
Abstract
Genetic variants responsible for Maturity-Onset-Diabetes of the Young (MODY) in Kuwait were investigated. A newly established a National Referral Clinic, the Dasman Diabetes Institute (DDI-NRC), assessed forty-five members from 31 suspected MODY families by whole exome sequencing. Thirty-three of the 45 samples were independently sequenced at the DDI-NRI, Exeter University, UK (https://www.diabetesgenes.org/) using targeted 21-gene panel approach. Pathogenic mutations in GCK, HNF1A, HNF1B, HNF4A, and PDX1 confirmed MODY in 7 families, giving an overall positivity rate of 22.6% in this cohort. Novel variants were identified in three families in PDX1, HNF1B, and HNF1B. In this cohort, Multiplex Ligation-dependent Probe Amplification assay did not add any value to MODY variant detection rate in sequencing negative cases. In highly selected familial autoantibody negative diabetes, known MODY genes represent a minority and 77.3% of the familial cases have yet to have a causal variant described.
Similar content being viewed by others

Introduction
Maturity-onset diabetes of the young (MODY) is a rare form of monogenic diabetes caused by dominantly acting heterozygous variants in genes necessary for the development or function of pancreatic β-cells1. To date, multiple distinct forms of MODY (MODY1-14) have been identified with varied clinical and genetic heterogeneity2. MODY frequently represents a diagnostic challenge for clinicians as it is a rare condition that shares clinical features with both type 13,4, and type 2 diabetes mellitus5,6 (T1D and T2D) and is often misdiagnosed as such. While clinicians often learn about MODY from its autosomal dominant inheritance pattern, many individuals lack a family history7. However, unlike T1D and T2D, molecular genetic testing is sensitive and specific for diagnosing MODY, and it provides essential information on therapeutic management and follow-up8,9,10.
The 14 known MODY genes account for ~ 1–6% of pediatric diabetes cases worldwide11,12. Since dozens of disease alleles have been reported for most of these loci in European and non-European populations13, next generation sequencing (NGS) based genetic testing is the preferred diagnostic approach as it provides cost-effective screening of the entire exome or a selected panel of MODY genes14. The genetic changes associated with MODY in European populations have expanded to include copy number variation15. As a result, the introduction of additional technologies, such as DNA microarrays or Multiplex Ligation-dependent Probe Amplification (MLPA), can complement NGS in establishing a definitive diagnosis of MODY.
To date, most studies have searched for genetic causes of MODY in individuals with European ancestry, while only a small number of studies have been conducted in Arabia and the Middle East16,17,18,19,20. Given the high frequency of consanguineous marriages and the large burden of genetic homozygosity21, studies in this region are crucial to identify novel genetic variants and new MODY-associated genes. Recently, we highlighted the MODY knowledge gap to healthcare providers in the Middle East region to rectify this niche16. To the best of our knowledge, this is the first study from the region reporting on an initiative to diagnose patients with MODY based on a referral system to a specialized National Referral Clinic at the Dasman Diabetes Institute (DDI-NRC). We present data from the initial phase of the study, which describes 31 families characterized by whole exome sequencing (WES) and MLPA performed at the DDI-NRC and by a model sequencing panel performed independently at the Royal Devon and Exeter NHS Foundation Trust Genetics Laboratory (“Exeter”).
Materials and methods
Family data and diagnosis
This study reports data collected between January 2013 to June 2017. Patients with suspected MODY (31 families, 45 individuals), based on the International Society of Pediatric and Adolescent Diabetes (ISPAD) clinical criteria9, were referred from all over Kuwait to the DDI-NRC. At the DDI-NRC, the patients and their family members completed a standardized questionnaire and were evaluated by a pediatric endocrinologist, who examined the patients, collected detailed information on their phenotype, and coordinated with the treating physicians to compile clinical data. Body mass index (BMI) measures for children younger than the age of 19 years were expressed as standard deviation scores (SDS) determined by the World Health Organization growth standards22.
MODY was suspected if: (1) the patient was < 25 years of age at the time of diabetes diagnosis; (2) had a family history suggestive of autosomal dominant inheritance; (3) lacked evidence suggesting a diagnosis of T1D (evidence of endogenous insulin production outside the honeymoon period with detectable C peptide level (> 200 nmol/L) when glucose is > 8 mmol/L and absence of pancreatic islet autoantibodies); and/or (4) lacked evidence supporting a diagnosis of T2D (normal body weight, absence of acanthosis nigricans, and no evidence of insulin resistance with normal fasting C peptide level).
The index patient and/or their parents were informed about the benefits of testing first-degree relatives: if consent was provided, family members were also enrolled. Genetic testing was performed in Kuwait (DDI) and at Exeter. The DDI laboratory and bioinformatic staff were blinded to the Exeter results. Only the Principal Investigator of the study had full access to the laboratory results. Patients with positive test results were offered genetic counselling and susceptibility testing of first-degree relatives, if not already performed.
The study was approved by the Ethical Review Committees at DDI and the Ministry of Health in Kuwait and carried out in accordance with the principles of the Declaration of Helsinki as revised in 2008. Written informed consent was obtained from all patients or their parents if the index patient was a minor.
Extraction of DNA from peripheral blood
Blood samples were collected from the index patient and his/her first-degree relatives. Genomic DNA was extracted using a QIAamp Blood DNA kit (Qiagen, Germany) and quantified spectrophotometrically using a Qubit Fluorometer (Thermofisher, USA), following the manufacturer’s protocol.
Whole exome sequencing and analysis
Exome libraries were prepared using Nextera Rapid Capture Exome kits (Illumina Inc., USA) following the manufacturer’s protocol. Sequencing was carried out on a HiSeq 2500 using Illumina’s Sequence by Synthesis technology with 100 bp paired-end reads at a mean depth of 100X.—BCL to FASTQ conversion was carried out using bcl2fastq v.2.20 software. Sequence reads are aligned to the reference human genome build hg19 using BWA v.0.7.17, and variant calling was carried using GATK v.3.7 HaplotypeCaller following the best practices. The variant call file of the affected individual was analyzed using VSClinical v2.2.2 software (Golden Helix, MT, USA) with default setting of genotype quality of ≥ 20, depth ≥ 25, and MAF OF < 0.01. Of the variants that pass the filter, only those that are predicted to cause loss-of-function or missense mutations were analyzed. The pathogenicity of identified variants was further assessed using ACMG criteria. Allelic frequencies were estimated, within Varseq, based on public databases including gnomAD v2.1.1, dbSNP, 1000 Genomes Project, our local Arab genome database, and Iranome23. Clinical interpretation of the variants was based on Polyphen, ClinVar, Leiden Open Variation Database (LOVD), Human Gene Mutation Database (HGMD), Online Mendelian Inheritance in Man (OMIM), and American College of Medical Genetics (ACMG) recommendations. The quality of the sequence reads was assessed using Integrative Genomics Viewer (IGV) Software (Broad Institute, United States).
Targeted validation by sanger sequencing
Shortlisted variants were validated by Sanger sequencing on an ABI 3730 xl DNA sequencer (Applied Biosystems, Foster City, USA) following standard protocol.
Multiplex Ligation-dependent Probe Amplification (MLPA) assay
MLPA assays were carried out following the manufacturer’s protocol (MRC-Holland, Amsterdam, The Netherlands) using P241-E1 MODY Mix-1 and P357-A3 MODY Mix-2. A total of 100 ng of DNA extracted from peripheral blood of patients, and reference samples were subjected to denaturation, hybridization, ligation, and amplification as per protocol instructions. Capillary electrophoresis was performed on ABI-3730xl DNA Analyser (Applied Biosystems, USA), and data were analyzed using Coffalyser (MRC Holland).
Results
Defining the MODY phenotypic spectrum
During the study period, 31 families (45 individuals) were recruited and referred for genetic screening. Table 1 shows the clinical characteristics of the index patients (16 males and 15 females); mean age was 13.6 (SD ± 7.48) years, mean age at diagnosis of diabetes was 10.02 (SD ± 5.85) years, and mean BMI z scores was 1.37 (SD ± 1.42). The mean HbA1c for the index patients was 8.26% (SD ± 1.73).
The MODY exome and validation
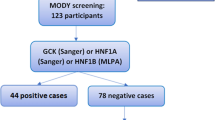
Forty-five patients contributed DNA for exome sequencing (31 index patients and 15 first-degree family members). Seven out of 31 index patients (22.6%) were confirmed as MODY with variants in genes encoding GCK, HNF1A, HNF1B, HNF4A, and PDX1 (Table 2). In most cases (24 families; 77%), no variant of significance was detected in any monogenic diabetes-linked genes (Table 2). To exclude large deletion/duplication events as a possible cause of MODY in these 24 families, we subjected the DNA to MLPA but found no copy number variant in any of the MODY genes (Fig. S1). To confirm our sequencing and MLPA data accuracy, 35 samples from 28 families were independently validated by comparing the DDI-NRC results with panel-based genetic testing performed at Exeter (adopting a blinded experimental approach).
Identification of causal variants in MODY
A likely causal variant was identified in 21.8% of the studied families, spanning 36 genes that have been reported previously to cause monogenic diabetes. Each of the seven affected individuals carried a monoallelic variant in one of the 36 monogenic diabetes genes, and together represented five different subtypes of MODY (Table 2).
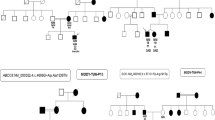
In Family 1, two male siblings presented with diabetes at the ages of 16 and 12 years, respectively, requiring insulin replacement therapy (Fig. 1A). Exome sequences from both siblings showed a novel missense variant in PDX1, which was confirmed by Sanger sequencing in both siblings (Fig. 1B,C). The maternal grandfather, as well as two maternal uncles, had diabetes, indicating a likely possibility of maternal transmission of the variant. Indeed, the variant was maternally inherited, although the mother was not diabetic, suggesting incomplete penetrance of this variant (Fig. 1C). Incomplete penetrance of pathogenic mutations in the PDX1 gene has been described previously24, particularly in the absence of MLKL genetic mutation, a key necroptosis protein25. PDX1 is an essential gene in pancreatic development: homozygous PDX1 variants have been previously described in two children with pancreatic agenesis (one who had consanguineous parents and one whose both parents had MODY4);. In contrast, heterozygous PDX1 variants have been shown to cause MODY4 in a small number of studies26. Our finding thus reports a novel PDX1 variant. Although we used the objective ACMG scoring system, which designated it as “likely pathogenic”, we need to be cautious with this label because the variant has not been observed previously, and functional studies are lacking. Clinvar reports six known pathogenic missense variants in PDX1, indicating that missense alterations are a common disease-causing mechanism in this gene. The c.461C > G, p.Thr154Arg variant is located in the DNA-binding homeodomain and is two amino acids downstream of T151, which is phosphorylated by Per-Arnt-Sim Kinase (PASK) and is essential for the regulation of insulin promoter factor-1 activity.

(A) Represents pedigree of a family positive for Maturity Onset Diabetes of the Young (MODY4). The square symbol represents male, and the circle represents female. The filled in symbols indicate affected individuals and the blue arrow indicates the index case. Next generation sequencing revealed a novel heterozygous missense variant in Pancreatic and duodenal homeobox 1 (PDX1) c.461C > G resulting in p.Thr154Arg. (B) Exome sequencing summary layout depicting part of the PDX1 gene location on chromosome 13 at the top and the exons coverage in green and blue. The location of the variants in exon 2 is represented by a vertical green bar and the reference sequence and the altered codon is highlighted. (C) Chromatogram represents Sanger sequencing results confirming the segregation of heterozygous variants in the mother, the index case and his brother.
The rare clinical subtype, MODY5, was confirmed by identifying variants in HNF1B for two index cases whose clinical presentations were consistent with the diagnosis of renal cyst and diabetes syndrome (RCAD) (Table 2). The HNF1B exon 4 p.E138K variant, in family 2, was detected in a Kuwaiti boy diagnosed shortly after birth with chronic renal failure due to bilateral cystic kidney disease. After renal transplantation at the age of 3 years, he started to show intermittent symptomatic hyperglycemia at the age of 9 years. His hyperglycemia became persistent, requiring insulin therapy at the age of 10 years. Unfortunately, other family members refused to consent for genetic testing. Three variants within six amino acid positions of the variant p.E138K have been shown to be pathogenic, while none are benign. The second HNF1B variant, c.494G > A, was identified in 6 years and nine-month-old Pakistani boy who was found to have a high creatinine level (108.0 µmol/L) at the initial diagnosis of diabetes at the age of around 4- years. The child was obese with a BMI of 23.9 kg/m2 (z score + 3.84 SD) and was managed by multiple daily injections of insulin (Table 2, Family 27). The c.494G > A (p.R165H) variant has been reported as “pathogenic” and “likely pathogenic” in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/12647/). Nine variants within six amino acids of p.R165H have been shown to be pathogenic, while none are benign. HNF1B contains 74 pathogenic missense variants, indicating that missense variants are a common disease mechanism in this gene. It encodes a member of the homeodomain-containing superfamily of transcription factors. The protein binds to DNA as either a homodimer or a heterodimer with the related protein hepatocyte nuclear factor 1-alpha (HNF1A). HNF1B has been shown to function in nephron development and to regulate the development of the embryonic pancreas. Variants in this gene result in renal cysts and MODY5.
The diagnosis of MODY3 (HNF1A) was established in two index cases, one with a well-known frameshift insertion c.872dupC (p. G292Argfs*25) in exon 4 and one with a novel heterozygous missense variant c.8C > A (p.S3Y) in exon 1 of the HNF1A (Table 2). The frameshift c.872dupC variant was detected in a 30-year-old Jordanian woman diagnosed with T1D at the age of 16 years (Fig. 2). She was initially treated with insulin (0.5 U/kg/day). Following the diagnosis of MODY3, the insulin therapy was replaced with Gliclazide (60 mg, twice daily), resulting in optimal glycemic control (HbA1c = 6.7%). This specific variant has been reported previously in at least 22 families and is listed in LOVD database 322 times as pathogenic. The variant causes a frameshift with loss of HNF1α protein activity. The pathogenic duplication variant detected in this family is estimated to account for approximately 20% of HNF1A-MODY3 families27. The other heterozygous c.8C > A variant in HNF1A was identified in a 26-year-old Kuwaiti woman diagnosed with T2D who had a strong family history of diabetes (Fig. 3). She was slightly overweight with a BMI of 25.5 kg/m2 and had poor glycemic control (HbA1c = 9.0%) while being treated with oral agents (Tables 1 and 2, Family 9). The missense variant p.S3Y in HNF1A has not been reported previously as a pathogenic variant nor as a benign variant, to our knowledge, but has been previously described in a patient with hepatocellular carcinoma28. The variant co-segregates with the disease in multiple affected family members (Fig. 3).

(A) Represents pedigree of a family positive for Maturity Onset Diabetes of the Young (MODY3). The square symbol represents male, and the circle represents female. The filled in symbols indicate affected individuals and the blue arrow indicates the index case. (B) Exome sequencing summary layout depicting part of the HNF1A gene location on chromosome 12 at the top and the exons coverage in green and blue. The location of the c.872dupC, (p.Gly292fs) frameshift mutation is represented by a vertical yellow bar and the reference sequence and the altered codon is highlighted. (C) Chromatogram represents Sanger sequencing of the HNF1A gene in control family member (top) and confirming the heterozygous frameshift mutation in the index (bottom arrow-line).

(A) Represents pedigree of a family positive for Maturity Onset Diabetes of the Young (MODY3). The square symbol represents male, and the circle represents female. The filled in symbols indicate affected individuals and the blue arrow indicates the index case. Next generation sequencing revealed a novel heterozygous missense variant in HNF1A, c.8C > A, resulting in p.Ser3Tyr. (B) Chromatogram represents Sanger sequencing results confirming the segregation of heterozygous variant in the index case, affected and unaffected family members.
In family 8, the index patient was a 6-year-old girl who had easily controlled diabetes since 2.5 years (Table 1). Her mother had a history of gestational diabetes. We identified a heterozygous missense variant in HNF4A, confirming MODY1 (Table 2). To the best of our knowledge, the missense variant p.I463V in HNF4A has not been reported previously as a pathogenic variant nor as a benign variant. Polyphen In silico analysis predicts a Tolerated/benign effect on protein function when isoleucine is substituted for a valine amino acid at codon 463. However, SIFT analysis predicts a damaging change. HNF4A contains 11 pathogenic missense variants and a low rate of benign missense variation as indicated by a high missense variant Z-score of 1.81 (Table 2, ACMG, PP2). The p.I463V variant has been reported as rare in all populations with -minor allele frequency of 0.00037 or 0.037% except for the Latino population in gnomAD where it is observed in 0.07% of alleles (ACMG, BS1). For these reasons, we classified the variant as VUS (Table 2).
Family 12 was diagnosed with MODY2 (Table 2). The index patient was a 5-year-old Egyptian boy with a maternally inherited missense variant in GCK gene c.572G > A (p. R191Q). The child was born to a mother with T2D managed on oral medications. The child’s fasting blood glucose ranged from 7.0–7.7 mmol/L, while his post-prandial blood glucose did not exceed 10.0 mmol/L. Neither mother nor the child required any additional treatment upon diagnosis. The p.R191Q variant occurs at the same amino acid position as the previously classified pathogenic variant p.R191W. GCK contains 122 pathogenic missense variants and three variants within six amino acid positions of the variant p.R191Q, while none have been shown to be benign. The missense variant has been identified in three families from Chile29 and Italian children with MODY230.
Discussion
The genetic basis of MODY in Middle Eastern populations is unknown. In the Gulf Cooperation Council (GCC) region, a study from Oman examined 20 patients with suspected MODY but found no variants in three MODY genes (HNF4A, GCK, and HNF1A) sequenced at Exeter20. Similarly, two gene-specific studies conducted in Tunisia identified two variants, one in HNF4A (from 12 patients with diabetes) and the other in the GCK gene (from 23 unrelated patients with diabetes) respectively17,18. More recently, a third study from Tunisia utilized targeted NGS and identified four variants from 11 patients suspected to have MODY in ABCC8, HNF1A, and GCK, improving the positive diagnostic rate significantly19. There are no studies on MODY published from Kuwait, Bahrain, or Qatar to date. Yet the Middle East and North Africa (MENA) region has the second highest prevalence of diabetes world-wide; and furthermore 14.8% of the adult Kuwaiti population are estimated to live with diabetes31. Kuwait also has one of the highest incidence rates of T1D in children32,33. This challenge is further confounded in countries like Kuwait and other Gulf Cooperation Council (GCC) by familial clustering of T2D and high rates of consanguineous marriages. It is unclear why academically and clinically lucrative studies on MODY have not been pursued in the Middle East. However, we can postulate three possible reasons for the lack of information on MODY and its genetics in the Middle East; The first may be due to the fact that the diagnosis of MODY is often challenging due to its shared clinical features with T1D and T2D5. The second reason may pertain to the technically demanding infrastructure required for the diagnosis of MODY, which is just being addressed in this area of the world. The third reason may be linked to the high propensity of familial clustering in T1D and T2D. For example, a panoply of locally conducted studies has established strong links between positive family history and T2D, with 71% of Arab patients with diabetes in Qatar and 80% of Omani patients having first degree relatives with diabetes34; compared to a lower frequency of 33% among Europeans35. Lastly, with the extremely high prevalence of obesity in the Middle East and the GCC, specifically in children and young adults36,37, the differentiation between MODY and T2D becomes even more clinically challenging. Since ISPAD guidelines suggest that the diagnosis of MODY should be suspected in cases that lack phenotype characteristics of type 2 diabetes, including obesity, the increasing prevalence of obesity may lead to fewer people with MODY being investigated using genetic tests9.
In this first-of-its-kind study from the region, we sequenced whole exomes of index patients and first-degree family members from 31 families with suspected MODY based on phenotype and clinical presentation (including negative T1D autoantibodies). We detected 7 MODY gene variants accounting for a positive detection rate of 22.6%. As our genetic testing was not done systematically in the population but focused on highly selected patients with clinically suspected MODY, we cannot establish a reliable estimate of the prevalence of MODY in the Kuwaiti population. In addition, we expect that the prevalence will vary between different ethnic groups, and more population or registry-based studies across the region are needed.
Recent data from a Norwegian nationwide population-based registry14 suggests that the prevalence of MODY in children with antibody-negative diabetes may reach 6.5%. Importantly, one-third of these cases with MODY had not been recognized by clinicians. Nationwide screening programs in Europe show similar prevalence for MODY: Poland38 has reported 7% MODY cases with a GCK/HNF1A ratio of 21, while Germany and Austria39 have reported a diagnosis rate of 97% with GCK/HNF1A ratio of 2. The United Kingdom40 has a MODY diagnosis rate of 27% with GCK/HNF1A ratio of 0.61, which closely resembles our data (positive detection rate of 21.8% and GCK/HNF1A ratio of 0.5).
GCK-MODY2 is the most common subtype of MODY identified in pediatric diabetes clinics41. Since it is rarely associated with microvascular or macrovascular complications42, affected individuals do not require diabetes treatment or regular follow-up visits 9,43. Similarly, MODY 3 cases resulting from variants in HNF1A are of significant clinical relevance as they provide a basis for individualized “precision” treatment and management of complications associated with glycemic control. The ISPAD guidelines9 recommend that HNF1α-MODY be the first diagnostic possibility considered in patients with symptomatic autosomal dominant diabetes. Variants in HNF1α show high penetrance44 with 79% of carriers developing diabetes before the age of 35. Consistent with this, the two cases diagnosed with MODY3 in our study presented diabetes before the age of 32 years. Exposure to maternal diabetes in utero is considered to accelerate the development of diabetes in the offspring by about 12 years45. One of the two MODY3 cases diagnosed in our study shows maternal inheritance of the disease before the age of 14 years. In both families with MODY3 the mothers of the index patients had diabetes. Our analysis also detected two cases of MODY5 driven by the presence of HNF1β variants, who had evidence of renal dysfunction (renal cysts and dysplasia requiring renal transplantation in one case and increased levels of serum creatinine in the other). Though diabetes secondary to HNF1β variant develops typically during adolescence or early adulthood46,47; two of the MODY5 patients in our study developed diabetes at the age of 4 and 9 years, respectively. The accelerated onset of diabetes could be due to modifiable risk factors such as post-transplantation medications or obesity.
A major challenge in interpreting genetic test results, not limited to MODY, is defining variant pathogenicity. Although we strictly followed the ACMG-AMP variant classification criteria48, our study as well as other data sets remain deficient in functional data. Also, there is currently one expert group in ClinGen that has recently started the process of ACMG-based MODY variant classification. Therefore, given the current lack of comprehensive MODY-variant classification expertise, our ACMG-based classification should be interpreted cautiously and may be subject to change in the future.
Despite our success in diagnosing different forms of MODY and thus enabling personalized treatment for patients locally, the refusal of family members to be screened remains a challenge. Another major caveat in this study is that it was based on referrals from physicians rather than utilizing a systematic, nationwide screening of suspected cases based on the ongoing “Childhood Diabetes Registry” in Kuwait. Therefore, our study does not claim to report the prevalence of MODY in the Kuwaiti population.
Moreover, our study shows that, even in highly selected families with suspected MODY that were autoantibody negative, 77.3% did not harbor known MODY-causative genes, although the samples were analyzed using state-of-the-art methodologies of NGS and MLPA at two reputable and independent centers. This suggests that either additional new genes play a role in the MODY-like families presented here or that they are T2D that mimics MODY in terms of familial clustering. Further studies based on broader population screening and deeper whole genome sequencing are being planned to address this vital niche and to unravel the influence of intronic or gene regulatory mutations in the pathogenesis of MODY, if any.
To circumvent these challenges and to contribute more to our understanding of MODY, we established a highly specialized MODY clinic that is appropriately tailored to educate and serve clinicians from Kuwait and the region centrally. Implementing a nationwide national diabetes registry in Kuwait will allow us to address MODY at the population level within the near future with active screening protocols, including more in-depth information on the phenotype of patients and their pedigrees. Increased awareness and improved access to next generation genetic testing will provide interesting new findings to improve our understanding of the genetic makeup of MODY.
In summary, our study provides the first data set on the genotype and phenotype of patients with suspected MODY in Kuwait, based on referrals to a specialized MODY clinic. We describe seven cases representing five MODY gene variants, including three novel variants. Our results demonstrate the importance of addressing MODY by genetic testing to enable precision diabetes management for the affected patients. We plan to integrate MODY screening into the future National Diabetes Registry to capture comprehensive data on MODY in Kuwait, including genotype, phenotype, and long-term outcome in affected patients. The social and ethical implications associated with genetic testing within families and the broader community must be given careful consideration before the implementation of a nationwide project.
Data availability
The data set generated during and/or analyzed during the current study are available on reasonable request from the corresponding author.
References
Fajans, S. S. & Bell, G. I. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 34, 1878–1884. https://doi.org/10.2337/dc11-0035 (2011).
Oliveira, S. C., Neves, J. S., Pérez, A. & Carvalho, D. Maturity-onset diabetes of the young: From a molecular basis perspective toward the clinical phenotype and proper management. Endocrinol. Diabetes Nutr. 67, 137–147. https://doi.org/10.1016/j.endinu.2019.07.012 (2020).
Lambert, A. P. et al. Identifying hepatic nuclear factor 1alpha mutations in children and young adults with a clinical diagnosis of type 1 diabetes. Diabetes Care 26, 333–337. https://doi.org/10.2337/diacare.26.2.333 (2003).
Møller, A. M. et al. Mutations in the hepatocyte nuclear factor-1alpha gene in Caucasian families originally classified as having Type I diabetes. Diabetologia 41, 1528–1531. https://doi.org/10.1007/s001250051101 (1998).
Awa, W. L. et al. Reclassification of diabetes type in pediatric patients initially classified as type 2 diabetes mellitus: 15 years follow-up using routine data from the German/Austrian DPV database. Diabetes Res. Clin. Pract. 94, 463–467. https://doi.org/10.1016/j.diabres.2011.09.011 (2011).
Kleinberger, J. W. et al. Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: the TODAY clinical trial. Genet. Med. 20, 583–590. https://doi.org/10.1038/gim.2017.150 (2018).
Stanik, J. et al. De novo mutations of GCK, HNF1A and HNF4A may be more frequent in MODY than previously assumed. Diabetologia 57, 480–484. https://doi.org/10.1007/s00125-013-3119-2 (2014).
De Franco, E. et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: An international cohort study. Lancet 386, 957–963. https://doi.org/10.1016/s0140-6736(15)60098-8 (2015).
Hattersley, A. T. et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 19(Suppl 27), 47–63. https://doi.org/10.1111/pedi.12772 (2018).
Pearson, E. R. et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362, 1275–1281. https://doi.org/10.1016/s0140-6736(03)14571-0 (2003).
Hattersley, A. T. & Patel, K. A. Precision diabetes: learning from monogenic diabetes. Diabetologia 60, 769–777. https://doi.org/10.1007/s00125-017-4226-2 (2017).
Sanyoura, M., Philipson, L. H. & Naylor, R. Monogenic diabetes in children and adolescents: Recognition and treatment options. Curr. Diab. Rep. 18, 58. https://doi.org/10.1007/s11892-018-1024-2 (2018).
Landrum, M. J. & Kattman, B. L. ClinVar at five years: Delivering on the promise. Hum. Mutat. 39, 1623–1630. https://doi.org/10.1002/humu.23641 (2018).
Johansson, B. B. et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia 60, 625–635. https://doi.org/10.1007/s00125-016-4167-1 (2017).
Bellanné-Chantelot, C. et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54, 3126–3132. https://doi.org/10.2337/diabetes.54.11.3126 (2005).
Al-Kandari, H., Al-Abdulrazzaq, D., Davidsson, L. & Al-Mulla, F. Maturity-onset diabetes of the young (MODY): A time to act. Lancet Diabetes Endocrinol. 8, 565–566. https://doi.org/10.1016/s2213-8587(20)30150-9 (2020).
Amara, A. et al. Familial early-onset diabetes is not a typical MODY in several Tunisian patients. Tunis Med 90, 882–887 (2012).
Ben Khelifa, S. et al. Maturity Onset Diabetes of the Young (MODY) in Tunisia: Low frequencies of GCK and HNF1A mutations. Gene 651, 44–48. https://doi.org/10.1016/j.gene.2018.01.081 (2018).
Dallali, H. et al. Genetic characterization of suspected MODY patients in Tunisia by targeted next-generation sequencing. Acta Diabetol. 56, 515–523. https://doi.org/10.1007/s00592-018-01283-5 (2019).
Woodhouse, N. J. et al. Clinically-Defined Maturity Onset Diabetes of the Young in Omanis: Absence of the common Caucasian gene mutations. Sultan Qaboos Univ. Med. J. 10, 80–83 (2010).
Hebbar, P. et al. Genome-wide association study identifies novel risk variants from RPS6KA1, CADPS, VARS, and DHX58 for fasting plasma glucose in Arab population. Sci. Rep. 10, 152. https://doi.org/10.1038/s41598-019-57072-9 (2020).
WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr Suppl 450, 76-85, https://doi.org/10.1111/j.1651-2227.2006.tb02378.x (2006)
Fattahi, Z. et al. Iranome: A catalog of genomic variations in the Iranian population. Hum. Mutat. 40, 1968–1984. https://doi.org/10.1002/humu.23880 (2019).
Doddabelavangala Mruthyunjaya, M. et al. Comprehensive maturity onset diabetes of the Young (MODY) gene screening in pregnant women with Diabetes in India. PLoS ONE 12, e0168656. https://doi.org/10.1371/journal.pone.0168656 (2017).
Hildebrand, J. M. et al. A family harboring an MLKL loss of function variant implicates impaired necroptosis in diabetes. Cell Death Dis. 12, 345. https://doi.org/10.1038/s41419-021-03636-5 (2021).
Wright, N. M., Metzger, D. L., Borowitz, S. M. & Clarke, W. L. Permanent neonatal diabetes mellitus and pancreatic exocrine insufficiency resulting from congenital pancreatic agenesis. Am. J. Dis. Child 147, 607–609. https://doi.org/10.1001/archpedi.1993.02160300013005 (1993).
Ellard, S. Hepatocyte nuclear factor 1 alpha (HNF-1 alpha) mutations in maturity-onset diabetes of the young. Hum. Mutat. 16, 377–385. https://doi.org/10.1002/1098-1004(200011)16:5%3c377::aid-humu1%3e3.0.co;2-2 (2000).
Paradiso, V. et al. Diagnostic targeted sequencing panel for hepatocellular carcinoma genomic screening. J. Mol. Diagn. 20, 836–848. https://doi.org/10.1016/j.jmoldx.2018.07.003 (2018).
Codner, E. et al. Glucokinase mutations in young children with hyperglycemia. Diabetes Metab. Res. Rev. 22, 348–355. https://doi.org/10.1002/dmrr.622 (2006).
Massa, O. et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia 44, 898–905. https://doi.org/10.1007/s001250100530 (2001).
40(th) EASD Annual Meeting of the European Association for the Study of Diabetes: Munich, Germany, 5–9 September 2004. Diabetologia 47, A1-a464, https://doi.org/10.1007/bf03375463 (2004).
Shaltout, A. A. et al. Further evidence for the rising incidence of childhood Type 1 diabetes in Kuwait. Diabet. Med. 19, 522–525. https://doi.org/10.1046/j.1464-5491.2002.00703.x (2002).
Shaltout, A. A. et al. High incidence of childhood-onset IDDM in Kuwait. Kuwait Study Group of Diabetes in Childhood. Diabetes Care 18, 923–927. https://doi.org/10.2337/diacare.18.7.923 (1995).
Bener, A., Yousafzai, M. T., Al-Hamaq, A. O., Mohammad, A. G. & Defronzo, R. A. Parental transmission of type 2 diabetes mellitus in a highly endogamous population. World J. Diabetes 4, 40–46. https://doi.org/10.4239/wjd.v4.i2.40 (2013).
Cheţa, D. et al. A study on the types of diabetes mellitus in first degree relatives of diabetic patients. Diabete Metab. 16, 11–15 (1990).
Alhyas, L., McKay, A. & Majeed, A. Prevalence of type 2 diabetes in the States of the co-operation council for the Arab States of the Gulf: A systematic review. PLoS ONE 7, e40948. https://doi.org/10.1371/journal.pone.0040948 (2012).
Elkum, N., Al-Arouj, M., Sharifi, M., Shaltout, A. & Bennakhi, A. Prevalence of childhood obesity in the state of Kuwait. Pediatr. Obes. 11, e30–e34. https://doi.org/10.1111/ijpo.12090 (2016).
Małachowska, B. et al. Monogenic diabetes prevalence among Polish children-Summary of 11 years-long nationwide genetic screening program. Pediatr. Diabetes 19, 53–58. https://doi.org/10.1111/pedi.12532 (2018).
Schober, E. et al. Phenotypical aspects of maturity-onset diabetes of the young (MODY diabetes) in comparison with Type 2 diabetes mellitus (T2DM) in children and adolescents: Experience from a large multicentre database. Diabet. Med. 26, 466–473. https://doi.org/10.1111/j.1464-5491.2009.02720.x (2009).
Shields, B. M. et al. Maturity-onset diabetes of the young (MODY): How many cases are we missing?. Diabetologia 53, 2504–2508. https://doi.org/10.1007/s00125-010-1799-4 (2010).
Carmody, D. et al. GCK-MODY in the US National Monogenic Diabetes Registry: Frequently misdiagnosed and unnecessarily treated. Acta Diabetol. 53, 703–708. https://doi.org/10.1007/s00592-016-0859-8 (2016).
Steele, A. M. et al. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 311, 279–286. https://doi.org/10.1001/jama.2013.283980 (2014).
Chakera, A. J. et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 38, 1383–1392. https://doi.org/10.2337/dc14-2769 (2015).
Murphy, R., Ellard, S. & Hattersley, A. T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 4, 200–213. https://doi.org/10.1038/ncpendmet0778 (2008).
Stride, A. et al. Intrauterine hyperglycemia is associated with an earlier diagnosis of diabetes in HNF-1alpha gene mutation carriers. Diabetes Care 25, 2287–2291. https://doi.org/10.2337/diacare.25.12.2287 (2002).
Edghill, E. L., Bingham, C., Ellard, S. & Hattersley, A. T. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J. Med. Genet. 43, 84–90. https://doi.org/10.1136/jmg.2005.032854 (2006).
Raile, K. et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J. Clin. Endocrinol. Metab. 94, 2658–2664. https://doi.org/10.1210/jc.2008-2189 (2009).
Amendola, L. M. et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am. J. Hum. Genet. 98, 1067–1076. https://doi.org/10.1016/j.ajhg.2016.03.024 (2016).
Acknowledgements
This work was supported by Kuwait Foundation for the Advancement of Sciences Kuwait Familial Diabetes grant number RA HM 2019-009. We also thank all referring physicians, patients and their family members for their interest and willingness to contribute to the study. We thank Dr. Rob Sladek and Dr. Graham Taylor for their critical feedback on the manuscript. We extend our sincere gratitude to National Dasman Biobank staff Ms. Daisy Thomas, Mr. Sriraman Devarajan and Ms. Betty Chandy for their extended support throughout the study. We also thank Dr. Azza Shallout for her initial support.
Author information
Authors and Affiliations
Contributions
H.A.K. designed and implemented the study. She was responsible for the clinical assessment of the patients, organized for genetic testing and reviewed the results. D.A.A. and L.D. reviewed the data and contributed to the drafting of the manuscript. F.A.M. was responsible for genetic testing at DDI, assisted by R.N., S.J., M.M. & S.E.J. He led the interpretation of data and prepared the first draft of the manuscript. The document was finalized based on comments from all co-authors. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Kandari, H., Al-Abdulrazzaq, D., Davidsson, L. et al. Identification of Maturity-Onset-Diabetes of the Young (MODY) mutations in a country where diabetes is endemic. Sci Rep 11, 16060 (2021). https://doi.org/10.1038/s41598-021-95552-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95552-z
- Springer Nature Limited