
Abstract
Chitosanase plays a pivotal role in the production of chitooligosaccharide. Nevertheless, there is untapped potential for enhancing both its catalytic efficiency and thermostability, which could significantly bolster its therapeutic and biotechnological applications. In this study, two computer-aided protein design methods, namely Fireprot and PROSS, were utilized to pinpoint 6 single Bacillus subtilis chitosanase BsCsn46A mutants (S126A, D191A, K70A, L159I, K104P, and A129L) as well as 4 multiple mutants (K70A/S126A, K70A/S126A/K104P, K70A/S126A/L159I, and K70A/S126A/K104P/L159). The wild-type (WT) and all 10 BsCsn46A mutants displayed robust adaptability across a broad pH range, exhibiting peak activity within the pH spectrum of 5.5 to 9.5. The results demonstrated that, compared to the WT, 9 out of 10 mutants exhibited significantly heightened chitosanase activity, with the sole exception being the D191A mutant, which displayed activity levels nearly identical to the WT. Notably, the A129L displayed an impressive 20% increase in the enzyme activity at elevated temperatures, specifically in the range of 55–80°C. Assessing protein stability, results indicated that all samples maintained stability when incubated at 30°C for 1 h. However, when subjected to a higher temperature of 40°C for 1 h, only the A129L mutant retained stability, which persisted even after an extended incubation period of 3 h at 40°C. Furthermore, a thermal stability analysis revealed noteworthy differences between the WT and the mutants. The WT chitosanase activity diminished by 50% after brief 30-min incubation at 50°C, whereas the K70A/S126A, K70A/S126A/K104P, and A129L mutants maintained 50% of their activity for approximately 2 h under the same conditions. In summary, the study provides valuable insights into the thermostability and catalytic activity of chitosanase, highlighting promising candidates for industrial chitosanase applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chitosanase (EC. 3.2.1.132), a class of glycoside hydrolase (GH), primarily produced by bacteria and fungi, is involved in the hydrolysis of chitosan to produce chitooligosaccharide [1–3]. Chitooligosaccharide has gained extensive attention due to its outstanding biological properties, including anti-microbial [4], anti-tumor [5], antioxidizing [6], anti-inflammatory [7], immune enhancing [8], anti-Alzheimer’s [9], and free radical scavenging [10]. According to structural and amino acid sequence comparisons, chitosanases are differentiated into 6 families: GH 5, 7, 8, 46, 75, and 80 [11]. GH46 has been the most intensively examined in terms of its structure and function [2].
Catalytic activity and thermal stability are two critical aspects of enzymatic viability for industrial applications [2]. Due to production costs and industrial production conditions, there is increasing interest in improving the activity and thermal stability of industrially applied enzymes. Nature can generate new functionalities, however, it takes millions of years to evolve proteins with new functions, thus, humans must rely on their ingenuity to obtain the best-performing enzymes [12]. Rational design or directed evolution has significantly improved the properties of chitosanase. Guo et al. [2] optimized the catalytic activity of chitosanase from Bacillus subtilis using site-saturation mutagenesis of Pro 121. Sheng et al. [13] enhanced the thermostability of Bacillus ehimensis chitosanase by introducing disulfide bonds.
While these strategies are effective, over 30% of mutations have a deleterious impact on protein activity and stability, and when multiple random mutations are introduced to a protein simultaneously, it often causes a loss of protein function [14]. Recently, computational approaches have offered attractive alternatives to protein modifications. Software programs may be employed to improve protein activity and thermostability. However, rational design of chitosanase based on diverse computer-aided protein design methods has not yet been reported.
The aim of this study was to improve the activity and thermostability of the B. subtilis chitosanase BsCsn46A (BsCsn46A). Using Fireprot, a robust computational strategy that combines evolution- and energy-based approaches to predict highly stable multiple-point mutants [15] alongside PROSS, an automated sequence, and structure-based design method, for optimizing protein stability and heterologous expression levels [16], the essential amino acids of BsCsn46A were identified. Subsequently, the conservation of the identified loci was examined using the ConSurf server [17]. Ultimately, of the 10 mutants, 9 had the higher enzyme activity than that in the wild-type (WT). Mutants A129L, K70A/S126A, and K70A/S126A/K104P were identified to have significantly enhanced chitosanase thermal stability. The A129L mutant exhibited a 20% increase in activity compared to the WT at a broad range of temperatures. The protein modification strategy employed in the study may be appropriate for the rational modification of other microbial enzymes that have industrial applications.
MATERIALS AND METHODS
Strains, plasmids, and materials. Escherichia coli Top10 and E. coli BL21 (DE3) chemically competent cells were acquired from WEIDI (China). The coding sequence of BsCsn46A (GenBank: SNY67994.1) lacking the signal peptide sequence (N-terminal 35 amino acids) was ligated into the pET-28b (+) vector and synthesized by Sangon Biotech. (China). The chitosan substrates were obtained from Sangon Biotech. (China). The 3,5-dinitrosalicylic acid (DNS) was purchased from Solarbio (China).
Bioinformatics analysis. The theoretical isoelectric point and molecular weight of BsCsn46A and its mutants were obtained using Expasy (http://web.expasy.org/compute_pi) [18]. The signal peptide was predicted using Signal P 5.0 (http://services.healthtech.dtu.dk/services/SignalP-5.0) [19]. The three-dimensional protein structures of BsCsn46A and its mutants were projected using Fast AF2 (https://cloud.zelixri.com/fastaf2/#/). SAVES (https://saves.mbi.ucla.edu) was employed for structure validation. All structure-related figures were generated using PyMOL (http://pymol.org/2/) [20]. Fireprot (https://loschmidt.chemi.muni.cz/Fireprot/) [15] and PROSS (http://PROSS.weizmann.ac.il/step/PROSS-terms/) [16] were utilized for mutation site selection. Protein sequence conservation analysis was performed using the ConSurf server (https://consurf.tau.ac.il/index_proteins.php) [17]. Stabilizing substitutions in proteins were estimated using Consensus Finder (http://kazlab.umn.edu/) [21].
Site saturate mutagenesis. All site-directed mutagenesis of BsCsn46A was performed using the standard QuickChange PCR method [22]. Mutation conditions consisted of 25 cycles of 98°C for 40 s, 55°C for 1 min, and 72°C for 2 min. The final PCR products were digested using the restriction endonuclease DpnI at 37°C for 2 h to eliminate the parental methylated plasmid DNA. The digested DNA was transformed into E. coli Top10 cells and transformants were grown in Luria-Bertani (LB) agar medium (Sangon Biotech., China) supplemented with kanamycin (50 μg/mL). After verification through sequencing, correctly sequenced plasmids were transformed into E. coli BL21 (DE3) cells and transformants were grown in the same medium. All recombinant plasmids were confirmed by sequencing at Sangon Biotech. (China).
Protein expression and purification. E. coli BL21(DE3) containing pET-28b-BsCsn46A or its mutants was grown overnight in 100 mL LB medium supplemented with kanamycin (50 μg/mL) at 37°C with shaking (200 rpm). Twenty percent (vol/vol) of the overnight culture was transferred into 1 L the same fresh medium and cultured at 37°C and 220 rpm until the OD600 reached 0.8. The culture was then cooled to 18°C, 0.3 mM IPTG was added to induce target protein production, and the culture grew at 18°C and 200 rpm for 20 h. Cells were harvested by centrifugation (6000 g, 4°C, 15 min), and the supernatant was discarded. The wet cells were resuspended in 50 mM Tris-HCl buffer pH 8.0, 500 mM NaCl, 10 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol (equilibration buffer). Following high-pressure fragmentation by high-pressure homogenizer (JNBIO, China) for 15 min, cellular debris was removed by centrifugation at 18 000 g for 60 min at 4°C. The supernatant was then applied onto the Ni-affinity column (1 × 5 mL, Sangon Biotech., China) pre-equilibrated with equilibration buffer. The target protein was eluted with 50 mM Tris-HCl buffer pH 8.0, 500 mM NaCl, 250 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol (elution buffer). The eluted fraction was concentrated to approximately 1 mL using 10 kDa cut-off ultrafiltration tubes (Millipore, USA), and the protein was purified using Superdex 75 10/300 column (GE Healthcare, USA) in 20 mM Tris-HCl buffer with 100 mM NaCl, 2 mM DTT, pH 7.4 (gel filtration buffer). The purity of the purified proteins was assessed using 12.5% SDS-PAGE.
Chitosanase activity assay. The activity of BsCsn46A and its mutants was determined utilizing the modified DNS method [23] using chitosan as substrate. The reaction mixture was comprised of 5 μL of purified protein (5 mg/mL), 900 μL 50 mM MES buffer (pH 6.5), and 100 μL 2% (wt/vol) colloidal chitin dissolved in 1% glacial acetic acid. Following incubation at 50°C for 15 min, the mixture was centrifuged at 5000 g for 5 min to remove the residual chitosan. Then, 200 μL of supernatant was mixed with 200 μL of DNS reagent, boiled for 5 min, and subsequently cooled on ice. Enzyme activity was examined at 540 nm, and one unit (U) of enzyme activity was defined as the amount of enzyme that produced 1 µmol of reducing sugars per min under the above reaction conditions.
Temperature, optimal pH, and kinetic parameters. The optimum pH was measured in three 0.1 M buffers ranging from 5.5 to 9.5 (MES pH 5.5–6.5, Tris-HCl pH 7.0–8.5, and CHES pH 9.0–9.5). The effect of temperature on BsCsn46A or its mutant activity was assessed at temperatures ranging from 55 to 80°C in 0.1 M MES buffer (pH 6.5). Across all test groups, the relative enzyme activity under various pH and temperatures was determined by defining the highest enzyme activity rate as 100%.
Thermostability analysis. To assess the thermostability of BsCsn46A and its mutants, the half-life (t1/2) of the enzyme was measured. The t1/2 was established by preincubating the enzyme in size-exclusion chromatography buffer (20 mM Tris-HCl pH 7.4, 100 mM NaCl, 2 mM DTT) at 30–50°C over 10°C intervals for time from 0 to 2 h. The relative enzyme activity was computed by defining the highest enzyme activity rate as 100%.
RESULTS
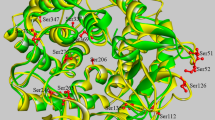
Homology modeling and identification of critical mutation sites. BsCsn46A encodes 277 amino acids with a signal peptide at its N-terminus (Figs. S1, Met1-Ala35). The molecular weight of the protein following removal of the signal peptide is approximately 28 kDa. The amino acid sequence after the removal of the signal peptide was renumbered, with Ala36 representing the starting amino acid, labeled Ala1, and the subsequent amino acids were sequentially ordered. The homology model of BsCsn46A was assembled using Fast AF2 by inputting the amino acid sequence of the enzyme. The model is composed of 9 α-helices and 2 β-folds (Fig. 1). The quality of the predicted 3D model was examined based on non-bonded atomic interactions and the Ramachandran plots. The results of the analysis exhibited that 91.2% of the amino acid residues were located within the Ramachandran favorable region, and 8.8% were within the Ramachandran allowed region (Table S1), with the overall quality factor being 100 (Figs. S2). These results suggest that the predicted model was of high quality and could be utilized as a structural model for the identification of key mutation sites.

3D model of BsCsn46A generated using FastAF2. The identified key mutation sites are exhibited as sticks, and the potential catalytic residues are shown as spheres.
The predicted model of BsCsn46A was uploaded to the Fireprot and PROSS servers for calculation. According to the Fireprot algorithm, 34 mutations based on evolution and free energy were recognized (Table S2). Of these, D191A is the only mutation site identified by the energy-based approaches FoldX and Rosetta (Table S2). According to the PROSS server, 9 multi-mutation combinations based on the structure and sequence were identified. Of these, K70A and S126A appear in 9 mutation combinations, L159I and K104P in 8, and A129L in 7 (Table S3). The following 6 single mutation sites were identified: K70A, K104P, S126A, A129L, L159I, and D191A (Fig. 1). We obtained 4 well-expressed multiple mutation sites K70A/S126A, K70A/S126A/K104P, K70A/S126A/L159I, and K70A/S126A/K104P/L159I based on single mutation site combinations. All screened amino acid sites were more than 15 Å away from the previously reported catalytically active amino acids Glu19 and Asp35 [2]. Sequence conservation results were examined by the ConSurf server and are shown in Figs. S3. Of all the identified mutation sites, S126 was the most conserved. Consensus Finder was used to predict the substitution of S126 (Table S4), which showed that 73% of similar proteins contained Ala instead of Ser at this site.
Expression and purification of BsCsn46A and its mutants. The BsCsn46A and all mutants were expressed as soluble proteins in E. coli BL21 (DE3) cells. High-purity target proteins were obtained by Ni-affinity chromatography and size exclusion chromatography. Similar to BsCsn46A, the molecular mass of all mutant proteins was approximately 30 kDa (Fig. 2).

SDS-PAGE analysis of BsCsn46A and its mutants. M, protein markers; lane 1, WT (Epizyme Biotech Company, cat.no.WJ101); lane 2, K70A; lane 3, S126A; lane 4, K70A/S126A; lane 5, K104P; lane 6, A129L; lane 7, L159I; lane 8, D191A; lane 9, K70A/S126A/K104P; lane 10, K70A/S126A/L159I; lane 11, K70A/S126A/K104P/L159I.
Determination of the specific activities of BsCsn46A and its mutants. The optimal pH of BsCsn46A and its mutants were assessed in three buffers ranging from 5.5 to 9.5, as described above. As illustrated in Fig. 3, the enzymes exhibited a wide range of pH adaptability in all groups, with strong enzyme activity at pH 6.5 and pH 8.0 or pH 8.5. The mutant K104P had stronger activity under alkaline conditions, with no significant difference between the remaining 10 recombinant enzymatic activities under acid-alkaline conditions (Figs. S3a–3b). Finally, we selected the BsCsn46A optimum pH condition in 0.1 M MES buffer pH 6.5 as the pH condition for thermal stability assessment.

Effects of pH on BsCsn46A and its single (a) and multiple (b) mutants.
Previously, Guo et al. [2] reported the optimum temperature of BsCsn46A to be 60°C. To determine if the thermal stability of the 10 selected mutants was enhanced, we performed an enzyme activity assay at a temperature 5°C higher than the optimal temperature of the WT, i.e. 65°C. Results of the enzyme activity assay in 0.1 M MES buffer at pH 6.5 and 65°C demonstrated that all remaining 9 mutants, aside from the D191A mutant, increased enzyme activity by approximately 20% over the WT BsCsn46A (Fig. 4a). No significant alteration in enzyme activity of the D191A mutant was observed compared to the WT. Further, we examined the enzyme activity under alkaline conditions. The results of the enzyme activity assay in 0.1 M Tris-HCl buffer at pH 8.0 also demonstrated that, except for the D191A mutant, the enzyme activity of the remaining 9 mutants was enhanced compared to the WT, but the enhancement was lower than that of the assay at pH 6.5 (Fig. 4b). Subsequently, the D191A mutant was removed, and we further compared the enzyme activities of the WT and the remaining 9 mutants in 0.1 M MES buffer (pH 6.5) at 55, 70, and 80°C (Figs. S4c–4e). The results exhibited the same trend at the 65°C assay, with the mutant enzyme’s activity being increased compared to the WT. The A129L mutant had the most significant increase in enzyme activity at an elevated temperature than the optimal reaction temperature of the WT (60°C).

Effects of temperature on the activity of BsCsn46A and its mutants. The values in X axes represent the sample. (a, b): 1, WT; 2, K70A; 3, S126A; 4, K70A/S126A; 5, K104P; 6, A129L; 7, L159I; 8, D191A; 9, K70A/S126A/K104P; 10, K70A/S126A/L159I; 11, K70A/S126A/K104P/L159I. (c–e): 1, WT; 2, K70A; 3, S126A; 4, K70A/S126A; 5, K104P; 6, A129L; 7, L159I; 8, K70A/S126A/K104P; 9, K70A/S126A/L159I; 10, K70A/S126A/K104P/L159I.
Thermal stability analysis of BsCsn46A and its mutants. The protein thermal stability results demonstrated that all 10 samples, including the WT, were stable after 1 h of incubation at 30°C. K70A/S126A (termed: M2), K70A/S126A/K104P (termed: M3) precipitated slightly after 1 h of incubation at 40°C, K70A and S126A had a small amount of precipitation, A129L was stable with no precipitation, and the remaining 4 mutants, K104P, L159I, K70A/S126A/L159I, K70A/S126A/K104P/L159I, and WT precipitated heavily. A129L remained stable even after 3 h of incubation at 40°C. A129L, M2, and M3 were chosen from the above mutants to examine the stability of the protein at 50°C. Complete precipitation of WT and thermally stable A129L, M2, and M3 mutants was observed following 10 min incubation at 50°C (Figs. S4a–4c). We examined the enzyme activity of heat-stable mutants A129L, M2, M3 and the WT after incubation at 40 and 50°C for 0–2 h. The results illustrated that although the WT protein partially precipitated after 1 h of incubation at 40°C, when the sample was mixed and added to the reaction system, it did not impact its enzymatic activity even after 2 h of incubation (Fig. 5a). The WT exhibited 50% reduction in enzymatic activity after 40 min of incubation at 50°C, while heat-stable mutants A129L, M2, M3 retained about 50% activity even after 2 h of incubation (Fig. 5b).

Enzyme activity of heat-stable mutants A129L (1), M2 (2), M3 (3) and WT (4) after incubation at 40 (a) and 50°C (b) for 0–2 h.
DISCUSSION
Improving enzymes’ activity or thermal stability to attain higher catalytic efficiency has attracted increasing attention [2]. Modification in thermostability is the key method of increasing the industrial value of enzymes [24]. Computer-aided design of stability-enhancing mutations is a reliable alternative to screening-based approaches [25]. Many recent breakthroughs have been achieved in the study of the thermal stability of enzymes. Rosetta and FoldX have been utilized in combinatorial strategies such as Fireprot, supplemented by protein structure analysis, and evolutionary analysis to acquire mutants with enhanced stability [15, 26]. However, it is necessary to employ a combination of forecasting methods to enhance the accuracy of the predictions. Zhao et al. [24] improved the thermal stability of pepsin by examining structural features and using a combination of multi-prediction software programs. Li et al. [27] reduced the experimental screening process of fungal lipases through rational design using multiple computational design methods. Li et al. [28] employed 4 computational methods combined with saturation mutations to optimize the multiple properties of β-glucosidase Bgl6.
To enable the thermostability and catalytic activity of BsCsn46A by combining the prediction results of Fireprot, PROSS, ConSurf, and Consensus Finder, 10 key combination mutations were identified. The mutation sites were scattered on the surface of BsCsn46A (Fig. 1), meeting our requirements for thermodynamic weakness analysis. Meanwhile, the lowered activity of heat-stable mutants is a common activity-stability trade-off in enzymatic design [29]. Often, the increased stability of an enzyme is accompanied by a decrease in catalytic activity [30]. However, in our study, we did not observe a similar trend. The results exhibited that 9 mutants had higher activity than BsCsn46A, while the mutant D191A, which was identified using energy-based Fireprot did not enhance enzyme activity. As previously reported by Zhao et al. [24], mutants predicted by energy-based programs HoTMuSiC, PoPMuSiC, DeepDDG, and ETSS resulted in the complete loss of enzyme activity. Among all mutants, the A129L mutant had the highest enzymatic activity and protein thermal stability. HotSpot Wizard is a web server for the automated design of smart libraries and mutations according to the sequence input information [31]. A129L was identified as a key amino acid site based on HotSpot Wizard analysis (Table S5). This provides novel evidence for A129 as a critical amino acid site for BsCsn46A.
In summary, using Fireprot and PROSS, we identified 6 single mutants and 4 multiple mutants. The WT and the 10 mutants had strong pH adaptability with activity at pH region of 5.5–9.5. All 9 mutants had higher activity than the WT, except for the D191A mutant. The protein thermal stability of A129L, M2, and M3 increased, especially A129L. A129L exhibited a 20% increase in activity compared to the wild type at 55–80°C. Overall, our results identified key amino acids impacting the thermostability and catalytic activity of BsCsn46A.
DATA AVAILABILITY
All data that support the findings from this study are available from the corresponding author SYD and RJ upon reasonable request.
REFERENCES
Su, H., Sun, J., Guo, C., Jia, Z., and Mao, X., J. Agric. Food Chem., 2022, vol. 70, pp. 6168–6176. https://doi.org/10.1021/acs.jafc.2c01577
Guo, J., Wang, Y., Zhang, X., Gao, W., Cai, Z.; Hong, T., et al., J. Agric. Food Chem., 2021, vol. 69, pp. 11835–11846. https://doi.org/10.1021/acs.jafc.1c04206
Shinya, S. and Fukamizo, T., Int. J. Biol. Macromol., 2017, vol. 104, pp. 1422–1435. https://doi.org/10.1016/j.ijbiomac.2017.02.040
Yue, L., Li, J., Chen, W., Liu, X., Jiang, Q., and Xia, W., Carbohydr. Polym., 2017, vol. 176, pp. 356–364. https://doi.org/10.1016/j.carbpol.2017.07.043
Zhang, W., Xu, W., Lan, Y., He, X., Liu, K., and Liang, Y., Int. J. Nanomed., 2019, vol. 14, pp. 5287–5301. https://doi.org/10.2147/IJN.S203113
Ma, C., Li, X., Yang, K., and Li, S., Mar. Drugs, 2020, vol. 18. https://doi.org/10.3390/md18020126
Yang, Y., Ao, H., Wang, Y., Lin, W., Yang, S., Zhang, S., et al., Bone Res., 2016, vol. 4. https://doi.org/10.1038/boneres.2016.27
Rahimnejad, S., Yuan, X., Wang, L., Lu, K., Song, K., and Zhang, C., Fish Shellfish Immunol., 2018, vol. 80, pp. 405–415. https://doi.org/10.1016/j.fsi.2018.06.025
Eom, T.K., Ryu, B.M., Lee, J.K., Byun, H.G., Park, S.J., and Kim, S.K., J. Enzyme Inhib. Med. Chem., 2013, vol. 28, pp. 214–217. https://doi.org/10.3109/14756366.2011.629197
Park, P.J., Je, J.Y., and Kim, S.K., J. Agric. Food Chem., 2003, vol. 51, pp. 4624–4627. https://doi.org/10.1021/jf034039+
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P.M., and Henrissat, B., Nucleic Acids Res., 2014, vol. 42, pp. 490–495. https://doi.org/10.1093/nar/gkt1178
Lynch, M. and Conery, J.S., Science, 2000, vol. 290, pp. 1151–1155. https://doi.org/10.1126/science.290.5494.1151
Sheng, J., Ji, X., Zheng, Y., Wang, Z., and Sun, M., Biotechnol. Lett., 2016, vol. 38, pp. 1809–1815. https://doi.org/10.1007/s10529-016-2168-2
Clifton, B.E., Kozome, D., and Laurino, P., Biochemistry, 2023, vol. 62, pp. 210–220. https://doi.org/10.1021/acs.biochem.1c00757
Musil, M., Stourac, J., Bendl, J., Brezovsky, J., Prokop, Z., Zendulka, J., et al., Nucleic Acids Res., 2017, vol. 45, pp. 393–399. https://doi.org/10.1093/nar/gkx285
Weinstein, J.J., Goldenzweig, A., Hoch, S.Y., and Fleishman, S.J., Bioinformatics, 2020, vol. 37, pp. 123–125. https://doi.org/10.1093/bioinformatics/btaa1071
Ashkenazy, H., Abadi, S., Martz, E., Chay, O., Mayrose, I., Pupko, T., et al., Nucleic Acids Res., 2016, vol. 44, pp. 344–350. https://doi.org/10.1093/nar/gkw408
Duvaud, S., Gabella, C., Lisacek, F., Stockinger, H., Ioannidis, V., and Durinx, C., Nucleic Acids Res., 2021, vol. 49, pp. 216–227. https://doi.org/10.1093/nar/gkab225
Almagro Armenteros, J.J., Tsirigos, K.D., Sønderby, C.K., Petersen, T.N., Winther, O., Brunak, S., et al., Nat. Biotechnol., 2019, vol. 37, pp. 420–423. https://doi.org/10.1038/s41587-019-0036-z
O’Donoghue, S.I., Goodsell, D.S., Frangakis, A.S., Jossinet, F., Laskowski, R.A., Nilges, M., et al., Nat. Methods, 2010, vol. 7, pp. 42–55. https://doi.org/10.1038/nmeth.1427
Jones, B.J., Kan, C.N.E., Luo, C., and Kazlauskas, R.J., Methods Enzymol., 2020, vol. 643, pp.129–148. https://doi.org/10.1016/bs.mie.2020.07.010
Hogrefe, H.H., Cline, J., Youngblood, G.L., and Allen, R.M., Biotechniques, 2002, vol. 33, pp. 1158–1160, 1162, 1164–1165. https://doi.org/10.2144/02335pf01
Guo, N., Sun, J., Wang, W., Gao, L., Liu, J., Liu, Z., et al., Food Chem., 2019, vol. 286, pp. 696–702. https://doi.org/10.1016/j.foodchem.2019.02.056
Zhao, Y., Miao, Y., Zhi, F., Pan, Y., Zhang, J., Yang, X., et al., Front. Physiol., 2021, vol. 9, pp. 1–11. https://doi.org/10.3389/fphy.2021.755253
Xing, H., Zou, G., Liu, C., Chai, S., Yan, X., Li, X., et al., Enzyme Microb. Technol., 2021, vol. 143, https://doi.org/10.1016/j.enzmictec.2020.109720
Li, G., Fang, X., Su, F., Chen, Y., Xu, L., and Yan, Y., Appl. Environ. Microbiol., 2018, vol. 84, pp. e02129—17. https://doi.org/10.1128/aem.02129-17
Li, G., Fang, X., Su, F., Chen, Y., Xu, L., and Yan, Y., Appl. Environ. Microbiol., 2018, vol. 84. https://doi.org/10.1128/AEM.02129-17
Li, S., Cao, L., Yang, X., Wu, X., Xu, S., and Liu, Y., Bioresour. Technol., 2023, vol. 374, p. 128792. https://doi.org/10.1016/j.biortech.2023.128792
Stimple, S.D., Smith, M.D., and Tessier, P.M., AIChE J., 2020, vol. 66, e16814. https://doi.org/10.1002/aic.16814
Zhu, F., He, B., Gu, F., Deng, H., Chen, C., Wang, W., et al., J. Biotechnol., 2020, vol. 309, pp. 68–74. https://doi.org/10.1016/j.jbiotec.2019.12.014
Sumbalova, L., Stourac, J., Martinek, T., Bednar, D., and Damborsky, J., Nucleic Acids Res., 2018, vol. 46, pp. 356–362. https://doi.org/10.1093/nar/gky417
ACKNOWLEDGMENTS
This work was supported by the doctoral research fund of Zaozhuang University, China.
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This work does not contain any studies involving human and animal subjects.
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
DECLARATIONS COMPETING INTEREST
The authors declare that there are no financial/personal interests that could affect the objectivity of the study.
Additional information
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Fig. S1. The predicted signal of BsCsn46A. Signal peptide (Sec/SPI). The cleavage site between pos. 35 and 36: VFA-AG. Probability: 0.9189.
Fig. S2. The overall quality factor of the model identified by errat is 100. On the error axis, two lines are drawn to indicate the confidence with which it is possible to reject regions that exceed that error value.
Fig. S3. Sequence conservation analysis of BsCsn46A. The sequence is colored according to the conservation score.
Fig. S4. Effects of temperature on the thermal stability of BsCsn46A and its mutants. (a). Thermal stability analysis of BsCsn46A and its mutants at 30°C in size-exclusion chromatography buffer (20 mM Tris-HCl pH7.4, 100 mM NaCl, 2 mM DTT). Upper level from left to right: WT, K70A, S126A, M2, K104P, Lower level from left to right: A129L, L159I, M3, K70A/S126A/L159I, K70A/S126A/K104P/L159I. (b) Thermal stability analysis of BsCsn46A and its mutants at 40°C with the same sample order as at 30°C. Heat-stable samples are labeled with red circles. (c) Thermal stability analysis of BsCsn46A and its mutants at 50°C. Left to right: WT, A129L, M2, M3.
Table S1. Ramachandran plot of the model
Table S2. Key amino acids identified by Fireprot
Table S3. Key amino acids identified by PROSS
Table S4. The mutations predicted by Consensus Finder
Table S5.
Rights and permissions
About this article

Cite this article
Duan, S.Y., Zhang, X.S., Yuan, Y.Q. et al. Improving the Thermostability and Catalytic Performance of the Bacillus subtilis Chitosanase BsCsn46A via Computational Design. Appl Biochem Microbiol 60, 889–896 (2024). https://doi.org/10.1134/S0003683823603207
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0003683823603207