followed by decrease in the activity of K/Na-ATPase in the next few minutes and violation of the membrane ion gradients with excessive entry into the cell of sodium and exit from it of potassium and glutamate ions. An increase in membrane permeability affects the generation of the mitochondrial membrane potential, promotes the exit of NAD+ and potassium ions from mitochondria, and, as a result, leads to excessive intake of calcium ions into the cell and mitochondria. These processes are accompanied by an acceleration of the electron flow, an increase in free radical activity, and the development of cytotoxic edema [5, 9, 10].
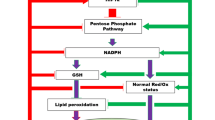
The lack of oxygen leads to difficulties in recycling of the NADH/NAD+ pair in the Krebs cycle, without which ATP generation is impossible. The level of reduced NADH and other reducing equivalents in mitochondria increases with the development of reductive stress. Adaptive processes that support electron transport through complex II of the respiratory chain via the ketoglutarate dehydrogenase complex with the participation of succinyl-CoA can provide oxidative phosphorylation under moderate hypoxia [11–13]. A decrease in the oxidation of sulfides involved in electron transport in the respiratory chain leads to their accumulation. An excess of sulfides during the prolongation of the hypoxic state promotes the activation of the production of NO and reactive oxygen species (ROS), which, together with sulfides, cause the uncoupling of oxidative phosphorylation and suppression of cellular respiration [14]. Oxidative stress during reoxygenation reduces the level of reduced glutathione, the main antioxidant in the cell, and suppresses the antioxidant system of mitochondria, followed by hyperoxidation of NAD+ and a decrease in its content in the tissue [15].
The resulting peroxide products cause, among other things, DNA damage. Degradation of DNA in cells leads to the activation of its repair through poly ADP-ribosylation, which entails a decrease in the level of NAD+ in the cell as a substrate for this reaction and, as a result, an increase in the content of nicotinamide as a breakdown product of NAD+. Accumulation of nicotinamide in the cell by the feedback mechanism can hinder the course of the reaction [8, 16–19]. Another mechanism of activation of poly (ADP-ribose)-polymerase 1 (PARP-1) is carried out in the body through the inositol (1,4,5)-triphosphate/calcium pathway of alpha1-adrenoagonists, primarily under the action of endogenous norepinephrine [20]. Thus, the manifestation of stress during acute cerebral hypoxia provokes the processes of poly ADP-ribosylation.
NADH and NADPH are key regulators of redox reactions and the state of the antioxidant system of tissues. Therefore, a decrease in the level of NAD+ in mitochondria leads to an imbalance in the antioxidant system, followed by an increase in ROS production and membrane depolarization, which leads to apoptosis or cell necrosis. A decrease in the content of NAD+ in the cytosol affects the activity of the sirtuin family, which is important for the prevention of excitotoxicity [21].
PARP-1, in addition to its important role in DNA repair, is a cofactor in the activation of the nuclear factor NF-kB and the family of pro-inflammatory cytokines associated with it; that is, it enhances inflammatory processes in cerebral ischemia and promotes degenerative processes in cerebral stroke [22]. Activation of PARP-1 in response to DNA damage leads to the suppression of mitochondrial function, and a decrease in the content of glutathione and ATP with increased oxidative stress. PARP activity in the body is limited by poly (ADP-ribose)-glycohydrolase involved in its metabolism, suppression of which intensifies apoptosis in damaged cells [20].
As an urgent adaptive response to a decrease in the ATP level in the cell and an increase in ROS during hypoxia [23], preceding the involvement of HIF-1α, a nuclear factor that triggers the genomic regulation of this process [24], activation of 5'-AMP-activated protein kinase occurs, which suppresses anabolism, reducing the need for ATP, and enhances catabolism in the cell by stimulating beta-oxidation of fatty acids through increased mitochondrial respiration and biogenesis; thereby, it supplies the necessary ATP to the cell [25]. At the same time, 5'-AMP-activated protein kinase activates the PGC-1α-induced antioxidant response to the growth of ROS during hypoxia through sirtuin-1 by increasing the NAD+/NADH ratio in the cell [26, 27]. Sirtuin-1 converts NAD to nicotinamide, which by a feedback mechanism suppresses the activity of sirtuins and enhances NAD synthesis [28].
POTENTIAL MECHANISMS OF THE NEUROPROTECTOR AND THERAPEUTIC EFFECT OF EXOGENOUS NICOTINAMIDE IN ACUTE BRAIN HYPOXIA
The neuroprotective effect of nicotinamide was discovered on a model of cerebral stroke at the turn of the late 1990s and early 2000s in a number of laboratories around the world, independently of each other [29–36]. M.R. Choane et al. [37–40] identified and studied the temporal parameters of the therapeutic effect of nicotinamide in stroke, including with repeated use. Nicotinamide mononucleotide had a similar neuroprotective and therapeutic effect [41–43].
The mechanism of the neuroprotective action of nicotinamide is still not clear. It is generally accepted that its neuroprotective effect is, firstly, due to the fact that, being a substrate for the synthesis of nicotinamide mononucleotide and NAD+, nicotinamide can regulate the NAD+ level in the cell, maintaining and preventing its decrease in hypoxic conditions. Secondly, nicotinamide, is a PARP-1 blocker and can thus also provide the necessary level of NAD+ in the cell and reduce its use in reactions with PARP. Thirdly, nicotinamide is a substrate for NAD+ synthesis and is important for the functioning of the respiratory mitochondrial chain under conditions of acute hypoxia [43]. Nicotinamide can suppress the generation of ROS involving NAD+ by activation of the antioxidant system through nuclear erythroid factor 2, which is responsible for the expression of antioxidant genes. Nuclear erythroid factor 2 limits oxidative stress by superoxide detoxification in a feedback mechanism. At the same time, this factor regulates the intensity of the inflammatory process by the blockade of the NF-kB pathway and the production of pro-inflammatory cytokines. It has been established that NAD+ increases the level of glutathione in cells through activation of the regulator of the antioxidant system, nuclear erythroid factor 2 [44]. This is also achieved by stimulating sirtuins, primarily sirtuin-1, which prevents mitochondrial depolarization and fragmentation, as well as subsequent caspase activation. Suppression of apoptosis by nicotinamide can also be achieved through the mTOR pathway. Nicotinamide reduces mitochondrial stress through the activation of 5'-AMP-activated protein kinase and stimulates mitochondrial biogenesis in the same way [43, 44]. Nicotinamide supports the functioning of mitochondria through these mechanisms by stimulating the antioxidant system of cells and reducing the generation of ROS [43, 45–47]. In this case, the effect of nicotinamide on the antioxidant system is also carried out through NAD+.
Nevertheless, there are a number of facts that require clarification with the established logic of substantiating the mechanism of action of nicotinamide. Firstly, nicotinamide is able to increase the content of NAD+ in the cytosol of the cell, but not in the mitochondria. Secondly, during short-term acute hypoxia, the level of NAD does not decrease in the cell, and only during subsequent reoxygenation with PARP-1 activation, which is necessary for the repair of damaged DNA, does it inevitable decrease occurs, when exogenous nicotinamide can realize its action of blocking PARP-1 or the synthesis of NAD+ [8]. Therefore, it is not clear by what mechanism nicotinamide can reduce animal mortality during exposure to short-term acute hypoxia, when there is a manifestation of reductive stress and the preservation of the level of NAD in the cell, and the pathophysiological changes noted above during reoxygenation, which could be influenced by nicotinamide, do not yet have time to fully manifest themselves through the support of the NAD function [48].
We can consider another possibility of the direct action of nicotinamide through GABA/benzodiazepine receptors, the stimulation of which leads to the suppression of hyperactivation of neurocyte glutamate receptors observed during acute hypoxia with a decrease in excitotoxicity and cell death. This phenomenon, specific for nervous tissue, is caused by excessive release of the neurotransmitter glutamate from neurocytes and neuroglia under hypoxic conditions, followed by hyperactivation of glutamate receptors, which enhances the entry of calcium ions into neurocytes with an excessive increase in all calcium-related processes in the cell, including the cascade of degenerative enzymes and cellular anoxia, leading to the death of neurocytes through the development of apoptosis or necrosis [49, 50]. The functioning of the inhibitory GABAergic system is violated during acute hypoxia, which impairs the excitation/inhibition balance in the brain and provokes excitotoxicity [51, 52]. The death of animals during acute hypoxia is accompanied by the manifestation of tonic-clonic seizures, which are associated with hyperactivation of CA3 pyramidal neurons in the hippocampus [52, 53]. Nicotinamide inhibits the overexcitation of neurons, acting through benzodiazepine receptors, which leads to a decrease in cell death from excitotoxicity [54–56]. GABA agonists realize part of their neuroprotective properties in this way by preventing excessive activation of glutamate receptors and the development of excitotoxicity [57]. Nicotinamide prevents the decrease in the NAD content in the cell in the second phase of the development of the consequences of acute hypoxia.
CONCLUSIONS
Potential mechanisms for the realization of the neuroprotective action of nicotinamide during acute lethal short-term hypoxia were evaluated on the basis of modern knowledge about its effect on metabolic processes. The pathophysiological regularities of the development of acute hypoxia of the brain were considered. Attention was drawn to the role of oxidative stress, mitochondrial dysfunction, and excitotoxicity, followed by axonal degeneration and apoptosis of neurocytes and neuroglia in the pathogenesis of an inflammatory response in trauma or cerebral ischemia. The main symptom of tissue hypoxia is a decrease in the level of ATP in the cells, which affects the generation of the mitochondrial membrane potential, and promotes membrane permeability, release of NAD+ from mitochondria, sodium entry into the cell, and the development of intracellular edema. Processes associated with the development of stress and activation of poly ADP-ribosylation during tissue reoxygenation are of particular importance in the pathophysiology of acute cerebral hypoxia. PARP-1 causes dysfunction of the respiratory mitochondrial complex by reducing the level of NAD in the cell; it is a cofactor in the activation of the NF-kB nuclear factor and enhances inflammatory processes in cerebral ischemia and promotes degenerative processes in stroke. Nicotinamide at high doses has neuroprotective properties in traumatic brain injury, ischemia and stroke, as well as in neurodegenerative Alzheimer’s, Parkinson’s and Huntington’s diseases. The first pioneering studies on the discovery of the neuroprotective properties of nicotinamide was noted. It is generally accepted that the neuroprotective effect of nicotinamide, firstly, is due to the fact that, being a substrate for the synthesis of nicotinamide mononucleotide and NAD+, it can regulate the NAD+ level in the cell; thus, it maintains the NAD+ level and prevents its decrease under hypoxic conditions. Secondly, nicotinamide, being a PARP-1 blocker, can also provide the necessary level of NAD+ in the cell by reducing its use in the reactions with PARP. Thirdly, nicotinamide is a substrate for the synthesis of NAD+ and is important for the functioning of the antioxidant system in the mitochondrial respiratory chain under conditions of acute hypoxia. All noted potential mechanisms of the neuroprotective action of nicotinamide are associated with the functioning of NAD, the level of which does not decrease during the development of reductive stress under conditions of short-term lethal hypoxia. This calls into question the role of the mechanisms noted above in the effect of nicotinamide. A variant of the mechanism of the effect of nicotinamide through GABA/benzodiazepine receptors, which causes blocking of the activation of glutamate receptors of neurocytes during acute hypoxia by stabilizing the plasma membrane potential, which leads to a decrease in the death of neurocytes from excitotoxicity, was considered. This phenomenon is due to excessive release of glutamate during hypoxia and hyperactivation under its influence of tissue respiration of neurocytes with the development of acute cellular hypoxia, which leads to cell death. This occurs when the efficiency of the inhibitory GABAergic system of the brain is decreased, which is noted during ischemia of various origins. The death of animals occurs during the development of tonic-clonic seizures provoked by hyperactivity of pyramidal neurocytes of the hippocampus, where GABAergic receptors are located, when excitotoxicity occurs during acute hypoxia. GABA agonists prevent excessive activation of glutamate receptors and the development of excitotoxicity; thus, they realize their neuroprotective properties. Nicotinamide prevents the decrease in the NAD content in the cell in the second phase of the development of the consequences of acute hypoxia.
REFERENCES
R. Moretti and C. Peinkhofer, Int. J. Mol. Sci. 20, 5797 (2019). https://doi.org/10.3390/ijms20225797
R. A. Fricker, E. L. Green, S. I. Jenkins, and S. M. Griffin, Int. J. Tryptophan Res. 11, 1178646918776658 (2018). https://doi.org/10.1177/1178646918776658
E. S. Hwang and S. B. Song, Biomolecules 10, 687 (2020).https://doi.org/10.3390/biom10050687
J. R. Tribble, A. Otmani, S. Sun, et al., Redox Biol. 43, 101988 (2021). https://doi.org/10.1016/j.redox.2021.101988
T. A. Voronina, Rev. Clin. Pharmacol. Drug Ther. 14, 63 (2016). https://doi.org/10.17816/RCF14163-70
M. E. Watts, R. Pocock, and C. Claudianos, Front. Mol. Neurosci. 11, 216 (2018). https://doi.org/10.3389/fnmol.2018.00216
S. Y. Ng and A. Y. W. Lee, Front. Cell. Neurosci. 13, 528 (2019). https://doi.org/10.3389/fncel.2019.00528
G. Qi, Y. Mi, and F. Yin, Front. Physiol. 10, 1531 (2020). https://doi.org/10.3389/fphys.2019.01531
M. S. Sekhon, P. N. Ainslie, and D. E. Griesdale, Crit. Care 21, 90 (2017). https://doi.org/10.1186/s13054-017-1670-9
K. Shetty, F. Galeffi, and D. A. Turner, Neurobiol. Dis. 62, 469 (2014). https://doi.org/10.1016/j.nbd.2013.10.025
C. Chinopoulos, Exp. Neurol. 327, 113218 (2020). https://doi.org/10.1016/j.expneurol.2020.113218
M. V. Vasin, I. B. Ushakov, and I. V. Bukhtiyarov, Biol. Bull. 45, 73 (2018). https://doi.org/10.1134/S1062359017060115
P. Belenguer, J. M. N. Duarte, P. F. Schuck, and G. C. Ferreira, Neurotoxic. Res. 36, 219 (2019). https://doi.org/10.1007/s12640-019-00061-7
E. Marutani, M. Morita, S. Hirai, et al., Nat. Commun. 12, 3108 (2021). https://doi.org/10.1038/s41467-021-23363-x
N. Klimova, A. Fearnow, and T. Kristian, Brain Sci. 10, 449 (2020). https://doi.org/10.3390/brainsci10070449
O. P. Mishra, W. Akhter, Q. M. Ashraf, and M. Delivoria-Papadopoulos, Neuroscience 119, 1023 (2003). https://doi.org/10.1016/s0306-4522(03)00166-0
P. Jagtap and C. Szabo, Nat. Rev. Drug Discovery 4, 421 (2005). https://doi.org/10.1038/nrd1718
R. P. Strosznajder, K. Czubowicz, H. Jesko, and J. B. Strosznajder, Mol. Neurobiol. 41, 187 (2010). https://doi.org/10.1007/s12035-010-8124-6
K. Erdélyi, P. Pacher, L. Virag, and C. Szabo, Int. J. Mol. Med. 32, 339 (2013). https://doi.org/10.3892/ijmm.2013.1397
S. Tanuma, A. Sato, T. Oyama, et al., Curr. Protein Pept. Sci. 17, 668 (2016). https://doi.org/10.2174/1389203717666160419150014
W. Ying, Antioxid. Redox Signal. 10, 179 (2008). https://doi.org/10.1089/ars.2007.1672
T. Neira-Pena, E. Rojas-Mancilla, V. Munoz-Vio, et al., Neurotoxic. Res. 27, 453 (2015). https://doi.org/10.1007/s12640-015-9517-0
B. M. Emerling, F. Weinberg, and C. Snyder. Free Radic. Biol. Med. 46, 1386 (2009).
J. M. Marti, A. Garcia-Diaz, and D. Delgado-Bellido, Redox Biol. 41, 101885 (2021). https://doi.org/10.1016/j.redox.2021.101885
E. Dengler, Int. J. Mol. Sci. 21 (7), 2428 (2020). https://doi.org/10.3390/ijms21072428
R. C. Rabinovitch, B. Samborska, B. Faubert, et al. Cell Rep. 21, 1 (2017). https://doi.org/10.1016/j.celrep.2017.09.026
C. Cantó, Z. Gerhart-Hines, J. N. Feige, et al., Nature 458 (7241), 1056 (2009). https://doi.org/10.1038/nature07813
J. Brandauer, S. G. Vienberg, M. A. Andersen, et al. J. Physiol. 591, 5207 (2013). https://doi.org/10.1113/jphysiol.2013.259515
A. Y. Sun and D. S. Cheng, Zhongguo Yaoli Xuebao 19, 104 (1998).
M. V. Vasin, T. V. Ryasina, and Yu. N. Chernov, Tsitologiya 41, 812 (1999).
T. V. Ryasina, M. V. Vasin, L. D. Smirnov, et al., Usp. Gerontol., No. 6, 67 (2001).
J. Yang, L. K. Klaidman, A. Nalbandian, et al., Neurosci. Lett. 333, 91 (2002). https://doi.org/10.1016/s0304-3940(02)01005-4
J. Yang, L. K. Klaidman, M. L. Chang, et al., Pharmacol., Biochem. Behav. 73, 901 (2002). https://doi.org/10.1016/s0091-3057(02)00939-5
M. L. Chang, J. Yang, S. Kem, et al., Neurosci. Lett. 322, 137 (2002). https://doi.org/10.1016/s0304-3940(01)02520-4
L. Klaidman, M. Morales, S. Kem, et al., J. Pharmacol. 69, 150 (2003). https://doi.org/10.1159/000072668
M. R. Hoane, S. L. Akstulewicz, and J. J. Toppen, Neurotrauma 20, 1189 (2003). https://doi.org/10.1089/089771503770802871
M. R. Hoane, J. L. Pierce, M. A. Holland, and G. D. Anderson., Neuroscience 154, 861 (2008). https://doi.org/10.1016/j.neuroscience.2008.04.044
M. R. Hoane, J. L. Pierce, N. A. Kaufman, and J. E. Beare, Oxid. Med. Cell Longevity 1, 46 (2008). https://doi.org/10.4161/oxim.1.1.6694
A. M. Goffus, G. D. Anderson, and M. R. Hoane, Oxid. Med. Cellul. Longevity 3, 145 (2010). https://doi.org/10.4161/oxim.3.2.11315
C. Vonder Haar, G. D. Anderson, and M. R. Hoane, Behav. Brain Res. 224, 311 (2011). https://doi.org/10.1016/j.bbr.2011.06.009
J. H. Park, A. Long, K. Owens, and T. Kristian, Neurobiol. Dis. 95, 102 (2016). https://doi.org/10.1016/j.nbd.2016.07.018
C. C. Wei, Y. Y. Kong, G. Q. Li, et al., Sci. Rep. 7, 717 (2017). https://doi.org/10.1038/s41598-017-00851-z
N. Klimova, A. Fearnow, A. Long, and T. Kristian, Exp. Neurol. 325, 113144 (2020). https://doi.org/10.1016/j.expneurol.2019.113144
J. Zhang, Y. Hong, W. Cao, et al., Front. Mol. Neurosci. 12, 108 (2019). https://doi.org/10.3389/nmol.2019.00108
Y. F. Lai, L. Wang, and W. Y Liu, Eur. Rev. Med. Pharmacol. Sci. 23, 1797 (2019). https://doi.org/10.26355/eurrev_201902_17143
K. Maiese, Curr. Neurovasc. Res. 17, 765 (2020). https://doi.org/10.2174/1567202617999201111195232
N. Klimova, A. Long, and T. Kristian, J. Neurosci. Res. 97, 975 (2019). https://doi.org/10.1002/jnr.24397
Yu. N. Chernov, M. V. Vasin, and I. B. Ushakov, Eksp. Klin. Farmakol. 84, 32 (2021). https://doi.org/10.30906/0869-2092-2021-84-3-8-10
D. Belov Kirdajova, J. Kriska, J. Tureckova, and M. Anderova, Front. Cell Neurosci. 14, 51, (2020). https://doi.org/10.3389/fncel.2020.00051
D. W. Choi, Front. Neurosci. 14, 579953 (2020). https://doi.org/10.3389/fnins.2020.579953
C. Chen, X. Zhou, J. He, et al., Oxid. Med. Cell Longevity 2019, 4028394 (2019). https://doi.org/10.1155/2019/4028394
C. Vinnakota, K. Govindpani, W. P. Tate, et al., Int. J. Mol. Sci. 21, 3284 (2020). https://doi.org/10.3390/ijms21093284
H. Song, S. M. Mylvaganam, J. Wang, et al., Front. Cell Neurosci. 12, 278, (2018). https://doi.org/10.3389/fncel.2018.00278
H. Mohler, P. Polc, R. Cumin, et al., Nature 278, 563 (1979).
J. Prousky, J. Orthomol. Med. 19, 104 (2004).
M. Slomka, E. Zieminska, E. Salinska, and W. Lazarewicz, Folia Neuropathol. 46, 69 (2008).
D. Mayor and M. Tymianski, Neuropharmacology 134 (Pt B), 178 (2018)https://doi.org/10.1016/j.neuropharm.2017.11.050
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest. The authors declare that they have no conflicts of interest.
Statement on the welfare of humans or animals. This article does not contain any studies involving animals or human subjects performed by any of the authors.
Additional information
Translated by E. Puchkov
Abbreviations: ROS, reactive oxygen species; PARP-1, poly (ADP-ribose) polymerase 1.
Rights and permissions
About this article

Cite this article
Vasin, M.V., Ilyin, L.A. & Ushakov, I.B. Analysis of the Effect of Exogenous Nicotinamide on Bioenergetic Processes in the Brain During Acute Hypoxia. BIOPHYSICS 67, 637–641 (2022). https://doi.org/10.1134/S0006350922040224
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006350922040224