
Abstract—In the first part of this review (Biophysics, 63, 858 (2018)), the structure of H1 family linker histones, their posttranslational modifications, as well as the role of H1 histone in the formation of compact transcriptionally inactive chromatin, were considered. The second part is devoted to the role of H1 family linker histones in the structural organization of chromatin at different levels: from nucleosomes to metaphase chromosomes. The mechanisms of interaction of H1 histone with other elements of chromatin, including with DNA and nuclear proteins, are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In recent years, a large number of works devoted to the study of the structural organization of chromatin and based on a combination of various molecular biological, biochemical, physical, and mathematical approaches have been published [1–11]. Data on electron and atomic force microscopy [12–14], X-ray analysis [3, 15–18], methods of molecular spectroscopy [19–24], including circular dichroism [25–33], fluorescence spectroscopy [34, 35], methods of bioinformatics [11], etc. are among them. According to modern ideas, DNA–protein and protein–protein interactions play a key role in the structural organization of chromatin. In the first part of the review [36], we addressed issues regarding the structure of linker histones, posttranslational modifications of H1 histone, and their role in the functioning of chromatin. Special attention was paid to the peculiarities of the structure of H1 linker histones in nuclei of transcriptionally inactive cells characterized by a high degree of DNA compaction. In the second part of the review, we made an attempt to reflect the most interesting (from our point of view) results illustrating the role of H1 family linker histones in the formation of different levels of the structural organization of chromatin.
NUCLEOSOME
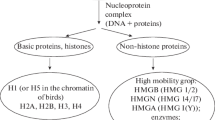
The chromatin structure is characterized by fairly complex multi-level organization, which is provided by various DNA–protein and protein–protein interactions (Fig. 1). The proteins of the histone group, which are characterized by a high content of positively charged amino-acid residues of lysine and arginine, are the most numerous of the nuclear proteins involved in the formation of chromatin structure. The proteins of this type are divided into two functionally different groups: so-called core (H2A, H2B, H3, and H4) and H1 family linker histones (a variant called H5 exists in red blood cells of birds). The interaction of histone proteins with nuclear DNA provides the formation of the first and best-studied level of the structural organization of chromatin, that is, nucleosomes, which are traditionally considered elementary structural units of chromatin.

Levels of the structural organization of chromatin.
Significant progress in the study of the mechanism of DNA packaging in chromatin was made as far back as 1974 after the discovery of the subunit structure of the chromatin fibril. Using the method of electron microscopy it was established that the elementary chromatin fibril in all eukaryotes is a linearly ordered chain of periodically repeating subunits (nucleosomes) that are connected by linker DNA [12]. DNA localization in the nucleosome outside the protein particle was then demonstrated by the neutron diffraction method.
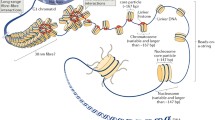
The nucleosome consists of a core that resembles a cylinder with a diameter of 11 nm and a height of 5.7 nm, around which a 146 bp DNA fragment is laid in a spiral (Fig. 1) [17]. DNA on the surface of a protein particle is stabilized by multiple electrostatic interactions and hydrogen bonds [15]. The protein core is formed during the interaction of H2A, H2B, H3, and H4 histones with each other [3, 5, 6, 15, 38] from which the term “core histones” originates. The core histones are small (102–135 aa) highly conserved globular proteins with a molecular weight of 11–16 kDa [3, 15, 38, 39]. The core particle contains two molecules of each of four histones: the central tetramer (H3/H4)2 and two H2A–H2B heterodimers located on the periphery. X-ray analysis data with a resolution of 1.9 Å [40] and 2.8 Å [15] confirmed that the 145–147 bp double-stranded DNA region is wound onto a protein particle with the formation of a left-handed superhelix (80 bp per turn); at the same time, the side groups of amino-acid residues of core histones interact with DNA.
Experimental confirmation of the possibility of structural rearrangements inside the core particle [41–43], which play a key role in the processes of chromatin remodeling, was recently received. Some plasticity of the H2A–H2B dimer is needed for unfolding a DNA molecule during transcription and for the stability of the nucleosome itself. When winding DNA onto the surface of the histone octamer, 121 and 146 base pairs that are part of the nucleosome bind to globular domains of histones. The remaining 25 bp interact with elongated N-terminal regions of core histones that travel to the surface of the nucleosome through turns of DNA approximately every 20 bp [15, 38. 44–47]. The high level of evolutionary conservation of the amino-acid sequences of core histones provides, among other things, efficient self-assembly of nucleosomes, while multiple posttranslational modifications of core histones make this process reversible and controlled [4, 10, 48, 49].
In all eukaryotic cells nucleosomes are interconnected with a short DNA fragment, a so-called “linker” region that forms a “bead-on-a-string” structure [38]. The nucleosome with linker DNA forms a complete nucleosome. Unlike the invariant structure of the nucleosome, which is identical in the chromatin of all eukaryotes, complete nucleosomes in the nuclei of cells of different species can differ significantly, both due to differences in the length of linker DNA and in the structure of H1 histone bound to it. The length of a DNA fragment in the composition of the complete nucleosome can vary in the cells of different tissues from 195–200 bp in active chromatin to 230–270 bp in its transcriptionally inactive regions. Recently, it was found [50] that it is possible to obtain two new intermediate particles when treated with exonuclease III; the authors called them “proto-chromatosomes”; these contain 7 bp linker DNA, respectively, from one or both ends of the nucleosome. According to the authors of [50], these particles can determine the properties of the histone H1 binding site; however, the role of the proto-chromatosome particle in the structural organization of chromatin is not yet clear.
Compared with core histones, the structural and functional peculiarities of the H1 interaction with other chromatin elements, as well as the structural characteristics of this protein in a free state, have been significantly less studied [36]. The interaction of H1 histone with linker DNA at the entrance/exit of the nucleosome [47] leads to stabilization of the nucleosome complex by the “contraction” of adjacent nucleosomes [51, 52] and contributes to the formation of higher (supranucleosomal) levels of the structural organization of chromatin, such as the 30-nanometer fibril [40].
THE POST-NUCLEOSOME LEVEL OF THE STRUCTURAL ORGANIZATION OF CHROMATIN
The following level of structural organization of chromatin (the formation of the 30-nanometer fibril) provides an additional decrease in linear DNA sizes by 40–50 times. The formation of a 30-nanometer fibril occurs in two stages: the formation of a nucleosome chain with a diameter of 10 nm (10-nanometer fibril) due to their contraction by H1 histone [6, 7] and subsequent folding of this 10-nanometer fibril to more compact supernucleosome structures with a diameter 30 nm [2, 5, 6, 53]. At the same time, the processes of fibril formation are reversible, and in fact chromatin is a system, which is in dynamic equilibrium between an “open” “bead-on-a-string” type conformation and a compact 30-nanometer fibril [54]. Different variants (subtypes) of histones, their posttranslational modifications, as well as different non-histone chromatin proteins, have a significant effect on the formation of these proteins. The chromatosome, which contains a histone octamer and a single H1 histone molecule bound with 165 DNA nucleotide pairs, is the fundamental structural unit of the 30-nanometer fibril [55].
Despite the fact that the structure of the nucleosome was solved years ago, the question of chromatosome structure is still open. In 2015, the first crystal structure of a nucleosome complex with the globular domain of the H5 histone and 167 bp DNA [56] was obtained with near-atomic resolution, which clearly demonstrates H1 histone binding to DNA at the entrance/exit from the nucleosome. The interaction of the linker histone with the nucleosome occurs due to the binding of Lys and Arg of the protein globular domain with phosphate groups of the main DNA chain. At the same time, DNA remains in the B-form [25, 56]. In addition, five key Ile amino-acid residues, which are responsible for the binding of this region with the nucleosome, are located in the globular domain of the H5 histone [57]. The use of the nuclear magnetic resonance method and cryoelectron microscopy allowed the determination of the fact that the terminal domains of H1 histone are not involved in direct interaction with the nucleosome [57, 58].
In recent years, such methods as numerical simulation and small angle X-ray and neutron scattering (SAXS and SANS) are also successfully used for the study of the nucleosome structure. The small angle scattering method is a powerful instrument for studying the spatial structure of chromatin and mitotic chromosomes, which allows the detection of periodic structures in biological samples and solutions. The use of new tool made it possible to question the hypothesis about the presence of stable levels of the structural organization of chromatin of a higher order than the 30-nanometer fibril. Using this method, it was recently demonstrated that the chromatin in interphase nuclei and mitotic chromosomes mainly consists of irregularly folded fibers without a strict structure of the 30-nanometer fibril [59]. Thus, the authors concluded that the compaction and irregular folding of the chromatin fibril in interphase and mitotic chromatin can occur without the formation of structures with a diameter of 30 nm. These issues were discussed in recently published reviews on the chromatin structure (for example, [1, 2, 9, 11, 60–62].
At present, two types of models of the secondary chromatin structure are considered in the literature [13]: a solenoid model (the “one-start” helix) and zigzag model (the “two-start” helix). In the first case, nucleosomes in the fibril interact with the fifth and sixth adjacent nucleosomes, while linker DNA bends, forming a helix [62, 64]. In the zigzag model, the nucleosomes are located in the form of a zigzag so that linker DNA region remains straight [2].
The length of the linker DNA, which varies from 20 bp in yeast to 35–50 bp in somatic cells of vertebrates [2, 5] and to 70–100 bp in echinoderm sperm [5, 6], plays a primary role in the formation of the 30-nanometer chromatin fibril. Variation in the length of linker DNA provides the diversity of chromatin structure in cells of various types.
Comparative analysis of chromatin in transcriptionally inactive and active cells indicates that the presence of arginine-enriched linker proteins and the large length of linker DNA provide dense packing of nucleosomes and a supercompact state of chromatin in transcriptionally inactive cells. In contrast, short linker DNA is responsible for the less compact location of DNA in the nucleosome chain, which supports the chromatin of neurons in a state capable of transcription. Thus, the length of linker DNA is an important parameter that significantly affects higher levels of the organization and functional activity of chromatin. Variation in the length of linker DNA provides the diversity of the chromatin structure in cells of various types.
There is still no clear idea about the structure of higher levels of chromatin organization. The role of the 30-nanometer chromatin fibril in the nucleus is not limited to DNA compaction. It can also modulate the accessibility of specific DNA sequences for regulatory factors. It is considered that the 30-nanometer fibril is organized in loop domains (50–150 kb) that are fixed on the nuclear matrix. At the same time, each domain is a cluster of genes that are functionally linked between themselves. The nature of such domains is not yet completely clear; however, they are apparently built on the principle of a superhelix of higher order.
PECULIARITIES OF THE INTERACTION OF H1 LINKER HISTONE WITH DNA
The interaction of DNA with H1 family histones occurs first of all between the negatively charged sugar phosphate backbone and the positively charged amino-acid residues of the protein [19, 20, 65]. At the same time, interaction of the globular domain of the H1 histone and DNA bases in the major groove has been observed in some experiments [14, 20–22, 24]. It has been demonstrated that H1 histone binding leads to some violation of the conformation of the DNA double helix [25, 26, 28, 32]. However, there is no direct structural data to characterize H1 histone complexes with linker DNA. Indirect structurally sensitive methods (such as circular dichroism in the ultraviolet region) are not informative when studying supramolecular structures due to the high level of scattering of the solutions of such complexes, which is manifested in the form of the so-called ψ-type (polymer and salt induced, PSI) spectra of DNA circular dichroism [14, 19–24, 66]. Such a state of DNA occurs when binding not only to lysine-rich H1 histones, but also to cationic polypeptides and some other nuclear proteins [5, 14, 24–26, 28–30, 32, 67]. In the case of the interaction of DNA with nuclear proteins, the emergence of ψ-type spectra of DNA in circular dichroism is caused by the formation of supramolecular structures and the effects of polarized light scattering on large condensed particles. At the same time, experiments on X-ray scattering demonstrated that DNA in this cases remains in the B-form [25, 68, 69].
The H1 and H5 histones are able to interact selectively and with high affinity with DNA regions that already have structural violations: various cross structures [70], Holliday junction type structures [71], DNA binding sites with drugs [72, 73], etc. The degree of preference of H1 histone to binding to nucleosome DNA is higher compared with its affinity to free DNA, according to various sources, from two to ten [74] to 150 [1] times. Such significant variation can be explained by the fact that different authors used DNA regions with different lengths as a model binding target in their studies. Thus, the affinity of H1 to 4H/4WJ (four-way junction) type DNA cross structures is approximately 100 times higher than to linear DNA [1].
The positively charged C-terminal region of H1 histone plays an important role in the interactions with DNA [1, 75]. As noted previously, the main functional differences between H1 protein subtypes, according to a number of authors, are caused by variability of the primary structure of their C-terminal fragment. This was confirmed based on the example of the interaction of DNA with sperm specific H1 family histones [26–28, 32, 67]. The fact that the C-terminal region also persists in proteins related to H1 histone that lack the canonical globular domain, appears to be important [81]. A large number of lysine residues fairly evenly distributed along the polypeptide chain is a characteristic of the C-terminal fragment [81, 82]; this contributes to the stabilization of DNA–H1 complexes in the chromatin. According to the literature data, H1 histone devoid of its N-terminal sequence condenses DNA in the same way as in the intact molecule [83]. This is an indirect confirmation of the fact that it is the C-terminal H1 region that is actively involved in chromatin condensation [36]. This is the protein fragment that interacts with DNA and is required for strong binding of linker histones to the nucleosome [84, 85]. At the same time, two regions separated by a short linker are found within the C-terminal fragment of the H1 molecule [51, 78, 79]: region I, from the 97th to 121st aa (which is located directly behind the H1 globular domain), and region II, from the 145th to the 169th aa. It is interesting to note that deletion of the 122–144 and 170–196 aa fragments does not lead to changes in the functions of C-terminal domain. In addition, the study of mutant proteins, in which the substitution of amino acids from the 122nd to 144th and from the 170th to 196th amino acids with the 97–121 sequence was performed, demonstrated that the modified protein behaves in the same way as the wild type protein [51, 76].
Recent studies demonstrated that the DNA-binding properties of H1 histone also persist with a change in the order of the amino-acid location in the C-terminal domain [84, 86, 87]. Thus, specific amino-acid residues in this region are the main factors in the regulation of the interaction of the H1 C-terminal domain with linker DNA (that is, a specific set of amino-acid residues that form it, but not any specific amino-acid sequence) [51, 75–77, 84, 85, 88]. This means that the presence of certain amino acids, but not their location, is important for DNA compaction.
In addition to the C-terminal region of H1, the globular domain is also involved in the interaction with DNA. Based on the conducted experiments, several models of binding of linker histones to the nucleosome were suggested (Fig. 2). All of the suggested models can be divided into models of symmetrical and asymmetrical binding. These models differ from each other by the globule position relative to the nucleosome axis of symmetry and the length of DNA region, which is protected by the protein from the effect of micrococcal nuclease. In the symmetrical model [89], the globular domain is located in the center of the nucleosome from the outside, while N- and C-terminal fragments of the protein interact with nucleosome DNA and protect 10 bp from each side of the location of its entrance/exit from the nucleosome (Fig. 2). According to the asymmetrical model [90, 91], DNA is protected at the axis of symmetry by 15 and 5 bp from each side at the location of DNA entrance/exit from the nucleosome. At the same time, the globule is located at a distance 70 bp from the axis of symmetry so that it connects two turns of a DNA helix (Fig. 2a). According to the alternative variant of the asymmetrical model [92, 93], the globular domain is located between DNA and the nucleosome (Fig. 2b). In this model, 20 bp are protected only from one side of the axis of symmetry.

A schematic presentation of the models of the possible location of the globular H1 domain on the nucleosome: symmetric (a) and asymmetric (b, c) models; (1) globular domain of H1 histone; (2) DNA; (3) histone octamer.
Based on the latest structural studies using the methods of cryoelectronic microscopy and nuclear magnetic resonance, it was possible to confirm the existence of chromatin elements that satisfy the asymmetrical model of binding [84]. In addition to previously described variants of the asymmetric binding model, the globular domain of the linker histone, DNA, and core particle are connected in this case by bridges and are also protected asymmetrically by 10 bp DNA (the so-called “off–dyad” model, Fig. 3b). In the globular domain, three protein–DNA binding sites are found, whose exact position has not been determined thus far [84, 94]. However, it was demonstrated that Arg74 is present in each of them [95]. In addition, it was demonstrated that the C-terminal domain of the H2A histone is also involved in the formation of the H5 histone complex with the nucleosome [84].

Binding of the nucleosome DNA to the globular domain of the H5 histone isolated from chicken red blood cells [56]. The figure is presented with the kind consent of the authors: (a) the “on–dyad” model of binding corresponding to more condense state of chromatin; (b) the “off–dyad” model of binding corresponding to a less condensed state of chromatin.
Thus, multiple studies devoted to the interaction of H1 histone with the nucleosome indicate that the existence of different alternative structures of such a complex is possible. In particular, these configurations differ in the position and orientation of the globular domain of linker histone relative to the nucleosome itself. Recently, the interaction of a 165 bp nucleosome DNA with the H5 histone globular domain isolated from chicken red blood cells [56] was described using X-ray analysis and nuclear magnetic resonance with a resolution of 3.5 Å. It should be noted that this model does not contradict the models described previously. The authors demonstrated that the H5 histone globule is located on the axis of the nucleosome and overlaps both arms of linker DNA, demonstrating symmetrical (“on–dyad”) binding (Fig. 3a) [56].
The authors of the models of symmetrical “on-dyad” (Fig. 3a) and asymmetrical “off-dyad” (Fig. 3b) binding suggest that the first type relates to a loose state of chromatin, while the second describes condensed chromatin [56].
The structures of the H1 and H5 histones are very similar (the homology of chicken H5 with mouse H1.0 is ~88%) and it can be expected that the binding of these proteins to DNA will be similar. Despite the fact that terminal regions of linker histone do not play an important role in globule positioning in the chromatosome, the high positive charge of the N-terminal domain can stabilize protein binding with DNA. Most likely, there are several different mechanisms (similar to those described above) by which linker histones form chromatin structures of a higher order.
Despite the fact that both the globular domain of H1 histone and its C-terminal region are responsible for binding to DNA, their functions are completely different. The globular domain is an anchor during the interaction of the histone with linker DNA. This domain has at least two DNA binding sites: the main site is located on the side of the major groove of the DNA superhelix; the second is located on the inside of the minor groove close to the axis of symmetry. The presence of such DNA interaction sites separated in space is clearly manifested upon binding of H1 histone to cross or deformed DNA regions [70–73], as well as upon binding of linker histone at the point of entrance/exit from the nucleosome. S/TPKK motifs (Ser/Thr–Pro–Lys–Lys) found in C-terminal sequence are responsible for the ability of this region to bend DNA during its interaction with H1 and thus modulate the geometry of the DNA double helix in the chromatin. Apparently, the globular domain can be considered as an independent nucleosome-binding domain.
INTERACTION OF THE LINKER H1 HISTONE WITH OTHER PROTEINS
In addition to histones, a large number of diverse non-histone proteins are part of chromatin. The H1 histone is sometimes considered as a naturally disordered protein [36, 69, 82] that is capable of interacting with many diverse partners. The interactions of H1 with other proteins are significant for the formation of higher levels of the structural organization of chromatin [63, 96]. The proteins that interact with the H1 histone can be conditionally divided into several categories. Among them are proteins involved in splicing, in the processes associated with DNA damage recognition, in transcription and translation; and those that act as chaperones for core histones during the assembly/disassembly of the nucleosome particle [77].
A specific binding of H1 with the proteins was demonstrated using different methods. A combination of immunoprecipitation method and mass spectrometry analysis allowed us to detect proteins specifically interacting with H1 [97]. The components of the 40S and 60S ribosomal subunits, as well as some nuclear ribonucleoproteins, are among them. It was demonstrated that the interaction of the histone with these proteins causes the suppression of the transcription of some genes. More than 100 proteins that directly or indirectly interact with H1.0 were detected in the nucleus and nucleolus using mass spectrometry [77]. It was established that H1 histone is a key protein that is required for the formation of the canonical structure of the nucleolus and for its functioning [98]. In addition, H1 can be involved in larger macromolecular protein–protein complexes.
It is well known that the linker H1 histone can be a mediator of transcription [77, 99]. At the same time, it is able to act both as a repressor of transcription of specific genes [96, 97, 100, 101] and as an activator of transcription processes [77, 80, 102, 103]. Recent studies demonstrated that H1 plays an important role in epigenetic processes [104]. As an example, a decrease in the level of H1.2, H1.3, and H1.4 expression in the cells leads to a decrease in the DNA methylation level [62, 104]. According to the literature data, H1.0 binds to a number of proteins that are directly involved in the processes of DNA repair [62, 77]. These include the Ku70 and Ku86 proteins and the vasoline-containing VCP protein. The interaction of H1.0 histone with these proteins leads to strong chromatin compaction and thus contributes to the protection of damaged regions.
The members of a large family of nuclear proteins with high electrophoretic mobility (high mobility group (HMG) proteins) are among the first proteins for which the ability to interact with linker histones was demonstrated. The first group includes HMGN (previously known as HMG14/17) proteins. In forming a heterodimeric complex, these proteins interact with the nucleosome and facilitate transcription; at the same time, they are not a part of the transcription complex [105]. The close proximity of the globular domains of HMGN and H1 contributes to the interaction of linker H1 histone with the proteins of this group in the processes of remodeling chromatin structure [106]. It is possible that there is a competition between HMGN proteins and H1 for the DNA binding site [107]. In particular, it was demonstrated that the competition between H1 histone and HMGN proteins for binding to AT-rich regions affects the chromatin compaction [108]. In actively transcribed genes, the enrichment of some regions with the HMGN protein is accompanied by H1 depletion [109, 110]. The HMGN protein decreases the level of chromatin compaction caused by H1 histone and stimulates transcription [111]. Recently, it was established that the proteins of the HMGN group have a direct effect on the stabilization of highly ordered chromatin structure and on the interaction of terminal regions of histones with DNA [112].
The second group of HMG proteins that interact with linker histones includes the most common and the most studied non-histone proteins of chromatin (HMGB domain proteins, HMGB1/2) [113–115]. HMGB proteins are composed of homologous structurally conserved DNA binding domains (so-called HMGB domains). The TCF and LEF-1 transcription factors [116], SRY and SOX sex-determining proteins [117–120], BAF57 and PB1 chromatin rearrangement factors [121–123], etc. are site-specific single domain HMGB proteins. Among multi-domain proteins of this family, the HMGB1–4 proteins [113–115, 122, 124, 125], mtTF1 and ABF2 mitochondrial factors [122], UBF RNA polymerase I transcription factor [126], etc. can be allocated.
As well as H1 histones, the HMGB1 and HMGB2 proteins interact with the linker DNA region [113, 127] at the point of its entrance/exit from the nucleosome and can act both as competitors and as partners when binding to DNA [128]. The available experimental data indicate that binding of one protein to DNA stimulates binding of the second protein with it [14, 20, 22, 24, 66]. With their simultaneous presence in complex with DNA, the positively charged H1 histone mainly interacts with phosphate DNA groups and the C-terminal HMGB1 domain, screening their negative charges and stabilizing the formed supramolecular DNA–protein complexes [14, 24]. Studies on the structure of the complex formed by DNA with H1 histone in the presence of HMGB1 [14, 24, 66, 130] observed binding of individual proteins both to the sugar phosphate backbone and to DNA bases [14, 19, 66]. Using the method of atomic force microscopy it was demonstrated that the formation of fibril-like structures, each of which is formed by several DNA molecules, is observed in the complex of HMGB1/H1 with DNA [14, 66, 131]. At the same time, the protein molecules bind individual DNA molecules in the fibril with each other. Multiple protein–protein and DNA–protein interactions make these structures quite stable.
The interaction of H1 with HMGB1 (in the absence of DNA) in vitro was demonstrated in other works [130, 132, 133]. The direct interaction of two proteins was also demonstrated by circular dichroism and dynamic light scattering methods [134], as well as fluorescent methods [34, 35]. The interaction of two proteins leads to a change in the structure of at least one of them: the α-helix regions increase, which causes slight changes of the tertiary structure [35, 134]. The possibility of interaction between H1 histone and HMGB1 protein by their C-terminal regions was also demonstrated using small angle X-ray scattering and the nuclear magnetic resonance method [130]. Some works indicate that the result of interaction of two proteins can depend on the redox (reduction/oxidation) state of cysteine residues of the HMGB1 protein [114, 135]. The reduced form of HMGB1 easily displaces H1 histone from DNA, while binding of the protein in the oxidized state noticeably limits the mobility of both proteins in the chromatin [114].
The experimental data indicate that the interaction of H1 histone with other proteins mainly occurs due to its C-terminal region [60, 77, 97, 136]. In recent experiments it was demonstrated that deletion of the C-terminal region leads to the loss of approximately 25% of the H1.0 interactions with other proteins detected in the nucleus [60]. It is believed that in binding to other proteins, H1 histone screens the negative charge of a partner molecule and thus facilitates its interaction with DNA. Recent studies of the H1.0 protein demonstrated the possibility of the involvement of its globular fragment in interaction with other proteins [60]. The authors suggest that the effect of each of three functional H1 domains on its binding to other proteins is responsible for a specific interaction with most of them. The structural disorder of the N- and C-terminal domains can facilitate specific protein–protein interactions. However, in general, questions about the effects of these regions on H1 interaction with different proteins remain open.
The binding of various subtypes of H1 histone with chromatin is dynamic. The length of the C-terminal fragment of the protein is the main factor that determines the nature of this binding: the shortest duration on the nucleosome was detected for H1 with the shortest C-terminal region [87]. The magnitude of the time period when H1 is bound to the nucleosome regulates the degree of chromatin condensation and its availability for other proteins, as well as the possibility of chromatin remodeling processes [137]. In addition, the time of the H1 binding to the nucleosome also affects the possible access of transcription factors and other nuclear proteins to DNA [138]. HMGB proteins are also mobile, the time these proteins stay on the nucleosome (as well as H1 histone) is short [128]. However, H1 is located on the nucleosome much longer compared with all of the members of HMG family proteins [139–141]. Taking the fact into account that the interaction between H1 and HMG occurs in a fairly short time, it is quite difficult to establish what biological significance the specific interaction of HMG proteins with H1 histone has. It is likely that these interactions also have a certain effect on a specific gene or tissue [139, 140]. It has been suggested that a number of biological effects are directly associated with interactions between the H1 and HMG proteins [128]. However, the question of how the binding between specific variants of H1 and HMG proteins affect the structure and biological functions of the genome remains open.
We do not forget posttranslational modifications of H1 histone that can also modulate DNA–protein and protein–protein interactions (for details, see the first part of the review [36]). In addition, reversible changes in the states of histone modifications affect the chromatin structure in general [142]. As an example, methylation of lysine 26 in H1.4 contributes to its interaction with heterochromatin HP1 protein, which leads to the formation of chromatin regions with a reduced level of transcription [143]. Demethylation of lysine 26 in H1.4 contributes to blocking this protein–protein interaction and, consequently, leads to the activation of transcriptional activity, while the simultaneous phosphorylation of adjacent serine residue in 27th position nullifies it [144].
In terms of functional significance, the issues of H1 histone interaction with other proteins remain poorly studied and require further thorough study. Multiple contacts with other chromatin proteins suggest that the functions of H1 histone in the nucleus are much wider than only DNA compaction [60]. We can say with confidence that the multifunctionality of the linker histone is at least partially due to its interaction with many nuclear and nucleolar proteins.
CONCLUSIONS AND PROSPECTS
Despite the intensive studies on chromatin structure conducted over the past decades, we can say that a fairly clear idea has been formed only about the structure of the nucleosome (the first level of chromatin compaction). Current information about the structure of chromatin at the higher levels of structural organization are often quite controversial and still do not allow one to form an unambiguous picture. With the advent of new methods of studying biological molecules (fluorescence resonance energy transfer [145, 146], high-speed atomic force microscopy, spectroscopy of absorption and optical activity in the infrared region (vibrational circular dichroism and Raman optical activity) [88, 131, 147–150], small angle X-ray and neutron scattering [59, 145, 146, 150–152] and many other methods), the amount of structural information that is available is increasing rapidly. Some previous hypotheses require revision and additional experimental verification. In our opinion, this is most clearly illustrated by recent data on the structure of the 30-nanometer fibril [3, 56, 152–157].
The 30-nanometer fibril is one of the most intensively studied forms of compact chromatin in vitro [158]. According to generally accepted ideas, a 10-nanometer chain of nucleosomes is folded into helical structures with a diameter 30 nm and then is compacted until it reaches a size from 120 to 300–700 nm; as a result, mitotic chromosomes are formed. The authors of a number of works [17, 63, 159] have indicated that the formation of a 30-nanometer fibril is a necessary condition for the assembly and stabilization of condensed interphase and mitotic chromosomes. Clearly distinguishable 30-nanometer fibrils were detected in the nuclei of some terminally differentiated cells [160–162], which indicates the role of such structures in transcriptionally inactive chromatin. However, it should be noted that such fibrils have still not been described directly in vivo in many nuclei of eukaryotic cells [163–165], particularly, in intact mammalian cells [152, 163, 164]. The nucleosomes cannot be folded into a fibril with a strict structure with a diameter 30 nm and the interactions between distant nucleosomes can lead to the formation of less regular polymeric structure [41–43, 153, 165–168].
It is possible that 30- and 120-nanometer structures are specific to certain cell types. Recently, some authors have demonstrated that the chromatin of some cell types exists mainly in the form of fibers with a diameter of 10 nm [169] or heterogeneous groups of nucleosomes that are bound to linker histones [167]. In 2015, studies of chromatin fibrils were conducted in vivo at the level of a single cell with a resolution of 20 nm using the stochastic optical reconstruction microscopy method [167]. The authors demonstrated that nucleosomes assemble into groups of different sizes (“nucleosome muffs”). At the same time, approximately eight nucleosomes in the “muff” were found in human differentiated fibroblasts, while approximately four nucleosomes were found in stem cells. Thirty-nm-thick chromatin fibrils probably exist as short fragments [62], which is consistent with the results of studies of some authors that demonstrated that there are no regular 30-nanometer chromatin fibrils in natural chromatin [168–171]. Different interactions, including nucleosome–nucleosome, have a significant effect on the organization and dynamics of such a group of nucleosomes [8]. Interphase chromosomes can be organized in globular structures (topologically associating domains, TADs) that persist throughout the cell cycle [166]. A model according to which chromatin is in a “liquid drop” state in metaphase chromosomes has been considered in literature [166]. However, such a model cannot explain the existence of a structured metaphase chromosome. As an alternative to the existing ideas, a model of two-phase chromatin fractal structure, which allows one to describe both DNA compaction and chromatin dynamics, was recently suggested ([172], see details in the review [11]).
Based on the results obtained using the electron microscopy-assisted nucleosome interaction capture (EMANIC) method, it was concluded that there is no single type of helical organization of the 30-nanometer fibril in chromatin. This can be due to the fact that the structure of a chromatin fibril depends significantly on the length of linker DNA [3, 173]. Regular zigzag structures (a two-start helix) are formed in the case of short and medium-sized lengths of the linker DNA region (173–209 bp). At the same time, a solenoid model better corresponds to the structure of the 30-nanometer fibril in chromatin with a longer linker (218–226 bp). In the model of the heteromorphic 30-nanometer fibril discussed in the literature, alternation occurs of regions with a solenoid structure with the elements of the zigzag model [3, 63]. Thus, the problem of adequate description of higher levels of the structural organization of chromatin is still far from being solved and requires further thorough study.
REFERENCES
A. E. White, A. R. Hieb, and K. Luger, Sci. Rep. 6, 19122 (2016).
C. L. Woodcock and R. P. Ghosh, Cold Spring Harb. Perspect. Biol. 2 (5), a000296 (2010).
K. Luger, M. L. Dechassa, and D. J. Tremethick, Nat. Rev. Mol. Cell Biol. 13, 436 (2012).
T. L. Caterino and J. J. Hayes, Biochem. Cell Biol. 89, 35 (2011).
E. Chikhirzhina, G. Chikhirzhina, and A. Polyanichko, Biomed. Spectr. Imag. 3, 345 (2014).
E. V. Chikhirzhina and V. I. Vorob’ev, Tsitologiya 44, 721 (2002).
S. A. Grigoryev, G. Arya, S. Correll, et al., Proc. Natl. Acad. Sci. U. S. A. 106, 13317 (2009).
A. Kalashnikova, M. Porter-Goff, U. Muthurajan, et al., J. Roy. Soc. Interface 10, 20121022 (2013).
J. Ausio, Bioessays 37, 46 (2015).
A. Sadakierska-Chudy and M. Filip, Neurotox. Res. 27, 172 (2015).
A. V. Ilatovskii, D. V. Lebedev, M. V. Filatov, et al., Tsitologiya 54 (4), 298 (2012)
A. L. Olins and D. E. Olins, Science 184, 868 (1974).
H. Schiessel, J. Widom, R. F. Bruinsma, et al., Phys. Rev. Lett. 86, 4414 (2001).
A. M. Polyanichko and E. V. Chikhirzhina, J. Mol. Struct. 1044, 167 (2013).
K. Luger, A. W. Mader, R. K. Richmond, et al., Nature 389, 251 (1997).
R. K. Suto, R. S. Edayathumangalam, C. L. White, et al., J. Mol. Biol. 326, 371 (2003).
T. Schalch, S. Duda, D. F. Sargent, et al., Nature 436, 138 (2005).
A. Klug, Annu. Rev. Biochem. 79, 1 (2010).
A. Polyanichko and H. Wieser, Biopolymers 78, 329 (2005).
A. Polyanichko, E. Chikhirzhina, V. Andruschchenko, et al., Biopolymers 83, 182 (2006).
A. Polyanichko and H. Wieser, in Methods in Protein Structure and Stability Analysis: Vibrational Spectroscopy, ed. by E. Permyakov and V. Uversky (Nova Science Publ., New York, 2007), pp. 267–302.
A. Polyanichko and E. Chikhirzhina, Spectroscopy 27, 393 (2012).
A. M. Polyanichko, V. V. Andrushchenko, P. Bour, et al., Circular Dichroism: Theory and Spectroscopy, ed. by D. S. Rodgers (Nova Science Publ., New York, 2012), pp. 67–126.
A. M. Polyanichko, V. I. Vorob’ev, and E. V. Chikhirzhina, Mol. Biol. (Moscow) 47 (2), 412 (2013).
J. Zlatanova and J. Yaneva, DNA Cell Biol. 10, 239 (1991).
E. V. Chikhirzhina, T. Yu. Starkova, E. I. Kostyleva, et al., Tsitologiya 53 (10), 826 (2011).
E. I. Ramm, E. V. Chikhirzhina, E. I. Kostyleva, et al., Biokhimiya 60 (1), 150 (1995).
E. V. CHikhirzhina, E. I. Kostyleva, E. I. Ramm et al., Tsitologiya 40 (10), 883 (1998).
A. M. Polyanichko, S. G. Davydenko, E. V. Chikhirzhina, et al., Tsitologiya 42 (8), 787 (2000).
E. V. Chikhirzhina, A. M. Polyanichko, A. N. Skvortsov, et al., Mol. Biol. (Moscow) 36 (3), 525 (2002).
A. M. Polyanichko, E. V. Chikhirzhina, A. N. Skvortsov, et al., J. Biomol. Struct. Dyn. 19, 1053 (2002).
E. Chikhirzhina, T. Starkova, E. Kostyleva, et al., Spectroscopy: Int. J. 27, 433 (2012).
E. V. Chikhirzhina, A. M. Polyanichko, E. I. Kostyleva, et al., Mol. Biol. (Moscow) 45 (2), 318 (2011).
A. V. Fonin, O. V. Stepanenko, K. K. Turoverov, et al., Tsitologiya 52 (11), 946 (2010).
A. Fonin, O. V. Stepanenko, I. M. Kuznetsova, et al., Spectroscopy 24, 165 (2010).
E. Chikhirzhina, T. Starkova, and A. Polyanichko, Biophysics (Moscow) 63 (6), 858 (2018).
J. B. Baldwin, P. G. Boseley, E. M. Bradbury, et al., Nature 253, 245 (1975).
R. D. Kornberg, Science 184, 868 (1974).
C. L. Peterson and M. A. Laniel, Curr. Biol. 14, R546 (2004).
C. A. Davey, D. F. Sargent, K. Luger, et al., J. Mol. Bio-l. 319, 1097 (2002).
S. Bilokapic, M. Strause, and M. Halic, Nat. Struct. Mol. Biol. 25, 101 (2018).
S. Bilokapic, M. Strause, and M. Halic, Nat. Commun. 9, 1330 (2018).
S. Bilokapic, M. Strauss, and M. Halic, Sci. Rep. 8, 7046 (2018).
G. Arents, R. W. Burlingame, B. C. Wang, et al., Proc. Natl. Acad. Sci. U. S. A. 88, 10148 (1991).
J. M. Harp, B. L. Hanson, D. E. Timm, et al., Acta Cryst. D: Biol. Cryst. 56, 1513 (2000).
C. M. Wood, J. M. Nicholson, S. J. Lambert, et al., Acta Cryst. F: Struct. Biol. Cryst. Comm. 61, 541 (2005).
A. Jerzmanowski, in Chromatin Struture and Dynamics: State-of-the-Art, Ed. by J. Zlatanova and S. H. Leuba (Elsevier, London, 2004), pp. 75–102.
B. Sarg, W. Helliger, H. Talasz, et al., J. Biol. Chem. 281, 6573 (2006).
A. Kowalski and J. Palyga, Cell Biol. Int. 36, 981 (2012).
J. Ocampo, F. Cui, V. B. Zhurkin, and D. J. Clark, Nucleus 7, 382 (2016).
X. Lu and J. C. Hansen, J. Biol. Chem. 279, 8701 (2004).
C. Crane-Robinson, Biochim. Biophys. Acta 1859, 431 (2016).
D. J. Tremethick, Cell 128, 651 (2007).
G. Li and D. Reinberg, Curr. Opin. Genet. Dev. 21, 175 (2011).
D. Angelov, J. Vitolo, V. Mutskov, et al., Proc. Natl. Acad. Sci. U. S. A. 98, 6599 (2001).
B. R. Zhou, J. Jiang, H. Feng, et al., Mol. Cell 59, 628 (2015).
B. R. Zhou, H. Feng, R. Ghirlando, et al., J. Mol. Biol. 428, 3948 (2016).
J. Bednar, I. Garcia-Saez, R. Boopathi, et al., Mol. Cell, 66, 384 (2017).
K. Maeshima, R. Imai, T. Hikima, et al., Methods 70, 154 (2014).
A. A. Kalashnikova, D. D. Winkler, S. J. McBryant, et al., Nucleic Acids Res. 41, 4026 (2013).
P. Zhu and G. Li, IUBMB Life 68, 873 (2016).
D. V. Fyodorov, B. R. Zhou, A. I. Skoultchi, et al., Nat. Rev. Mol. Cell Biol. 19, 192 (2018).
P. J. Robinson and D. Rhodes, Curr. Opin. Struct. Biol. 16, 336 (2006).
P. J. Robinson, L. Fairall, A. T. Van Huynh, et al., Proc. Natl. Acad. Sci. U. S. A. 103, 6506 (2006).
V. Andrushchenko, J. H. van de Sande, and H. Wieser, Biopolymers 72, 374 (2003).
A. Polyanichko and E. Chikhirzhina, Adv. Biomed. Spectrosc. 7, 185 (2013).
E. Chikhirzhina, T. Starkova, E. Kostyleva et al., Adv. Biomed. Spectrosc. 7, 177 (2013).
T. Maniatis, J. H. Venable, Jr., and L. S. Lerman, J. Mol. Biol. 84, 37 (1974).
A. L. Turner, M. Watson, O. G. Wilkins, et al., Proc. Natl. Acad. Sci. U. S. A., 115 (47), 11964 (2018).
F. Totsingan and A. J. Bell, Jr., Prot. Sci. 22, 1552 (2013).
J. Zlatanova and K. van Holde, BioEssays 20, 584 (1998).
J. N. Yaneva, E. G. Paneva, S. I. Zacharieva et al., Z. Naturforsch. C 61, 879 (2006).
J. N. Yaneva, E. G. Paneva, S. I. Zacharieva et al., Z. Naturforsch. C 62, 905 (2007).
K. P. Nightingale, D. Pruss, and A. P. Wolffe, J. Biol. Chem. 271, 7090 (1996).
X. Lu and J. C. Hansen, Biochem. Cell Biol. 81, 173 (2003).
X. Lu, B. Hamkalo, M. H. Parseghian, et al., Biochemistry 48, 164 (2009).
A. A. Kalashnikova, R. A. Rogge, and J. C. Hansen, Biochim. Biophys. Acta 1859, 455 (2016).
A. Roque, I. Ponte, and P. Suau, Chromosoma 126, 83 (2017).
A. Roque, I. Ponte, and P. Suau, Biochim. Biophys. Acta 1859, 444 (2016).
A. Roque, I. Ponte, J. L. Arrondo, et al., Nucleic Acids Res. 36, 4719 (2008).
H. E. Kasinsky, J. D. Lewis, J. B. Dacks, et al., FASEB J. 15, 34 (2001).
J. C. Hansen, X. Lu, E. D. Ross, et al., J. Biol. Chem. 281, 1853 (2006).
J. Allan, D. Ram, N. Harborne, et al., J. Cell. Biol. 98, 1320 (1984).
B. R. Zhou, H. Feng, H. Kato, et al., Proc. Natl. Acad. Sci. U. S. A. 110, 19390 (2013).
M. J. Hendzel, M. A. Lever, E. Crawford, et al., J. Biol. Chem. 279, 20028 (2004).
N. Raghuram, G. Carrero, J. Th’ng, et al., Biochem. Cell Biol. 87, 189 (2009).
J. P. Th’ng, R. Sung, M. Ye, et al., J. Biol. Chem. 280, 27809 (2005).
H. Fang, D. J. Clark, and J. J. Hayes, Nucleic Acids Res. 40, 1475 (2012).
J. Allan, P. G. Hartman, C. Crane-Robinson, et al., Nature 288, 675 (1980).
Y. Zhou, S. Gershman, V. Ramakrishnan, et al., Nature 395, 402 (1998).
S. Lambert, S. Muyldermans, J. Baldwin, et al., Biochem. Biophys. Res. Commun. 179, 810 (1991).
J. Hayes, Biochemistry 35, 11931 (1996).
D. Pruss, B. Bartholomew, J. Persinger, et al., Science 274, 614 (1996).
D. T. Brown, T. Izard, and T. Misteli, Nat. Struct. Mol. Biol. 13, 250 (2006).
L. Fan and V. A. Roberts, Proc. Natl. Acad. Sci. U. S. A. 103, 8384 (2006).
S. J. McBryant, X. Lu, and J. C. Hansen, Cell Res. 20, 519 (2010).
J.-Q. Ni, L.-P. Liu, D. Hess, et al., Genes Dev. 20, 1959 (2006).
H. J. Szerlong, J. A. Herman, C. M. Krause et al., J. Mol. Biol. 427, 2056 (2015).
J. Zlatanova, Trends Biochem. Sci. 15, 273 (1990).
H. Lee, R. Habas, and C. Abate-Shen, Science 304, 1675 (2004).
P. J. Laybourn and J. T. Kadonaga, Science 254, 238 (1991).
Q. Lin, A. Inselman, X. Han, et al., J. Biol. Chem. 279, 23525 (2004).
Y. Zheng, S. John, J. J. Pesavento, et al., J. Cell Biol. 189, 407 (2010).
S. Yang, B. J. Kim, L. N. Toro, et al., Proc. Natl. Acad. Sci. U. S. A. 110, 1708 (2013).
Y. V. Postnikov, L. Trieschmann, A. Rickers, et al., J. Mol. Biol. 252, 423 (1995).
Y. Postnikov and M. Bustin, Biochim. Biophys. Acta 1799, 62 (2010).
H. Kato, H. van Ingen, B. R. Zhou, et al., Proc. Natl. Acad. Sci. U. S. A. 108, 12283 (2011).
R. D. Phair, P. Scaffidi, C. Elbi, et al., Mol. Cell. Biol. 24, 6393 (2004).
Y. V. Postnikov, V. V. Shick, A. V. Belyavsky, et al., Nucleic Acids Res. 19, 717 (1991).
T. Deng, Z. I. Zhu, S. Zhang, et al., Mol. Cell. Biol. 33, 3377 (2013).
H. F. Ding, M. Bustin, and U. Hansen, Mol. Cell. Biol. 17, 5843(1997).
K. J. Murphy, A. R. Cutter, H. Fang, et al., Nucleic Acids Res. 45, 9917 (2017).
M. Stros, Biochim. Biophys. Acta 1799, 101 (2010).
M. Stros, E. Polanska, M. Kucirek, et al., PLoS One 10, e0138774 (2015).
R. Reeves, DNA Repair 36, 122 (2015).
F. A. Atcha, A. Syed, B. Wu, et al., Mol Cell Biol. 27, 8352 (2007).
L. H. Pevny and S. K. Nicolis, Int. J. Biochem. Cell Biol. 42, 421 (2010).
P. Bernard and V. R. Harley, Int. J. Biochem. Cell Biol. 42, 400 (2010).
F. Oppel, N. Muller, G. Schackert, et al., Mol Cancer. 10, 137(2011).
O. Leis, A. Eguiara, E. Lopez-Arribillaga, et al., Oncogene 31, 1354 (2012).
T. Chi, Nat. Rev. Immunol. 4, 965 (2004).
M. Stros, D. Launholt, and K. D. Grasser, Cell. Mol. Life Sci. 64, 2590 (2007).
D. Lai, M. Wan, J. Wu, et al., Proc. Natl. Acad. Sci. U. S. A. 106, 1169 (2009).
R. Catena, E. Escoffier, C. Caron, et al., Biol. Reprod. 80, 358 (2009).
S. Park and S. J. Lippard, Biochemistry 51, 6728 (2012).
G. J. Sullivan and B. McStay, Nucleic Acids Res. 26, 3555 (1998).
V. Ramakrishnan, Annu. Rev. Biophys. Biomol. Struct. 26, 83 (1997).
Y. V. Postnikov and M. Bustin, Biochim. Biophys. Acta 1859, 462 (2016).
A. M. Polyanichko, Z. V. Leonenko, D. Kramb, et al., Biophysics (Moscow) 53 (3), 202 (2008).
L. Cato, K. Stott, M. Watson, et al., Mol. Biol. 384, 1262 (2008).
A. Polyanichko and H. Wieser, Spectroscopy 24, 239 (2010).
L. A. Kohlstaedt, E. C. Sung, A. Fujishige, et al., J. Biol. Chem. 262, 524 (1987).
L. A. Kohlstaedt and R. D. Cole, Biochemistry 33, 570 (1994).
A. M. Polyanichko, B. A. Dribinskii, I. B. Kipenko, et al., Strukt. Dinam. Mol. Sistem 8A, 3 (2010).
E. Polanska, S. Pospisilova, and M. Stros, PLoS One 9, e89070 (2014).
P. Widlak, M. Kalinowska, M. H. Parseghian, et al., Biochemistry 44, 7871 (2005).
S. W. Harshman, N. L.Young, M. R. Parthun, et al., Nucleic Acids Res. 41, 9593 (2013).
E. Cheung, A. S. Zarifyan, and W. L. Kraus, Mol. Cell. Biol. 22, 2463 (2002).
R. D. Phair and T. Misteli, Nature 404, 604 (2000).
M. Harrer, H. Luhrs, M. Bustin, et al., J. Cell Sci. 117, 3459 (2004).
F. Catez, H. Yang, K. J. Tracey, et al., Mol. Cell. Biol. 24, 4321 (2004).
A. Allahverdi, R. Yang, N. Korolev, et al., Nucleic Acids Res. 39, 1680 (2011).
P. Trojer, J. Zhang, M. Yonezawa, et al., J. Biol. Chem. 284, 8395 (2009).
T. K. Hale, A. Contreras, A. J. Morrison, et al., Mol. Cell 22, 693 (2006).
M. Brehove, T. Wang, J. North, et al., J. Biol. Chem. 290, 22612 (2015).
R. Morra, T. Fessl, Y. Wang, et al., Methods Mol. Biol. 1431, 175 (2016).
I. H. McColl, E. W. Blanch, A. C. Gill, et al., J. Am. Chem. Soc. 125, 10019 (2003).
F. Zhu, N. W. Isaacs, L. Hecht, et al., Structure 13, 1409 (2005).
L. D. Barron, Biomed. Spectr. Imag. 4, 223 (2015).
M. L. Mello and B. C. Vidal, PLoS One 7, e43169 (2012).
H. B. Stuhrmann, Acta Crystallogr. A 64, 181 (2008).
Y. Joti, T. Hikima, Y. Nishino, et al., Nucleus 3, 404 (2012).
I. Garcia-Saez, H. Menoni, R. Boopathi, et al., Mol. Cell 72, 1 (2018).
M. A. Ozturk, V. Cojocaru, and R. C. Wade, Structure 26, 1 (2018).
M. A. Ozturk, V. Cojocaru, and R. C. Wade, Biophys. J. 114, 2363 (2018).
B. R. Zhou, J. Jiang, R. Ghirlando, et al., J. Mol. Biol. 430, 3093 (2018).
M. A. Ozturk, G. V. Pachov, R. C. Wade, et al., Nucleic Acids Res. 44, 6599 (2016).
J. C. Hansen, Ann. Rev. Biophys. Biomol. Struct. 31, 361 (2002).
F. Song, P. Chen, D. Sun, et al., Science 344, 376 (2014).
C. L. Woodcock, J. Cell Biol. 125, 11 (1994).
C. Kizilyaprak, D. Spehner, D. Devys, et al., PLoS One 5, e11039 (2010).
M. P. Scheffer, M. Eltsov, and A. S. Frangakis, Proc. Natl. Acad. Sci. U. S. A. 108, 16992 (2011).
M. Eltsov, K. M. Maclellan, K. Maeshima et al., Proc. Natl. Acad. Sci. U. S. A. 105, 19732 (2008).
E. Fussner, M. Strauss, U. Djuric, et al., EMBO Rep. 13, 992 (2012).
H. D. Ou, S. Phan, T. J. Deerinck, et al., Science 357, pii: eaag0025 (2017).
T. Nozaki, R. Imai, M. Tanbo, et al., Mol. Cell 67, 282 (2017).
M. A. Ricci, C. Manzo, M. F. Garcı’a-Parajo, et al., Cell 160, 1145 (2015).
K. Maeshima, R. Rogge, S. Tamura, et al., EMBO J. 35, 1115 (2016).
K. Maeshima, R. Imai, S. Tamura, et al., Chromosoma 123, 225 (2014).
S. A. Grigoryev, G. Bascom, J. M. Buckwalter, et al., Proc. Natl. Acad. Sci. U. S. A. 113, 1238 (2016).
W. Li, P. Chen, J. Yu, et al., Mol Cell 64, 120 (2016).
A. Bancaud, S. Huet, N. Daigle, et al., EMBO J. 28, 3785 (2009).
A. Routh, S. Sandin, and D. Rhodes, Proc. Natl. Acad. Sci. U. S. A. 105, 8872 (2008).
Funding
This work was supported by the Russian Foundation for Basic Research (grant no. 18-08-01500 (structural studies of DNA–protein interactions) and grant no. 18-04-01199 (studying the role of nuclear proteins HMGB1/2, H1 in the structural organization of chromatin).
Author information
Authors and Affiliations
Contributions
All of the authors made equal contributions to preparing this manuscript.
Corresponding authors
Ethics declarations
The authors declare that they have no conflict of interest.
This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A. Barkhash
Rights and permissions
About this article

Cite this article
Chikhirzhina, E.V., Starkova, T.Y. & Polyanichko, A.M. The Role of Linker Histones in Chromatin Structural Organization. 2. Interaction with DNA and Nuclear Proteins. BIOPHYSICS 65, 202–212 (2020). https://doi.org/10.1134/S0006350920020049
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006350920020049