
Abstract
Data on the role of H1 family linker histones in the structural organization and functioning of chromatin that have accumulated to date are presented in this review. The structure of the H1 histone and its post-translational modifications are considered. Special attention is paid to the role of the H1 histone in the formation of the chromatin in transcriptionally inactive state with a high degree of DNA compaction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
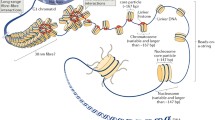
The chromatin of eukaryotic cells is a highly dynamic DNA–protein system with quite a complex organization, whose functioning is closely related to its multilevel structural organization. Reversible structural transformations in the chromatin allow regulation of the degree of its compactness and not only provide denser DNA packaging in the cell nucleus, but also underlie the functioning of the genome in general [1–8]. The length of the DNA molecule in each of the chromosomes exceeds the size of the cell nucleus, which is just a few microns, by thousands of times; consequently, DNA packaging in the nucleus is impossible without strong molecular compaction. First, DNA length is greatly decreased due to its interaction with histones and non-histone proteins. Subsequent stages of DNA compaction are associated with changes in the chromatin structure. As a result of these transformations, a high density of DNA packaging in the cell nucleus is reached. The nature of DNA packaging in the chromosomes plays an important role, since not only the chromatin structure, but also the possibility of the functioning of the genetic apparatus depends on it.
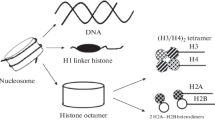
Histones are basic proteins. Five histone fractions (H1, H2A, H2B, H3, and H4) are distinguished according to their molecular weight and amino-acid composition. Four of them (H2A, H2B, H3, and H4) form a protein particle, around which DNA is wound. The fifth histone H1 (a variant named H5 exists in erythrocyte chromatin in birds) binds these DNA–protein particles together. This protein plays a key role in the development of the top levels of the structural organization of chromatin, which will be discussed in detail in a separate publication [9].
THE STRUCTURE OF H1 FAMILY LINKER HISTONES
The H1 histone molecule can be conventionally divided into three regions: a non-polar central domain (approximately ~80 amino-acid residues (a.a.)) and positively charged disordered N- and C-terminal regions (~20 and ~100 a.a., respectively) (Fig. 1) [10–12]. The central protein region in solutions with high ionic strength is able to form a globule, whose structure was initially solved for the H5 histone using X-ray crystallography (Fig. 2) [13] and then for the H1 histone using the NMR analysis [14]. Based on the data obtained, it was established that the globular domain consists of three α-helical regions that form a classical “helix–turn–helix” motif [11, 12]. It was also demonstrated that the globular domain structure is distinguished by a high level of interspecies conservation [15].

The main chromatin components.

The spatial organization of the globular domain of linker H5 histone (PDB:1HST). The domain consists of three α-helix regions (I (5–16 a.a.), II (24–34 a.a.), and III (42–56 a.a.)) and three regions with β-turn conformation (S1 (located behind the plane of the figure), S2, and S3). α-Helixes I and II are connected through the S1 β-turn. The S2 and S3 regions generate a β-hairpin and together with S1 develop a β-structure out of three antiparallel layers. The elongated W loop is generated in the interaction of the S2 and S3. This figure is formed based on the structure presented in the PDB database (1GHC Protein Data Bank).
The disordered N-terminal fragment of the H1 molecule consists of approximately 20–35 a.a. This fragment can be conditionally divided into two parts that significantly differ in their amino-acid compositions. One of them (the N-terminal) is enriched by alanine, proline residues, and hydrophobic amino-acid residues. As a consequence, this part of the polypeptide chain has no positive charge and is not actively involved in the binding to DNA [16]. In contrast, the second region located closer to the globular part of the molecule contains one arginine residue and five lysine residues, similar to the H3 histone sequence [16]. The high density of positively charged amino acids and location of this region close to the globular domain provides stronger binding of the latter with DNA at the entrance/exit of the nucleosome [17, 18]. The H1 histone N-terminal domain itself has no secondary structure in an aqueous solution [19]. However, part of the domain takes an α-helical conformation in trifluoroethanol [18]. In several papers, it was demonstrated that structurization of the N-terminal region of the protein is observed during the interaction of H1 with DNA [18, 20]; according to the authors, this can affect the nature of the interaction of the H1 histone with DNA.
The C-terminal fragment of the H1 histone molecule contains approximately 100 amino-acid residues (from 122 to 215 a.a.), is different in the composition in different protein subtypes [21, 22], and mainly consists of alternate lysine (~40%), alanine (~20–35%), and proline (~15%) residues [23]. Studies published by different authors demonstrated that this region is responsible for the ability of the protein to compact DNA [5, 6, 17, 24–29].
In the last decade, it was established that the C- and N-terminal H1 histone domains are internally disordered regions that take a certain conformation when interacting with DNA [30]. In addition, the secondary structure of the C-terminal segment can be modulated by phosphorylation of its a.a. [31]. Internal disorder of the protein molecule is quite common among eukaryotes [32–34], especially in architectural chromatin proteins such as histones, high mobility group (HMG) family proteins [35–37], and protamines. Variability of the spatial structure of linker proteins leads to some functional advantages, such as the ability of the protein to interact with different partners and an increase in the speed of interaction [23, 32, 38], which plays an important role in the regulation of the chromatin structure. This property of the polypeptide chain can play an important role in the regulation of the chromatin dynamics.
SPERM-SPECIFIC PROTEINS OF THE H1 FAMILY
Unlike other histones, the H1 family proteins are characterized by a high degree of species and tissue specificity. The structural and functional differences between the proteins of this family are especially pronounced in spermatogenic cells. In the process of spermatogenesis, the cells undergo a number of biochemical and morphological changes, as a result of which DNA is very tightly packed in the nucleus, while the chromatin is in a transcriptionally inactive state [5–7, 39]. Unlike somatic cells with a relatively conservative set of four core histones and linker H1 histone, other DNA-binding proteins are also present in sperm cells. H1 histones persist throughout the process of spermatogenesis in the members of many species of multicellular organisms; these proteins are structurally almost identical to the somatic H1. The mechanism is more complex in mammals. In the initial stages of spermatogenesis, the H1 histones are present in cells; they are gradually replaced by protamines and only protamines remain in sperm in the late stages. All of the varieties of changes in the protein composition of spermatogenic cells can be reduced to several main variants (Fig. 3): (1) H1 histone replacement by protamines or (2) thioprotamines, (3) the appearance of additional S-proteins, and (4) H1 replacement by sperm-specific variants.

Protamines are a special class of small (4–12 kDa) linker proteins found in the sperm of mammals, as well as some fish, birds, and cephalopods [39, 40]. These low-molecular-weight proteins do not form globular regions in the central part of the molecule. The latter can be associated with the absence of cysteine residues and with quite an even distribution of the positive charge throughout the polypeptide chain. In solution these proteins are characterized by a polyproline-II type conformation, which is most favorable for the interaction of protamines with DNA and during the development of intermolecular crosslinking in sperm DNA [41]. In the cells of some amphibians and insects, the H1 histone is replaced by thioprotamines or protamine-like proteins, which are an evolutionary intermediate between the H1 histones and protamines [7, 39]. These are short (50–60 a.a.) arginine-rich proteins containing six to nine cysteine residues per molecule [42]. Along with a complete set of histones, so-called S-proteins (sperm-specific proteins with a high content of arginine, lysine, serine, and alanine) were found in the sperm chromatin of some organisms [43]. These proteins are characterized by a high positive charge density, which contributes to their strong interaction with DNA. The H1 histone [39, 44] and sometimes the core H2A and H2B histones [45, 46] are replaced by sperm-specific variants in the chromatin of marine invertebrates, some amphibians, and fishes.
Out of 11 mammalian H1 histone variants described in the literature, 4 are found only in germ cells (H1t, H1T2, H1oo, and HILS1) [47]. The replacement of somatic H1 histones with proteins specific for certain cells indicates that they play a critical role in the development and maturation of sperm and oocytes. Thus, a decrease in the HILS1 expression in sperm correlates with a decrease in their mobility in men, which is directly associated with male fertility [48]. The H1 replacement with H100 in oocytes probably plays an important role in the organization of the chromatin structure during oocyte maturation. However, the mechanisms of the chromatin structure regulation in early embryogenesis remain in general insufficiently studied and require further research.
Sperm chromatin is unique in its extremely high density of genomic DNA packaging. The chromatin fibers that contain sperm-specific proteins of the H1 family have a nucleosome structure (similar to somatic cell chromatin). These proteins bind to linker DNA and stabilize the structure of the 30-nanometer fibril, whose compaction provides a high density of DNA packaging in the sperm [5, 6, 39, 49]. Protamines bind to DNA [39] stimulating the integration of the obtained nucleoprotein complexes in denser fibrils. It should be noted that the diameter of the chromatin fibril always remains within 30–50 nm regardless of which proteins are involved in DNA compaction [39].
The main differences between sperm-specific proteins among themselves are first of all caused by different contents of lysine and arginine residues (that is, the lysine/arginine ratio). The increased arginine content in protamines leads to a strengthening of the DNA-binding ability of the H1 histone [50, 51] and, consequently, to greater chromatin condensation in the sperm nucleus [52]. The activation of regulatory pathways occurs in the fertilized egg via polyarginine clusters [52]. In addition to this, differences at the level of secondary and tertiary structures were also found in some members of sperm-specific H1 histones [45, 53–55]. Thus, the presence of additional regions with an α-helical conformation in C-terminal segments that are directly involved in DNA compaction is a distinctive feature of the H1 of echinoderm sperm [24, 53, 56]. The ability to generate left-helix structures due to a decrease in the portion of α-helical regions is typical for the H1 of bivalve mollusk sperm [53]. These structural peculiarities of the proteins significantly affect their interaction with DNA [5, 6, 26–29]. Thus, for example, when binding DNA to the H1 of mollusk sperm, no supramolecular DNA–protein complexes that are typical of DNA interaction with other H1 family histones are formed [26–29].
Several mechanisms responsible for the structural rearrangements of chromatin (accompanying genome repression in spermatogenesis) have been established [57]. Among these, it is possible to distinguish the effect of the protein composition of spermatogenic cells both on the peculiarities of the sperm itself (the shape and mobility of the sperm head [42, 52]) and on the peculiarities of early embryonic development (see references in [52]). It should be noted that the shape and mobility of the sperm head also has a direct effect on fertility [42, 49]. DNA regions that interact with different sperm-specific proteins will condense in the nucleus in different ways, which can lead to a functionally significant redistribution of nucleosomes and regulatory proteins associated with different chromatin regions. Post-translational modifications of histones (including linkers), which will be discussed in the next section, are another important mechanism of the regulation of chromatin activity.
H1 HISTONE SUBTYPES
As noted above, great tissue- and species specificity [5, 6] and in some cases even intrapopulation individual variability [58] is typical for linker histones. To date, 11 H1 histone subtypes have been detected, for which it was demonstrated that their functions can vary significantly [15, 30, 47, 58–60]. All 11 H1 histone subtypes are actively expressed at all stages of development: from germ cells and embryos to the tissues of the adult organism. Each of the H1 histone subtypes is encoded by its gene, while their total numbers vary from one in infusoria [61], slime mold [62], or in Drosophila [63] to 11 subtypes in mammalian cells [59]. The mammalian H1 histone subtypes can be divided into two groups, one of which consists of the proteins found in somatic cells, while another consists of the proteins that are typical for spermine cells [47, 59, 64]. Thus, the H1t, H1T2, H1oo, and HILS1 histones are found only in germ cells, while the other seven (H1.1–H1.5, H1.0, and H1x, or according to another nomenclature, H1a–e, H10, and H1x) are present in mammalian somatic cells. According to the literature data [65, 66], the H1.1, H1.0, and H1x proteins are tissue specific. H1.1 is found in the cells of the thymus, ovaries, spleen, lymphatic, and nervous tissues in significant amounts, while the H1x protein has only been found in cultivated cells, despite the fact that the expression of the H1FX gene (encoding H1x) is observed in most tissues [66]. The highest amount of linker H1 histone was found in completely differentiated vertebrate cells, while the lowest amount was seen in pluripotent embryonic stem cells [67]. It has been established that a sharp increase in the H1.0 and H1.5 during the maturation of some tissues (liver, kidneys, lungs, and brain cortex) is accompanied by a decrease in the levels of the H1.1, H1.3, and H1.4 and by slowing of cell division. As an example, H1.0 and H1.5 make up 9.5 and 19%, respectively, of the total amount of H1 in the liver of newborn mice, but their levels reach 29 and 40%, respectively, in the liver of adult individuals. Similarly, the contribution of H1.5 is the largest to the total amount of H1, while the level of H1.0 grows during the differentiation of brain neurons. The clear regulation of the expression of each of the H1 variants and the combination of the H1 histone subtypes specific for each tissue with the development of mammals indicates that the amount and relative contribution of each H1 variant are important for the correct development of tissue function. A knockout of even two variants of somatic H1 histones does not lead to death due to the high contents of the other H1 subtypes [68]. However, knockouts of three or more subtypes cause various violations from developmental delay up to death. Knockout of individual genes encoding somatic H1 histone subtypes does not lead to serious developmental pathologies, indicating that the total amount of H1, but not its specific subtypes, is important for normal mammalian embryogenesis (see references in [47]).
The H1 histone subtypes found in somatic mammalian cells are characterized by a high conservation of primary structures, both within one organism and when comparing the proteins isolated from the same tissue of different animal species (http://www.uniprot.org) [69, 70]. Despite this, differences in their evolutionary stability [11], affinity for DNA, and the degree of its compaction upon binding [71, 72] have been detected.
POST-TRANSLATIONAL MODIFICATIONS OF THE HISTONE H1
The H1 family histones are exposed to different post-translational modifications [73] at different stages of the cell cycle, such as acetylation, phosphorylation, methylation, ubiquitination, ADP-ribosylation, and N-formylation. Let us discuss some of them in more detail.
Phosphorylation is the most common and important protein modification. Many enzymes and recep-tors become active or inactive during phospho-rylation/dephosphorylation, which is associated with conformational changes of the polypeptide chain. The association of H1 histone phosphorylation with such cellular processes as apoptosis [74], cell proliferation and differentiation [75], and chromatin remodeling [30, 76, 77] was demonstrated. Phosphorylation of the C-terminal region of the linker histone can be also associated with chromatin decondensation in the S phase of the cell cycle [78].
The H1 histone phosphorylation mainly occurs for serine (Ser, S) and threonine (Thr, T) and depends on the phase of the cell cycle [79]. The process of the H1 histone phosphorylation can be conditionally divided into two stages [71]. Partial phosphorylation of the H1.2 and H1.4 histones in positions S173 and S187, respectively, occurs in interphase; this leads to chromatin relaxation and to the activation of transcription [80]. Consequently, DNA replication and accumulation of structural and functional proteins required for the cell division occur. Subsequently, total phosphorylation of H1 histones in S/TPXK motifs (X, any amino acid), leading to chromatin condensation and chromosome divergence to daughter cells, is observed at the stage of mitosis [71, 80, 81]. However, there are publications in which the possibility of chromosome compaction in the absence of the histone H1 was demonstrated [77, 82]; therefore, the question about the relationship between these processes remains open.
Methylation on lysine often occurs near serine or threonine residues, whose phosphorylation can block the histone binding to other proteins. This is a binary methylation–phosphorylation switch [73, 83–85]. Similar H1 histone domains were identified in human HeLa cells [85] and are peculiar to core histones, for example, for K9/S10 in the H3 histone. The mechanism of functioning of this regulatory H1 histone region is not yet completely understood. It is known that lysine (Lys, K) methylation in the ninth position (K9) is a result of the histone binding to heterochromatin HP1 protein, which in turn contributes to the chromatin condensation [60, 86, 87]. However, neighboring serine phosphorylation at position 27 blocks this binding [84]. As well, H3 phosphorylation at position S10 at the stage of mitosis leads to the release of the HP1 protein and to an increase in the transcription level [88].
Systematic mass spectrometry mapping of human and mouse histone H1 subtypes demonstrated a huge number of mono-, di-, and trimethylated lysines, many of which are located in the globular domain [89]; however, their specific functions still await further study.
It was demonstrated by mass spectrometry that the H1 family histones can be acetylated/methylated by lysine residues both in the globular domain of the proteins and in the N- and C-terminal regions [70, 89]. The biological roles of the overwhelming majority of detected modifications of this type remain unclear. The acK34 site in the H1.4 histone is one of the most studied acetylation sites. The acetylated state of this protein is typical for the promoters of active transcriptional genes. It is known that acK34-H1.4 is a condition of the recruitment of the TFIID transcription factor in the gene-promoter region. Thus, acK34-H1.4 is an example of modification facilitating the access of the transcription co-activators to the chromatin. Deacetylation of K34-H1.4 should probably lead to the violation of the TFIID transcription factor binding to the promoter regions of transcriptionally active genes and, as a consequence, lead to a decrease in the level of transcriptional activity. A superposition of most methylation and acetylation sites, for example, at positions K26, K34/35, and K46 indicates the presence of bivalent methylation/acetylation sites in these regions [70, 89]. Thus, the character of modifications in this region can determine the “active/inactive” state of the chromatin.
Approximately 1% of the H1 histones experience poly-ADP-ribosylation of glutamic acid (Glu, E) residues. The most intensive poly-ADP-ribosylation is observed in the S-phase of the cell cycle and is accompanied by decondensation of higher chromatin structures [90, 91]. It is assumed that ADP-ribosylation of H1 decreases its affinity to DNA and changes the chromatin structure, which undoubtedly affects the processes that are dependent on the chromatin state [73]. Thus, ADP-ribosylation of the H1 histone causes chromatin reorganization with the development of spermatids: the somatic protein is replaced by protamine-like protein and subsequently with protamine.
Using mass spectrometry, ubiquitination sites of histone H1 were identified both in human cell lines and in mouse tissues [92, 93]. However, the significance of these changes in the protein molecule for the H1 function is still not clarified. The functions and biological roles of the H1.2K63-K85 and K97 formylation (introduction of HCO formic acid residue, as a rule, by substituting a hydrogen atom) detected in tissues (this modification was not detected in the cell lines) also have not been clarified [89].
The ability to regulate the cell’s redox environment is one of the main characteristics of the cell nucleus. In order to facilitate proliferation and to protect DNA from damages caused by oxidative stress, the nucleus should be in the reduced state. In fact, oxidative posttranslational modifications of nuclear proteins (especially histones) play a critical role in these processes [73]. Carbonylation of lysine and serine residues (introduction of carbonyl C=O groups by interacting with carbon monoxide) [94], which can be caused both by active oxygen forms and be a result of glycation reaction, is one such modification. The H1 and H3 histones are mainly exposed to carbonylation [95]. Using mouse NIH/3T3 line fibroblasts, it was demonstrated that the level of H1 carbonylation changes throughout the cell cycle and is at a maximum during DNA synthesis. A decrease in the level of H1 carbonylation is observed in the liver during rat aging [96]. Finally, H1 carbonylation can shield positive charges of lysine and arginine residues and thus affect chromatin compaction.
The H1 histone ubiquitination sites were identified by mass spectrometry both in human cell lines and in mouse tissues [92, 93]. However, the significance of these changes in the protein molecule for H1 function has still not been clarified. One more lysine modification (H1.2K63-K85 and K97 formylation) was found in tissues [89]. It should be noted that this modification was not detected in cell lines. The functions and the mechanism of its occurrence are not yet known. It was suggested that a specific enzyme can catalyze formylation from formaldehyde formed during lysine demethylation via LSD1 amine oxidase. The alternative possibility is that LSD1 itself catalyzes histone formylation [97].
Arginine citrullination is one more post-translational modification with a direct effect on chromatin compaction. Arginine of both core and linker H1.2, H1.3, and H1.4 histones [98] can be replaced by citrulline (citrullination) [99] in the chromatin in the process of the functioning of tissue-specific vertebrate enzymes (peptidyl arginine deaminases, PADI). Citrulline is an amino acid that is not encoded in DNA by a specific codon, but is generated from arginine after protein synthesis. The replacement of arginine with citrulline affects the chemical properties of a protein and makes it more hydrophobic, which affects its spatial structure and, as a consequence, its DNA-binding properties. As an example, it was demonstrated that the chromatin of stem cells is characterized by the presence of linker H1.2 histone citrullination at position R54, which leads to a violation in the protein binding to DNA and to the development of regions with “looser” structural organization [98].
According to currently available data, a large number of post-translational modifications are observed in the C-terminal region of histone H1, whose variability apparently causes the main functional differences between tissue-specific protein subtypes [26–29, 100–103]. Wide use of mass spectrometry to calculate the post-translational modifications of proteins allowed detection of many modifications of linker H1 histone. However, the functions of most of them have not yet been determined. It is only clear that some modifications are unique for certain H1 subtypes and play an important role in chromatin compaction and remodeling. The molecular mechanisms that underlie the emergence of these modifications and the biological significance of each of them must be clarified.
CONCLUSIONS
Thus, a high level of structural diversity and a large number of subtypes and post-translational modifications are typical of the H1 family linker histones. Although the functions performed by the proteins of this family can noticeably differ, all linker histones play a key role during the development of supranucleosome chromatin organization. However, despite the abundance of accumulated experimental data, many aspects of their functioning still remain unclear. This is partly due to the broad involvement of linker histones in the development of complexes with different DNA regions and other nuclear proteins, and partly due to difficulties of the experimental determination of the structure of supramolecular complexes in the composition of chromatin. In the second part of this review, we will attempt to systematize the published results concerning the mechanisms of H1 histone interactions with DNA and other nuclear proteins.
REFERENCES
A. E. White, A. R. Hieb, and K. Luger, Sci. Rep. 6, 19122 (2016).
C. L. Woodcock and R. P. Ghosh, Gold Spring Harb. Perspect. Biol. 2, a000296 (2010). doi 10.1101/cshperspect.a000596
K. Luger, M. L. Dechassa, and D. J. Tremethick, Nat. Rev. Mol. Cell Biol. 13, 436 (2012).
T. L. Caterino and J. J. Hayes, Biochem. Cell Biol. 89, 35 (2011).
E. V. Chikhirzhina and V. I. Vorob’ev, Tsitologiya 44, 721 (2002).
E. Chikhirzhina, G. Chikhirzhina, and A. Polyani-chko, Biomed. Spectr. Imaging 3, 345 (2014).
J. Ausio, Bioessays 37, 46 (2015).
C. Crane-Robinson, Biochim. Biophys. Acta 1859, 431 (2016).
E. Chikhirzhina, T. Starkova, and A. Polyanichko, Biophysics (Moscow) 64 (2019) in press).
H. E. Kasinsky, J. D. Lewis, J. B. Dacks, et al., FASEB J. 15, 34 (2001).
I. Ponte, J. M. Vidal-Taboada, and P. Suau, Mol. Biol. Evol. 15, 702 (1998).
M. H. Parseghian, AIMS Biophys. 2, 724 (2015).
V. Ramakrishnan, J. T. Fich, V. Graziano, et al., Nature 362, 219 (1993).
C. Cerf, G. Lippens, V. Ramakrishnan, et al., Biochemistry 33, 11 079 (1994).
A. Izzo, K. Kamieniarz, and R. Schneider, Biol. Chem. 389, 333 (2008).
L. Bohm and T. C. Mitchell, FEBS Lett. 193, 1 (1985).
J. Allan, T. Mitchell, and N. Harborne, J. Mol. Biol. 187, 591 (1986).
R. Vila, I. Ponte, M. Collado, et al., J. Biol. Chem. 276, 46 429 (2001).
A. V. Lyubitelev, D. V. Nikitin, A. K. Shaytan, et al., Biochemistry (Moscow) 81 (3), 213 (2016).
R. Vila, I. Ponte, M. A. Jimenez, et al., Protein Sci. 11, 214 (2002).
I. Ponte, R. Vila, and P. Suau, Mol. Biol. Evol. 20, 371 (2003).
A. Roque, I. Ponte, and P. Suau, Biochim. Biophys. Acta 1859, 444 (2016).
J. C. Hansen, X. Lu, E. D. Ross, et al., J. Biol. Chem. 281, 1853 (2006).
R. Misselwitz, D. Zirwer, H. Damaschun, et al., Int. J. Biol. Macromol. 2, 194 (1986).
E. I. Ramm, G. S. Ivanov, and V. I. Vorob’ev, Biokhimiya 58, 1604 (1993).
E. V. Chikhirzhina, T. Yu. Starkova, E. I. Kostyleva, et al., Cell Tissue Biol. 5 (6), 536 (2011).
E. V. Chikhirzhina, E. I. Kostyleva, E. I. Ramm, et al., Tsitologiya 40 (10), 883 (1998).
E. Chikhirzhina, T. Starkova, E. Kostyleva, et al., Spectroscopy 27, 433 (2012).
E. Chikhirzhina, T. Starkova, E. Kostyleva, et al., Adv. Biomed. Spectrosc. 7, 177 (2013).
A. Roque, I. Ponte, and P. Suau, Chromosoma 126, 83 (2017).
A. Roque, I. Ponte, J. L. Arrondo, et al., Nucleic Acids Res. 36, 4719 (2008).
L. Breydo, J. M. Redington, and V. N. Uversky, Int. Rev. Cell. Mol. Biol. 329, 145 (2017).
V. N. Uversky, Cell Mol. Life Sci. 74, 3065 (2017).
A. L. Darling and V. N. Uversky, Molecules 22, E2027 (2017).
M. Stros, Biochim. Biophys. Acta 1799, 101 (2010).
M. Stros, E. Polanska, M. Kucirek, et al., PLoS One 10, e0138774 (2015).
R. Reeves, DNA Repair 36, 122 (2015).
V. N. Uversky, Prot. Sci. 22, 693 (2013).
J. M. Eirín-López and J. Ausió, Bioessays 31, 1062 (2009).
H. E. Kasinsky, J. M. Eirín-López, and J. Ausió, Prot. Pept. Lett. 18, 755 (2011).
E. I. Ramm, E. V. Karpova, and V. I. Vorob’ev, Mol. Biol. (Moscow) 17, 293 (1983).
I. A. Belokopytova, E. I. Kostyleva, A. N. Tomilin, et al., Mol. Reprod. Dev. 34, 53 (1993).
I. A. Zalenskaya, N. A. Odintsova, and V. I. Vorob’ev, FEBS Lett. 188, 243 (1985).
J. D. Lewis, Y. Song, M. E. De Jong, et al., Chromosoma 111, 473 (2003).
I. A. Zalenskaya, V. A. Pospelov, A. O. Zalensky, et al., Nucleic Acids Res. 9, 473 (1981).
E. Reyes, V. Morin, S. Schwager, et al., Comp. Biochem. B. Physiol. Biochem. Mol. Biol. 128, 451 (2001).
C. Pan and Y. Fan, Biochim. Biophys. Acta 1859, 496 (2016).
P. Jedrzejczak, B. Kempisty, A. Bryja, et al., Arch. Androl. 53, 199 (2007).
J. R. Daban, Biochem. Cell Biol. 81, 91 (2003).
L. J. Frehlick, J. M. Eirin-Lopez, A. Prado, et al., J. Exp. Zool. A: Comp. Exp. Biol. 305, 277 (2006).
J. Ausio, K. O. Greulich, E. Haas, et al., Biopolymers 23, 2559 (1984).
J. Ausio, J. M. Eirin-Lopez, and L. J. Frehlick, in Spermatology, Ed. by E. R. S. Roldan and M. Gomendio (Nottingham Univ. Press, Nottingham, 2007), pp. 63–79.
E. I. Ramm, E. V. Chikhirzhina, E. I. Kostyleva, et al., Biokhimiya 60 (1), 150 (1995).
J. C. Jenson, P. Chin-Lin, B. Gerber-Jenson, et al., Proc. Natl. Acad. Sci. U. S. A. 77, 1389 (1980).
H. Triebel, H. Bar, A. Walter, et al., Int. J. Biol. Macromol. 11, 153 (1989).
E. I. Ramm, G. S. Ivanov, and V. I. Vorob’ev, Mol. Biol. (Moscow) 27 (5): 1061 (1993).
A. A. Kalashnikova, R. A. Rogge, and J. C. Hansen, Biochim. Biophys. Acta 1859, 455 (2016).
A. Kowalski and J. Palyga, Cell. Biol. Int. 36, 981 (2012).
N. Happel and D. Doenecke, Gene 431, 1 (2009).
D. V. Fyodorov, B. R. Zhou, A. I. Skoultchi, et al., Nat. Rev. Mol. Cell Biol. 1 (2017). doi 10.1038/nrm.2017.94
M. Colavito-Shepanski and M. A. Gorovsky, J. Biol. Chem. 258, 5944 (1983).
H. Yasuda, R. D. Mueller, K. A. Logan, et al., J. Biol. Chem. 261, 2349 (1986).
S. Nagel and U. Grossbach, J. Mol. Evol. 51, 286 (2000).
P. B. Talbert, K. Ahmad, G. Almouzni, et al., Epigen. Chromatin 5, 7 (2012).
J. Zlatanova and D. Doenecke, FASEB J. 8, 1260 (1994).
N. Happel, E. Schulze, and D. Doenecke, Biol. Chem. 386, 541 (2005).
C. L. Woodcock, A. I. Skoultchi, and Y. Fan, Chromosome Res. 14, 17 (2006).
Y. Fan, A. Sirotkin, R. G. Russell, et al., Mol. Cel. Biol. 21, 7933 (2001).
B. Sarg, R. Lopez, H. Lindner, et al., J. Proteomics 113, 162 (2015).
T. Yu. Starkova, A. M. Polyanichko, T. O. Artamonova, et al., Phys. Biol. 14, 016 005 (2017).
H. Talasz, N. Sapojnikova, W. Helliger et al., J. Biol. Chem. 273, 32 236 (1998).
S. Khochbin, Gene 271, 1 (2001).
A. Izzo and R. Schneider, Biochim. Biophys. Acta 1859, 486 (2016).
S. Mishra, A. Saleh, P. S. Espino, et al., J. Biol. Chem. 281, 5532 (2006).
D. Yellajoshyula and D. T. Brown, Proc. Natl. Acad. Sci. U. S. A. 103, 18 568 (2006).
B. Sarg, W. Helliger, H. Talasz, et al., J. Biol. Chem. 281, 6573 (2006).
P. J. Horn, L. M. Carruthers, C. Logie, et al., Nat. Struct. Biol. 9, 263 (2002).
A. Stutzer, S. Liokatis, A. Kiesel, et al., Mol. Cell 61, 247 (2016).
B. Xhemalce, M. A. Dawson, and A. J. Bannister, in Encyclopedia of Molecular Cell Biology and Molecular Medicine, Ed. by R. Meyers (Wiley, New York, 2011).
M. G. Alexandrow and J. L. Hamlin, J. Cell Biol. 168, 875 (2005).
S. W. Harshman, N. L.Young, M. R. Parthun, et al., Nucleic Acids Res. 41, 9593 (2013).
M. Dasso, S. Dimitrov, and A. P. Wolffe, Proc. Natl. Acad. Sci. U. S. A. 91, 12 477 (1994).
W. Fischle, Y. Wang, and C. D. Allis, Nature 425, 475 (2003).
S. Daujat, U. Zeissler, T. Waldmann, et al., J. Biol. Chem. 280, 38 090 (2005).
J.-M. Terme, L. Millán-Ariño, R. Mayor, et al., FEBS Lett. 588, 2353 (2014).
Y. Li, J. R. Danzer, P. Alvarez, et al., Development 130, 1817 (2003).
K. Ayyanathan, M. S. Lechner, P. Bell, et al., Genes Dev. 17, 1855 (2003).
S. L. Berger, Curr. Opin. Genet. Dev. 12, 142 (2002).
J. R. Wisniewski, A. Zougman, S. Krüger, et al., Mol. Cell Proteom. 6, 72 (2007).
A. Frechette, Huletsky, and R. J. Aubin, J. Biochem. Cell. Biol. 63, 764 (1985).
R. S. Wu, H. T. Panuasz, L. Hatch, et al., CRC Crit. Rev. Biochem. 20, 201 (1986).
J. M. Danielsen, K. B. Sylvestersen, S. Bekker-Jensen, et al., Mol. Cell. Proteom. 10, M110 003590 (2011).
R.Y. Tweedie-Cullen, J. M. Reck, and I. M. Mansuy, J. Proteome Res. 8, 4966 (2009).
G. T. Wondrak, D. Cervantes-Laurean, E. L. Jacobson, et al., Biochem. J. 351, 769 (2000).
J. L. Garcia-Gimenez, A. M. Ledesma, I. Esmoris, et al., Free Radic. Biol. Med. 52, 1453 (2012).
R. Sharma, A. Nakamura, R. Takahashi, et al., Free Radic. Biol. Med. 40, 1179 (2006).
Y. Shi, F. Lan, C. Matson, et al., Cell 119, 941 (2004).
M.A. Christophorou, G. Castelo-Branco, R. P. Halley-Stott, et al., Nature 507, 104 (2014).
E. R. Vossenaar, A. J. Zendman, and W. J. Van Venrooij, Arthritis Res. Ther. 6, 1 (2004).
M. Suzuki, EMBO J. 8, 797 (1989).
M. Suzuki, J. Mol. Biol. 207, 61 (1989).
J. C. Hansen, X. Lu, E. D. Ross, et al., J. Biol. Chem. 281, 1853 (2006).
X. Lu, B. Hamkalo, M. H. Parseghian, et al., Biochemistry 48, 164 (2009).
ACKNOWLEDGMENTS
This work was supported by the Russian Foundation for Basic Research (grant nos. 18-04-01199 and 18-08-01500) and by the Russian Science Foundation (grant no. 14-50-00068).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A. Barkhash
Rights and permissions
About this article

Cite this article
Chikhirzhina, E., Starkova, T. & Polyanichko, A. The Role of Linker Histones in Chromatin Structural Organization. 1. H1 Family Histones. BIOPHYSICS 63, 858–865 (2018). https://doi.org/10.1134/S0006350918060064
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006350918060064