
Abstract
The relevance of most genetic polymorphisms beyond CYP3A5*1 on tacrolimus disposition remains unclear. We constructed a predictive mixed model for tacrolimus dose-corrected trough concentration (C0/dose) at months 3, 12 and 24 after transplantation in a retrospective cohort of 766 predominantly Causasian adult renal recipients (n=2042 trough concentrations). All patients were genotyped for 32 single-nucleotide polymorphisms with a proven or possible relevance to tacrolimus disposition based on the previous studies. Of these, ABCB1, ABCC2, OATP1B1, COMT, FMO, PPARA and APOA5 were analyzed as (functional) diplotype groups. Predictors of C0/dose were CYP3A5*1, hematocrit, age, CYP3A4*22, use of concomitant CYP3A4 inhibitor or inducer, ALT, estimated glomerular filtration rate, tacrolimus formulation (once vs twice daily), ABCB1 diplotype and time after transplantation. The effect of ABCB1 diplotype was small but strongly accentuated in CYP3A4*22 carriers and non-existent in CYP3A5 expressors. ABCC2 diplotype had a limited effect on C0/dose that was only statistically significant in CYP3A5 non-expressors.
Similar content being viewed by others


Introduction
The calcineurin inhibitor tacrolimus is the cornerstone of most immunosuppressive regimens in solid organ transplantation. It is, however, characterized by a narrow therapeutic window and high pharmacokinetic variability, necessitating therapeutic drug monitoring. Tacrolimus is a dual substrate for the cytochrome P450 isoenzymes CYP3A4 and CYP3A5, and the efflux pump P-glycoprotein (P-gp, ATP-binding cassette subfamily B member 1 (ABCB1), encoded by the multidrug-resistance-1 gene (MDR1)). CYP3A and P-gp are present in enterocytes (limiting intestinal uptake) and hepatocytes (determining systemic clearance). P-gp is also significantly expressed in many other tissues and epithelia such as the kidneys, placenta, lymphocytes and blood–brain barrier. In blood, tacrolimus is >95% bound to red blood cells, which shields it from hepatic extraction because equilibration with the free fraction in plasma (which is available for uptake into hepatocytes where it is metabolized) is relatively slow.1 When hematocrit is low, the free fraction will tend to increase but is immediately extracted by the liver, resulting in higher clearance and a higher-dose requirement to maintain a given whole-blood concentration. Tacrolimus bioavailability is reduced when ingested with food,2 10–15% lower for once daily tacrolimus compared with the twice daily formulation3 and generally increased in case of intestinal inflammation and diarrhea.4 Clearance is reduced with liver dysfunction,5, 6 older age7 and increasing time after transplantation,8, 9 the latter in part because of a decrease in intestinal and hepatic CYP3A4 activity early after transplantation that is likely related to steroid tapering.9
Several common genetic polymorphisms in enzymes, transporters and regulatory molecules have been reported to affect tacrolimus disposition. The single most influential polymorphism by far is in the CYP3A5 gene. Carriers of one or more CYP3A5*1 alleles (termed CYP3A5 expressors) have strongly increased CYP3A activity resulting in 50% higher tacrolimus dose requirement compared with CYP3A5*3/*3 patients (non-expressors).10 In addition, the CYP3A4*22 polymorphism, which is present almost exclusively in CYP3A5 non-expressors, is associated with lower tacrolimus dose requirement.11 The effect of ABCB1 polymorphisms is unclear and variable between studies.12 A large pharmacogenomic screening study by Jacobson et al. identified several polymorphisms other than CYP3A5*1 that influenced the first tacrolimus trough after transplantation, but none significantly predicted troughs after the first week after correcting for CYP3A5*1 and these polymorphisms have never been validated.13 Other studies have reported varying effects of single-nucleotide polymorphisms (SNPs) in the POR (P-oxidoreductase),14, 15, 16, 17 human pregane X receptor,18, 19, 20, 21 multidrug-resistance-associated protein 2 (MRP2, encoded by the gene ABCC2),17, 22, 23, 24 OATP1B3,25 OATP1B1 (ref. 26) and PPARA (peroxisome proliferator-activated receptor alpha).21, 27, 28
The aim of this study was to develop a pharmagenetic model for tacrolimus exposure in a large cohort of renal recipients and examine the potential added value of previously described genetic polymorphisms when systematically correcting for CYP3A5 genotype and relevant clinical factors.
Materials and methods
Patient population
This is a single center, retrospective cohort analysis. In the University Hospitals Leuven Renal Transplant Program, the majority (>95%) of renal recipients are included in a prospective Renal Transplant Biobank program for which they provide written informed consent (ClinicalTrials.gov identifier NCT01331668). DNA samples are stored immediately before tranplantation. At 3, 12 and 24 months after transplantation, patients are seen in the outpatient clinic at which time protocol biopsies are performed together with routine laboratory analysis and physical examination. Clinical information including drug dose, weight and laboratory results was collected in electronic clinical patient charts. Glomerular filtration rate was estimated using the CKD-EPI formula. This study was approved by the Ethics Committee of the University Hospitals Leuven (S53364; ML7499).
Tacrolimus measurements
Whole-blood samples were collected in 4-ml ethylenediaminetetraacetic acid tubes between 08:00 and 10:00 hours, before intake of the morning dose of tacrolimus. Tacrolimus trough concentrations (C0) were determined on the microparticulate enzyme immunoassay platform used by the central hospital laboratory at the time. Up to 16 December 2010, this was Dimension RxL (Siemens, Munich, Germany; analytical sensitivity 1.2 μg l−1; coefficient of variation 2.9–6.8%). From 17 December 2010 onwards, this was Architect i2000sr (Abbott Laboratories, Abbott Park, IL, USA; analytical sensitivity ⩽1.5 μg l−1; coefficient of variation 8%). Tacrolimus was orally administered. Dose-corrected trough concentrations (C0/dose) were determined using the total dose of tacrolimus ingested the day before C0 measurement.
Genotyping and haplotype inferrence
Genomic DNA was isolated from whole-blood samples using a salting out procedure.29 The quantity and quality of genomic DNA were verified with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) before being assayed on an OpenArray platform (Life Technologies, Carlsbad, CA, USA). Overall call rate was 99.8%. A list of all tested SNPs is provided in Supplementary Table S1. Hardy–Weinberg equilibrium and linkage disequilibrium (LD) between SNPs were assessed using Haploview.30 Haplotypes were inferred using the program PHASE version 2.1.31
Statistical methods
Data are presented as mean±s.d. except when stated otherwise. Normality was tested using the Shapiro–Wilk test. C0/dose, estimated glomerular filtration rate (eGFR), aspartate aminotransferase (AST), alanine aminotransferase (ALT), bilirubin, albumin and methylprednisolone dose were not normally distributed and log-transformed for analysis. A linear mixed model with random slopes, random intercepts and a first-order autoregressive covariance structure was used to estimate the effect of covariates on tacrolimus C0/dose at months 3, 12 and 24 after transplantation. This model estimates C0/dose separately for each time point (that is, three times for each patient) based on the values of covariates at these specific time points. Calculation of estimates was based on restricted maximum likelihoods. Fixed and random effects tested included (when appropriate) age, gender, diabetes mellitus, living vs deceased donor, weight, hematocrit, time after transplantation, serum albumin, AST, ALT, bilirubin, eGFR, use of moderate or strong CYP3A4 inhibitor or inducer, calcineurin inhibitor formulation (once daily vs twice daily), methylprednisolone dose, and use of induction therapy. Genetic predictors were tested as haplotypes and (functional groups of) diplotypes. In the final mixed model, we only included those terms that were statistically significant using F-test and improved the model according to Akaike’s information criterion. The Holm–Bonferroni correction for multiple testing was used. A two-sided P-value<0.05 was considered statisically significant. All analyses were performed using IBM SPSS Statistics version 22 (IBM, New York, NY, USA) except for calculation of semipartial R2s, which was done with the program R using the lme4 and MuMIn packages. Figures were generated using Graphpad Prism version 6 (San Diego, CA, USA).
Results
Study population
All patients included in the Renal Transplantation Biobank program who received a single kidney transplant from December 2006 through September 2013 and were started on standard triple immunosuppressive therapy (tacrolimus–mycophenolic acid (MMF)–steroids) were included in the analysis (n=766). Tacrolimus was either administered once daily or twice daily (respectively, Advagraf and Prograft, Astellas Pharma Europe, Staines, UK); MMF was either mycophenolate mofetil (Cellcept, Roche, Basel, Switzerland) or mycophenolate sodium (Myfortic, Novartis, Basel, Switzerland). Baseline recipient characteristics including selected genotype information and evolution of C0/dose and clinical covariates between month 3, year 1 and year 2 are shown in Table 1.
Over the 2-year follow-up period, 42 patients (5.5%) died or lost their graft. At month 3, year 1 and year 2, there were 24, 46 and 82 patients, respectively, without available C0/dose because they had either been switched to ciclosporin, sirolimus or everolimus, or were lost to follow-up. Target trough concentrations for tacrolimus were 10–12 ng ml−1 from month 1 to 3, 8–10 ng ml−1 from month 3 to 12 and 6–8 ng ml−1 after month 12.
Genotype and inferred haplotype
All SNPs were in Hardy–Weinberg equilibrium except CYP3A4*1b. There was significant LD between SNPs in COMT (r2=0.99), PPARA (r2=1), ABCB1 (r2 between all four SNPs ⩾0.78), ABCC2 (r2 between all three SNPs ⩾0.79) and APOA5 (r=0.99), and between FMO-1 and 3 (r2=1). CYP3A5*1 was in significant LD with CYP3A7 (r2=0.91), CYP3A4*1G (r2=0.97) and CYP3A4*22 (r2=1). Within the OATP1B1 gene, there was strong LD between g.-11187G>A and c.388A>G (r2=0.95), c.388A>G and c.521T>C (r2=0.68), but only limited LD between g.-11187G>A and c.521T>C (r2=0.27). Four heterozygous CYP3A5 expressors were also CYP3A4*22 carrier. Frequency distribution of all SNPs and corresponding inferred diplotypes are available in the Supplementary Table S1 and S2). Eight ABCB1 haplotypes were identified, forming 22 different diplotypes. These were grouped into four diplotype categories based on the presence of wild-type CGC and triple mutated (loss of function) TTT haplotypes: CGC–CGC, CGC–TTT, TTT–TTT and ‘other’ (containing one or more other haplotypes). For the triallelic ABCB1 G2677A/T SNP, the A allele was only present in 12 patients and grouped with the T allele for all analyses. Six ABCC2 haplotypes were identified, which were grouped into three functional diplotype groups (reference group, high activity and low activity) based on the impact of their individual SNPs on protein expression as described by Laechelt.32
Predictors of tacrolimus C0/dose
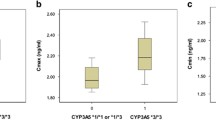
Independent predictors of C0/dose in multivariate mixed model analysis are shown in Table 2. Tacrolimus C0/dose increased with increasing age, time after transplantation, higher eGFR, higher ALT, use of CYP3A4 inhibitors, presence of the CYP3A4*22 allele and one or more ABCB1 TTT haplotypes. C0/dose was lower for CYP3A5 expressors, lower hematocrit, use of strong CYP3A4 inducers and use of the once daily formulation of tacrolimus. The final model explained 44.6% of interpatient variability in C0/dose. A scatterplot of model-predicted C0/dose vs observed C0/dose is shown in Figure 1. Observed C0/dose values stratified by CYP3A5 and CYP3A4 genotype are shown in Figure 2.

Scatterplot of predicted vs observed log-transformed C0/dose in final mixed model.

Observed log-transformed C0/dose stratified by CYP3A5 and CYP3A4 genotype. Boxplot whiskers indicate 95% confidence interval. The four CYP3A5*1/*3 patients who carried a CYP3A4*22 allele were categorized in the CYP3A5*1/*3 group.
The estimated effects reported in Table 2 are B-values (unstandardized coefficients) for the linear regression equation and indicate how much the C0/dose is expected to increase for each unit increase (or for each 100% increase, in the case of log-transformed predictors) in a continous predictor variable or for each increment of a categorical predictor variable. A significant interaction was identified between CYP3A4*22 and ABCB1 haplotype (P=0.017). Estimated mean C0/dose for the different CYP3A4*22–ABCB1 combinations are presented in Table 3. The C0/dose ratio was 11% higher in ABCB1 TTT–TTT carriers compared with CGC–CGC carriers if they were CYP3A4 wild type, but the difference was 62% if they were CYP3A4*22 carriers. Overall, there was no statistically significant difference in C0/dose between ABCC2 diplotype groups (P=0.076). In the subgroup of CYP3A5 non-expressors, the ABCC2 low-activity diplotype was associated with a lower C0/dose compared with the reference- and high-activity groups (mean difference 0.08±0.03 ng ml−1/mg per day; P=0.017).
Predictors of tacrolimus C0/dose in the subgroup of CYP3A5 expressors are presented in Supplementary Table S3. Together, these explained 21.1% of interindividual variability in C0/dose. For CYP3A5 non-expressors, predictors were identical to the overall model, explaining 35.1% of C0/dose variability (data not shown). An interaction was identified between CYP3A4*22 and age. The age-related increase in C0/dose was accelerated in CYP3A4*22 carriers (estimated difference 31.8%, P=0.030; for example, increase of C0/dose associated with aging 30 years: 0.73 ng ml−1/mg per day in CYP3A4*1/*1 and 1.07 ng ml−1/mg per day in CYP3A4*1/*22 patients).
Concomitant CYP3A4 inhibitors and inducers
Concomitantly used CYP3A4 inhibitors and inducers were categorized as being moderate or strong based on the Food and Drug Administration classification of their in vivo inhibitory/inductive potency33 and are listed in Supplementary Table S4. Model fit was significantly better when concomitant inhibitors/inducers were included as covariates (R2 0.446 vs 0.368 when they were not accounted for, see Figure 3). A significant interaction was identified between CYP3A5 genotype and the effect of CYP3A4 inhibitors (P<0.001), whereby moderate inhibitors had no effect in CYP3A5 expressors. Estimated mean C0/dose for patients using inhibitors depending on their CYP3A5 genotype are presented in Table 4. No genotype interactions were found with use of CYP3A4 inducers.

Scatterplot of predicted vs observed log-transformed C0/dose in mixed model where concomitant CYP3A4 inhibiting/inducing medication is not included as covariate (a). Including concomitant medication as covariate improves R2 by 0.078 (b).
Discussion
We demonstrate for the first time that the effect of ABCB1 diplotype on tacrolimus disposition is dependent on both CYP3A5 and CYP3A4 genotype. The P-gp transporter is subjected to considerable genetic variability with 3435C>T, 2677G>T/A and C1236C>T being the main polymorphisms. The functional implications of these polymorphisms are substrate-dependent and studies have reported conflicting results regarding their effect on tacrolimus disposition.34 In reports where an effect was observed, the TTT haplotype was generally associated with an increased C0/dose, compatible with reduced P-gp function. Several studies have noted that this effect was limited to CYP3A5 non-expressors.20, 35 Results of the current study are generally in line with these previous observations. The ABCB1 TTT allele was associated with a progressive increase in C0/dose in CYP3A5 non-expressors, but the overall effect was limited. Testing for interaction terms revealed that the increase in tacrolimus exposure related to the ABCB1 TTT haplotype was significantly more pronounced in CYP3A4*22 carriers. This would suggest that reduced P-gp function is most relevant when CYP3A activity is low, but has little to no impact when CYP3A activity is high (in CYP3A5 expressors), and also implies less interaction potential with P-gp inhibitors in CYP3A5 expressors. This is biologically plausible, as cells with a very high metabolic turnover of tacrolimus can be expected to depend little on efflux pumps. Conversely, when tacrolimus metabolism is slow, it becomes more important for efflux pumps to keep intracellular concentrations low so CYP3A4 capacity is not saturated. This interplay is expected to be most relevant in enterocytes, where a combination of low CYP3A and P-gp activity would result in saturation of metabolic capacity and strongly increased bioavailability. Indeed, the effect of loss-of-function ABCB1 alleles on whole-body disposition of tacrolimus is likely mainly the result of reduced intestinal efflux and higher bioavailability. Given that tacrolimus is a lipophilic BDDCS class II compound (low solubility and extensive metabolism), reduced function of hepatic efflux transporters is not expected to increase exposure.36 On the contrary, inhibition of hepatic P-gp in isolated rat liver experiments resulted in higher tacrolimus clearance, presumably because exposure to hepatic CYP3A enzymes increased.37
MRP2 is an apical efflux transporter that has an important role in biliary excretion of phase II conjugates, but is present on many physiological barriers, including the intestine. We observed a statistically significant but marginal (and clinically irrelevant) increase in tacrolimus exposure in CYP3A5 non-expressors possessing a low-activity ABCC2 diplotype. As for P-gp, this observation would be most easily explained by reduced intestinal efflux resulting in increased bioavailability.
The effect of CYP3A4 inhibitors on tacrolimus exposure depended both on their inhibitory potency and CYP3A5 genotype. In CYP3A5 non-expressors, moderate inhibitors increased C0/dose by 20%, whereas strong inhibitors increased it by 207%. In CYP3A5 expressors, moderate inhibitors increased C0/dose 13% (not statistically significant), but the strong inhibitor voriconazole increased it >20-fold. It must be noted that there was only one CYP3A5 expressor using voriconazole, which warrants caution in overinterpreting the precise differences in C0/dose between the groups. However, the data do suggest that CYP3A5 expressors are relatively resistant to the effect of moderate, but not of strong inhibitors. This is in agreement with the observation that the CYP3A5 enzyme is intrinsically resistant to inhibition in vitro.38 We have previously observed that the moderate CYP3A4 inhibitor fluconazole decreased tacrolimus dose requirements in CYP3A5 non-expressors, but not in CYP3A5 expressors.39 The potent inhibitor voriconazole, however, is known to significantly alter tacrolimus disposition even in CYP3A5 expressors.40 The observation that CYP3A5 genotype modulates the magnitude of interaction between tacrolimus and CYP3A4 inhibitors could have implications for the development of a predictive model for tacrolimus DDIs, which is an important, and currently unmet, clinical need. Predictably, CYP3A4 inducers decreased C0/dose. No interaction was identified between CYP3A4 inducers and CYP3A5 genotype groups, which may have been related to low patient numbers in several of these subgroups.
The effects of various clinical covariates on tacrolimus C0/dose in this report are generally in line with the previous studies and all but one are logical from a physiological point of view. Specifically, the higher-dose requirement observed with decreasing eGFR is counterintuitive, although others have reported the same association.19 The kidneys do not contribute meaningfully to overall clearance of tacrolimus. It has been demonstrated that end-stage renal disease can affect the disposition of nonrenally eliminated drugs, at least in part through the effects of uremic toxins, inflammatory cytokines and parathyroid hormone on hepatic and intestinal enzymes, and transporters.41 However, renal dysfunction results in the suppression of hepatic CYP3A4 and P-gp activity, which would be expected to result in lower tacrolimus dose requirements. At any rate, the in vivo effect of renal dysfunction on oral tacrolimus disposition seems to be minimal, as clearance of tacrolimus is similar between healthy volunteers and patients with end-stage renal disease.42 It is possible that the effect of renal dysfunction in our study was confounded by concomitant disease states such as anorexia and vomiting, and reverse causation might be present as high tacrolimus trough concentrations are nephrotoxic. Renal dysfunction is also often accompanied by anemia, which increases dose requirements, although no multicollinearity was identified between eGFR and hematocrit (variance inflation factor 1.22).
Polymorphisms in the genes POR, COMT, FMO-1 and -3, SLCO1B3, SLCO1B1, SCLO2B1, PPARA, GAN, SIM1, APOA5, and NR1I2 had no significant effect on tacrolimus C0/dose after correction for other genetic and clinical predictors, contrary to the previous reports. There are several possible explanations for these discrepancies. First, differences in ethnicity of the study population will likely have contributed. For example, CYP3A5*1 is much more prevalent in Afrian-American and Asian populations, studies reporting a significant effect of CYP3A4*1G were mainly performed in Asians,43 ethnic distribution of ABCB1 haplotypes varies widely,44, 45 CYP3A4*22 is very rare in Chinese patients46 and was not a predictor of tacrolimus disposition in Brazilian renal recipients.47 Second, between-study differences in time after transplantation are relevant when modeling C0/dose, as some predictors may only exert an effect within a certain time window. For example, the variability in observed hematocrit, albumin and eGFR values is highest early after transplantation, so their predictive effect on a population level is also expected to be highest at this point. We opted to analyze several fixed time points for every patient to accommodate time-related variability. Third, several pharmacogenomic studies did not correct for well-known clinical determinants such as hematocrit. Finally, there may have been a relatively high degree of clinical variability (concomitant medication, illness and time after transplantation) in our study population, which could have obscured the effect of genetic determinants that might be relevant in more controlled circumstances.
This study has several strengths. Sample size was large, multiple time points were evaluated and systematic correction for relevant clinical variables was performed. All tested SNPs have previously been reported to be relevant to tacrolimus disposition, reducing the probability of false-positive results. Analysis of (functional) diplotypes facilitates the interpretation of genotype–phenotype associations. In this case, the decrease in tacrolimus dose requirement associated with the loss-of-function ABCB1 and ABCC2 diplotypes is biologically plausible.
Several limitations must be noted. First, some potential sources of variability in tacrolimus exposure (diarrhea, non-compliance and food intake) were not registered systematically and cannot be accounted for in the model. Second, the population was unselected and encompasses the full range of variability encountered in clinical practice including anemia, hypoalbuminemia, DDI, hepatic and renal dysfunction. This clinical variability can obscure the effects of genetic polymorphisms that have a subtle impact. On the other hand, it reinforces the robustness of the predictors that did make it into the model and provides a model of a ‘real-life’ population. Third, although our transplant population is mainly Caucasian, information regarding ethnicity is not stored in medical files and exact numbers cannot be provided. Fourth, ABCB1 was highly genetically variable (22 individual diplotypes) and 33% of patients possessed haplotypes other than CGC or TTT that were generally too rare to allow reliable C0/dose estimates to be made. Finally, not every polymorphism with a reported possible relevance to tacrolimus disposition was tested, including some SNPs (SERPING1 rs4926 and EPS15 rs17567) reported by Jacobson et al. We did not test for SNPs that are very rare in Caucasian populations, such as CYP3A5*6 and *7 or SNPs in NR1I3.
In conclusion, the effect of ABCB1 diplotype on tacrolimus dose requirement was small and modulated by both CYP3A5 and CYP3A4 genotype. In CYP3A5 expressors, the effect of moderate CYP3A inhibitors on tacrolimus disposition was limited. This study demonstrates how the cooperative function of CYP3A and ABCB1 also translates into interdependence of the effects of genetic polymorphisms. This information could help facilitate future efforts to develop comprehensive pharmacokinetic models for tacrolimus that integrate clinical and genomic information.
References
Piekoszewski W, Chow FS, Jusko WJ . Disposition of tacrolimus (FK 506) in rabbits. Role of red blood cell binding in hepatic clearance. Drug Metab Dispos 1993; 21: 690–698.
Bekersky I, Dressler D, Mekki Q . Effect of time of meal consumption on bioavailability of a single oral 5 mg tacrolimus dose. J Clin Pharmacol 2001; 41: 289–297.
de Jonge H, Kuypers DR, Verbeke K, Vanrenterghem Y . Reduced C0 concentrations and increased dose requirements in renal allograft recipients converted to the novel once-daily tacrolimus formulation. Transplantation 2010; 90: 523–529.
Maes BD, Lemahieu W, Kuypers D, Evenepoel P, Coosemans W, Pirenne J et al. Differential effect of diarrhea on FK506 versus cyclosporine A trough levels and resultant prevention of allograft rejection in renal transplant recipients. Am J Transplant 2002; 2: 989–992.
Fukudo M, Yano I, Masuda S, Goto M, Uesugi M, Katsura T et al. Population pharmacokinetic and pharmacogenomic analysis of tacrolimus in pediatric living-donor liver transplant recipients. Clin Pharmacol Ther 2006; 80: 331–345.
Staatz CE, Willis C, Taylor PJ, Tett SE . Population pharmacokinetics of tacrolimus in adult kidney transplant recipients. Clin Pharmacol Ther 2002; 72: 660–669.
Jacobson P, Schladt D, Oetting WS, Leduc R, Guan W, Matas AJ et al. Lower calcineurin inhibitor doses in older compared to younger kidney transplant recipients yield similar troughs. Am J Transplant 2012; 12: 3326–3336.
Kuypers DRJ, de Jonge H, Naesens M, Lerut E, Verbeke K, Vanrenterghem Y . CYP3A5 and CYP3A4 but not MDR1 single-nucleotide polymorphisms determine long-term tacrolimus disposition and drug-related nephrotoxicity in renal recipients. Clin Pharmacol Ther 2007; 82: 711–725.
de Jonge H, Vanhove T, de Loor H, Verbeke K, Kuypers DR . Progressive decline in tacrolimus clearance after renal transplantation is partially explained by decreasing CYP3A4 activity and increasing haematocrit. Br J Clin Pharmacol 2015; 80: 548–559.
Terrazzino S, Quaglia M, Stratta P, Canonico PL, Genazzani AA . The effect of CYP3A5 6986A>G and ABCB1 3435C>T on tacrolimus dose-adjusted trough levels and acute rejection rates in renal transplant patients: a systematic review and meta-analysis. Pharmacogenet Genomics 2012; 22: 642–645.
Elens L, Hesselink DA, van Schaik RHN, van Gelder T . The CYP3A4*22 allele affects the predictive value of a pharmacogenetic algorithm predicting tacrolimus predose concentrations. Br J Clin Pharmacol 2013; 75: 1545–1547.
Staatz CE, Goodman LK, Tett SE . Effect of CYP3A and ABCB1Single nucleotide polymorphisms on the pharmacokinetics and pharmarcodynamics of calcineurin inhibitors: part II. Clin Pharmacokinet 2010; 49: 207–222.
Jacobson PA, Oetting WS, Brearley AM, Leduc R, Guan W, Schladt D et al. Novel polymorphisms associated with tacrolimus trough concentrations: results from a multicenter kidney transplant consortium. Transplantation 2011; 91: 300–308.
de Jonge H, Metalidis C, Naesens M, Lambrechts D, Kuypers DRJ . The P450 oxidoreductase *28 SNP is associated with low initial tacrolimus exposure and increased dose requirements in CYP3A5-expressing renal recipients. Pharmacogenomics 2011; 12: 1281–1291.
Gijsen VMGJ, van Schaik RHN, Soldin OP, Soldin SJ, Nulman I, Koren G et al. P450 oxidoreductase *28 (POR*28) and tacrolimus disposition in pediatric kidney transplant recipients—a pilot study. Ther Drug Monit 2014; 36: 152–158.
Kuypers DRJ, de Loor H, Naesens M, Coopmans T, de Jonge H . Combined effects of CYP3A5*1, POR*28, and CYP3A4*22 single nucleotide polymorphisms on early concentration-controlled tacrolimus exposure in de-novo renal recipients. Pharmacogenet Genomics 2014; 24: 597–606.
Pulk RA, Schladt DS, Oetting WS, Guan W, Israni AK, Matas AJ et al. Multigene predictors of tacrolimus exposure in kidney transplant recipients. Pharmacogenomics 2015; 16: 841–854.
Benkali K, Prémaud A, Picard N, Rérolle J-P, Toupance O, Hoizey G et al. Tacrolimus population pharmacokinetic-pharmacogenetic analysis and bayesian estimation in renal transplant recipients. Clin Pharmacokinet 2009; 48: 805–816.
Barraclough KA, Isbel NM, Lee KJ, Bergmann TK, Johnson DW, McWhinney BC et al. NR1I2 polymorphisms are related to tacrolimus dose-adjusted exposure and BK viremia in adult kidney transplantation. Transplantation 2012; 94: 1025–1032.
Li J-L, Liu S, Fu Q, Zhang Y, Wang X-D, Liu X-M et al. Interactive effects of CYP3A4, CYP3A5, MDR1 and NR1I2 polymorphisms on tracrolimus trough concentrations in early postrenal transplant recipients. Pharmacogenomics 2015; 16: 1355–1365.
Bruckmueller H, Werk AN, Renders L, Feldkamp T, Tepel M, Borst C et al. Which genetic determinants should be considered for tacrolimus dose optimization in kidney transplantation? A combined analysis of genes affecting the CYP3A locus. Ther Drug Monit 2015; 37: 288–295.
Renders L, Frisman M, Ufer M, Mosyagin I, Haenisch S, Ott U et al. CYP3A5 genotype markedly influences the pharmacokinetics of tacrolimus and sirolimus in kidney transplant recipients. Clin Pharmacol Ther 2007; 81: 228–234.
Zhao W, Elie V, Roussey G, Brochard K, Niaudet P, Leroy V et al. Population pharmacokinetics and pharmacogenetics of tacrolimus in de novo pediatric kidney transplant recipients. J Clin Pharmacol 2010; 50: 1280–1291.
Ogasawara K, Chitnis SD, Gohh RY, Christians U, Akhlaghi F . Multidrug resistance-associated protein 2 (MRP2/ABCC2) haplotypes significantly affect the pharmacokinetics of tacrolimus in kidney transplant recipients. Clin Pharmacokinet 2013; 52: 751–762.
Boivin A-A, Cardinal H, Barama A, Naud J, Pichette V, Hebert M-J et al. Influence of SLCO1B3 genetic variations on tacrolimus pharmacokinetics in renal transplant recipients. Drug Metab Pharmacokinet 2013; 28: 274–277.
Elens L, Capron A, Kerckhove V, Van, Lerut J, Mourad M, Lison D et al. 1199G>A and 2677G>T/A polymorphisms of ABCB1 independently affect tacrolimus concentration in hepatic tissue after liver transplantation. Pharmacogenet Genomics 2007; 17: 873–883.
Kurzawski M, Malinowski D, Dziewanowski K, Droździk M . Impact of PPARA and POR polymorphisms on tacrolimus pharmacokinetics and new-onset diabetes in kidney transplant recipients. Pharmacogenet Genomics 2014; 24: 397–400.
Lunde I, Bremer S, Midtvedt K, Mohebi B, Dahl M, Bergan S et al. The influence of CYP3A, PPARA, and POR genetic variants on the pharmacokinetics of tacrolimus and cyclosporine in renal transplant recipients. Eur J Clin Pharmacol 2014; 70: 685–693.
Miller SA, Dykes DD, Polesky HF . A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Barrett JC, Fry B, Maller J, Daly MJ . Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Stephens M, Smith NJ, Donnelly P . A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001; 68: 978–989.
Laechelt S, Turrini E, Ruehmkorf A, Siegmund W, Cascorbi I, Haenisch S . Impact of ABCC2 haplotypes on transcriptional and posttranscriptional gene regulation and function. Pharmacogenomics J 2011; 11: 25–34.
FDA. Drug development and drug interactions: table of substrates, inhibitors and inducers. Available at http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm (accessed on 18 November 2015).
Staatz C, Goodman L, Tett S . Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: part I. Clin Pharmacokinet 2010; 49: 141–175.
Wang J, Zeevi A, McCurry K, Schuetz E, Zheng H, Iacono A et al. Impact of ABCB1 (MDR1) haplotypes on tacrolimus dosing in adult lung transplant patients who are CYP3A5 *3/*3 non-expressors. Transpl Immunol 2006; 15: 235–240.
Wu C-Y, Benet LZ . Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res 2005; 22: 11–23.
Wu C-Y, Benet LZ . Disposition of tacrolimus in isolated perfused rat liver: influence of troleandomycin, cyclosporine, and gg918. Drug Metab Dispos 2003; 31: 1292–1295.
Yamazaki H, Nakamoto M, Shimizu M, Murayama N, Niwa T . Potential impact of cytochrome P450 3A5 in human liver on drug interactions with triazoles. Br J Clin Pharmacol 2010; 69: 593–597.
Kuypers DR, de Jonge H, Naesens M, Vanrenterghem Y . Effects of CYP3A5 and MDR1 single nucleotide polymorphisms on drug interactions between tacrolimus and fluconazole in renal allograft recipients. Pharmacogenet Genomics 2008; 18: 861–868.
Iwamoto T, Monma F, Fujieda A, Nakatani K, Katayama N, Okuda M . Hepatic drug interaction between tacrolimus and lansoprazole in a bone marrow transplant patient receiving voriconazole and harboring CYP2C19 and CYP3A5 heterozygous mutations. Clin Ther 2011; 33: 1077–1080.
Naud J, Nolin TD, Leblond Fa, Pichette V . Current understanding of drug disposition in kidney disease. J Clin Pharmacol 2012; 52: 10S–22S.
Astellas Pharma US. Tacrolimus (Prograf): prescribing information 2013. Available at https://www.astellas.us/docs/prograf.pdf; last accessed on 3 December 2015.
Miura M, Satoh S, Kagaya H, Saito M, Numakura K, Tsuchiya N et al. Impact of the CYP3A4*1G polymorphism and its combination with CYP3A5 genotypes on tacrolimus pharmacokinetics in renal transplant patients. Pharmacogenomics 2011; 12: 977–984.
Kroetz DL, Pauli-Magnus C, Hodges LM, Huang CC, Kawamoto M, Johns SJ et al. Sequence diversity and haplotype structure in the human ABCB1 (MDR1, multidrug resistance transporter) gene. Pharmacogenetics 2003; 13: 481–494.
Yi S-Y, Hong K-S, Lim H-S, Chung J-Y, Oh D-S, Kim J-R et al. A variant 2677A allele of the MDR1 gene affects fexofenadine disposition. Clin Pharmacol Ther 2004; 76: 418–427.
Shi Y, Li Y, Tang J, Zhang J, Zou Y, Cai B et al. Influence of CYP3A4, CYP3A5 and MDR-1 polymorphisms on tacrolimus pharmacokinetics and early renal dysfunction in liver transplant recipients. Gene 2013; 512: 226–231.
Santoro AB, Struchiner CJ, Felipe CR, Tedesco-Silva H, Medina-Pestana JO, Suarez-Kurtz G . CYP3A5 genotype, but not CYP3A4*1b, CYP3A4*22, or hematocrit, predicts tacrolimus dose requirements in Brazilian renal transplant patients. Clin Pharmacol Ther 2013; 94: 201–202.
Acknowledgements
We thank A Herelixka for managing the clinical database, T Coopmans, J de Loor, M Dekens and T Van Brussel for their technical assistance and our study nurses C Beerten, J De Vis, M Dubois and H Wielandt for their work in the Biobank program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
DK has received consulting fees and research grants from Astellas. The remaining authors declared no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Vanhove, T., Annaert, P., Lambrechts, D. et al. Effect of ABCB1 diplotype on tacrolimus disposition in renal recipients depends on CYP3A5 and CYP3A4 genotype. Pharmacogenomics J 17, 556–562 (2017). https://doi.org/10.1038/tpj.2016.49
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2016.49
- Springer Nature Limited
This article is cited by
-
Impact of single-nucleotide polymorphisms on tacrolimus pharmacokinetics in liver transplant patients after switching to once-daily dosing
Hepatology International (2023)
-
A New CYP3A5*3 and CYP3A4*22 Cluster Influencing Tacrolimus Target Concentrations: A Population Approach
Clinical Pharmacokinetics (2017)