
Abstract
Aims
Monogenic forms of diabetes that develop with autosomal dominant inheritance are classically aggregated in the Maturity-Onset Diabetes of the Young (MODY) categories. Despite increasing awareness, its true prevalence remains largely underestimated. We describe a Portuguese cohort of individuals with suspected monogenic diabetes who were genetically evaluated for MODY-causing genes.
Methods
This single-center retrospective cohort study enrolled patients with positive genetic testing for MODY between 2015 and 2021. Automatic sequencing and, in case of initial negative results, next-generation sequencing were performed. Their clinical and molecular characteristics were described.
Results
Eighty individuals were included, 55 with likely pathogenic/pathogenic variants in one of the MODY genes and 25 MODY-positive family members, identified by cascade genetic testing. The median age at diabetes diagnosis was 23 years, with a median HbA1c of 6.5%. The most frequently mutated genes were identified in HNF1A (40%), GCK (34%) and HNF4A (13%), followed by PDX1, HNF1B, INS, KCNJ11 and APPL1. Thirty-six unique variants were found (29 missense and 7 frameshift variants), of which ten (28%) were novel.
Conclusions
Our data highlights the importance of genetic testing in the diagnosis of MODY and the establishment of its subtypes, leading to more personalized treatment and follow-up strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Maturity-Onset Diabetes of the Young (MODY) includes most monogenic diabetes, which presents some classic features such as young age at diagnosis, autosomal dominant pattern of inheritance and insulin independence. MODY is responsible for approximately 1–2% of all cases of diabetes diagnosed in Europe, although its true prevalence remains largely underestimated [1, 2]. Misdiagnosis may result given some overlapping phenotypic characteristics with both type 2 diabetes, such as preserved β cell function or family history, and type 1 diabetes, such as young age at diagnosis and leanness [3]. Currently known MODY subtypes are caused by dominantly acting heterozygous variants in 14 genes that are crucial for the development or function of pancreatic-β-cells, namely HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, ABCC8, KCNJ11 and APPL1 [1,2,3]. However, three of these genes (BLK, PAX4 and KLF11) have recently been proposed for elimination based on recently either disputed or refuted gene-disease relationships [4]. Also, RFX6 has been proposed as an additional MODY gene based on multiple loss-of-function variants associated with a MODY “like” phenotype [5]. Heterozygous variants in GCK, HNF1A and HNF4A account for over 95% of the known genetic causes of MODY [1,2,3].
Genetic diagnosis is crucial to the diabetes management of these patients given that it can help to select the most appropriate treatment, stratify their prognosis and risk for vascular complications, alert to the existence of associated extra-pancreatic features and to guide family counseling [6]. MODY subtypes’ relative frequencies have been previously evaluated, with expected population-based differences between each European cohort, which also may result from the use of different criteria for individuals’ selection for genetic testing [7]. Next-generation sequencing (NGS) techniques have led to significant advancements in the understanding of numerous disorders within the field of endocrinology, allowing the parallel sequencing of multiple genes and providing rapid results to further increase diagnostic accuracy for monogenic forms of diabetes [8, 9].
The aim of our study was to identify the genetic variants in known MODY genes within a Portuguese cohort of individuals with suspected monogenic diabetes and to further characterize its subtypes and specificities.
Material and methods
Study design and population
This single-center retrospective study enrolled both children and adults with diabetes followed at our Pediatric and Adult Endocrinology Outpatient Clinic, between 2015 and 2021. All probands met the following criteria: (1) family history of diabetes in at least two generations with an autosomal dominant mode of inheritance; (2) the ability to control diabetes without insulin treatment for at least two years, or significant levels of fasting serum C-peptide (normal values > 0.8 ng/mL); (3) the absence of pancreatic islet autoantibodies including glutamic acid decarboxylase antibody (GAD), protein tyrosine phosphatase antibody (IA2), anti-zinc transporter protein 8 antibodies (ZnT8) and islet cell antibody (ICA); (4) no marked obesity or evidence of insulin resistance. Ancestry was participant-reported. Whenever possible, other affected and non-affected family members were studied.
Clinical data including age at diagnosis or enrollment, gender, body mass index (BMI) at diagnosis, family history of diabetes, diabetes-related complications and treatment options were collected from patient’s electronic records. Laboratory data at diagnosis such as plasma C-peptide, glycated hemoglobin (HbA1c), and β-cell autoantibodies were also obtained.
This study was conducted according to the principles of the Declaration of Helsinki and was approved by the Ethics Committee from Centro Hospitalar Universitário do Porto, Portugal. All participants or their guardians gave informed consent to genetic testing, according to national regulations. Due to the retrospective nature of the study and the absence of additional clinical procedures beyond those done in the delivery of usual care, consent to participate was waived by the local Ethics Committee. All data were anonymously collected and analysed.
Genetic testing
Nuclear deoxyribonucleic acid (DNA) was extracted from peripheral blood lymphocytes and used with custom-designed primers for polymerase chain reaction (PCR) amplification of the coding regions and exon–intron boundaries of the GCK and HNF1Agenes; HNF4A and HNF1B were also analyzed in some individuals, following specific clinical suspicion. Sanger sequencing analysis was undertaken for all individuals using the BigDye™Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. In addition, MLPA (multiplex ligation-dependent probe amplification) by Salsa® MLPA® (panel P297-C1, MRC-Holland, Amsterdam, The Netherlands) was performed to identify whole-exon deletions/duplications that might escape the automated sequencing described above. Those with no pathogenic variant identified by conventional sequencing underwent massive parallel sequencing through next-generation sequencing using Clinical Exome Solution V2® (Sophia Genetics SA, Saint Sulpice, Switzerland). Enriched libraries were sequenced on the NextSeq platform (Illumina Inc., San Diego, CA, USA) following the manufacturer’s recommendations using a multiplex system with 16 samples per run with the NextSeq 500/550 Mid Output V2 kit (Illumina Inc., San Diego, CA, USA). The genetic analysis strategy was performed with a virtual panel based on Human Phenotype Ontology consisting of 200 genes associated with familial hyperinsulinism, monogenic diabetes, neonatal diabetes and other disorders in which hypoglycemic/hyperglycemic events are a predominant sign [9]. Cascade testing was performed on the available family members using targeted Sanger sequencing of the respective mutation of the MODY-gene in which their relative had a likely pathogenic/pathogenic genetic variant.
To achieve a reliable clinical interpretation of the variants detected and to predict their pathogenicity, we considered prioritization criteria according to American College of Medical Genetics and Genomics (AMCG) guidelines [10]. We considered allele frequency using the Exome Aggregation Consortium database (ExAC), 1000 Genomes Project database and gnomAD [11,12,13]. Several pathogenicity algorithms were considered to predict disease by Mutation Taster and damaging by FATHMM (Functional Analysis through Hidden Markov Models) and DANN (Deleterious Annotation of genetic variants using Neural Networks) scores. According to Genomic Evolutionary Rate Profiling (GERP), PhyloP and PhastCons, variants were analyzed according to their positions in highly conserved regions through evolution. The clinical significance of variants was evaluated with ClinVar and Polymorphism database (dbSNP) [9].
Statistical analysis
Continuous and categorical variables are presented as mean ± standard deviation (SD) or median with interquartile range (IQR) and numbers with proportions, respectively. For continuous quantitative variables, distribution normality was tested through histogram observation and Kolmogorov–Smirnov test analysis. The Student's t-test and the Mann–Whitney U test were used to compare continuous variables with normal and non-normal distribution between groups, respectively. Pearson chi-square test was used to compare categorical data. All statistical tests were two-tailed and performed the IBM SPSS® computer statistics program, version 25. A p-value of < 0.05 was considered statistically significant.
Results
Characteristics of the study participants
This study included a total of 55 probands with a likely pathogenic/pathogenic heterozygous variant in one of the known MODY genes, from 138 individuals referred for genetic testing (Fig. 1). Moreover, cascade genetic testing in families identified additional 25 family members with MODY-causing variants, who were also included. Their clinical characteristics are summarized in Table 1. Approximately half of the participants (51%, n = 41) were male, with a median age at diabetes diagnosis of 23 years (min/max: 1–65) and median diabetes duration of 10 years (IQR 2–21). Sixty-four individuals (80%) had diabetes diagnosed up to 35 years of age and 45 (56%) were diagnosed before 25 years of age. Over three-quarters reported normal weight at diagnosis and 60 individuals (75%) had a positive first-degree family history of diabetes. At the study’s inclusion, the median age of the participants was 40 years (IQR 19–51). The median HbA1c was 6.4% (IQR 5.8–7.2), median fasting C-peptide at enrollment was 1.55 ng/mL (IQR 1.06–2.39) and 23% presented at least one diabetes-related complication. At MODY diagnosis, fifty-six individuals (70%) were under glucose-lowering treatment, of which 16 participants (20%) were under insulin therapy (Table 1).

Participant’s selection flowchart. Initial genetic screening was performed for 3 MODY subtypes, namely GCK, HNF1A and HNFB, according to clinical and laboratory characteristics. Next-generation sequencing (NGS) was performed in the negative cases. MLPA multiplex ligation-dependent probe amplification, NGS next-generation sequencing
Genetic diagnosis
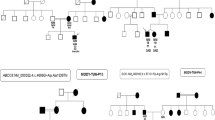
This two-step process for genetic testing resulted in a total of 80 individuals identified with pathogenic/likely pathogenic (P/LP) variants or variants of undetermined significance (VUS) in a known MODY gene, according to strict ACMG classification criteria [10]. A full description of the genetic variants found is available in Table 2. Thirty-six unique variants were found (29 missense and 7 frameshift variants), of which ten (28%) were novel, given that they have not been previously reported in the literature or ClinVar. Nine of the thirty-six variants (25%) were found by next-generation sequencing. The genes most frequently mutated were HNF1A (n = 32), GCK (n = 27) and HNF4A (n = 10). Specifically, five novel variants were found in HNF1A (c.305C > G, c.360G > C, c.1146_1156del, c.1133C > A and c.1422_1424delGCCinsCAG), three novel variants in HNF4A (c.354G > T, c.721C > G and c.850_860delinsCCT) and one novel variant in GCK (c.863 T > C). Family testing provided co-segregation data that was used in scoring the variant HNF4Ac.721C > G and it is represented in Fig. 2. The genes with a lower frequency of P/LP variants included HNF1B (n = 5), PDX1 (n = 2), INS (n = 2), KCNJ11 (n = 1) and APPL1 (n = 1). Within these rarer subtypes, novel variants were found in APPL1 (c.1433G > A). HNF1A c.305C > G, HNF1A c.1133C > A, INS (c.130G > A) and APPL1 c.1433G > A were classified as variants of uncertain significance and all the others were classified as either pathogenic or likely pathogenic variants (Table 2) [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. Lastly, one individual with PDX1-MODY presented a second missense mutation on exon 1, variant c-97C > A (p.Pro33Th3), classified as of uncertain significance.

HNF4A c.721C > G (p.Arg241Gly) family pedigree. Family testing provided co-segregation data that was used in scoring the variant HNF4A c.721C > G. Squares, circles and diamond symbols denote males, females and unspecified, respectively. Oblique lines through symbols represent deceased individuals. Arrow indicates the index case. The presence (x) of the mutation, when known, is shown. Black-filled symbols represent patients with diabetes, grey striped symbols represent individuals with the mutation but without diabetes and open symbols represent unaffected individuals. The age of diagnosis of diabetes (y, years) and HbA1c at enrollment (%) are presented
Specificities of MODY subtypes
When compared to GCK-MODY, HNF1A/HNF4A-MODY individuals were older at diabetes diagnosis (29 vs. 14 years, p < 0.001), with a higher median HbA1c at diagnosis (7.3% vs. 6.2%, p = 0.02). Only HNF1A/HNF4A-MODY individuals presented diabetes-related complications (29%) and were more frequently under glucose-lowering therapy (32 vs. 6, p < 0.001). Insulin therapy was only needed within HNF1A/HNF4A-MODY individuals. (Table 3). Moreover, one individual with HNF4A-MODY (10%) was diagnosed after congenital hyperinsulinemic hypoglycemia.
Regarding rarer MODY subtypes, all five unrelated probands with HNF1B-MODY presented any kidney structural abnormalities which lead to their diagnosis and only three of which (60%) have already developed diabetes, currently treated under insulin. Moreover, three individuals had a whole-gene deletion compatible with the diagnosis of 17q12 deletion syndrome, presenting multisystemic features such as neurodevelopmental disorders (developmental delay, intellectual disability and autism spectrum disorder), pancreatic dysgenesis and genital abnormalities. Table 4 presents clinical data and additional information for participants with pathogenic/likely pathogenic variants in these rarer MODY genes.
Impact of genetic diagnosis of MODY in diabetes treatment
We also evaluated the available information on possible treatment adaptation following the genetic diagnosis. Regarding HNF1A/HNF4A-MODY individuals (n = 42), half of them (n = 21) experienced a change in treatment after the genetic diagnosis, namely with the introduction of sulfonylureas, presenting an improved glycemic control during follow-up (median HbA1c decrease of 1.2%). In addition, six of ten participants were able to suspend insulin therapy after the introduction of targeted glucose-lowering therapy.
Within GCK-MODY individuals, six of twenty-seven participants (22.2%) with P/LP variants in GCK were initially treated under non-insulin hypoglycemic agents (NIHA). After genetic diagnosis, drug therapy was stopped in four of them (66.7%), without any deterioration in their glycemic control.
Discussion
With this unicentric cohort study, we intended to identify and characterize the genetic variants among individuals with suspected-monogenic diabetes. Within the study period, 80 individuals (55 probands and 25 relatives) were diagnosed with MODY. Of those, HNF1A and GCK were the genes most implicated, with a higher prevalence of HNF1A-MODY within our sample. Our results are concordant with large European series, such as from United Kingdom and Norway, which have found a higher frequency of variants in HNF1A- versus GCK-MODY [3, 34]. On the other hand, several series such as the ones from United States and Poland have noted predominance in the GCK-MODY subtype [35, 36]. Recent European series both from France and United Kingdom screened thousands of patients and propose the “ranking” of MODY genes frequency to be GCK first, then HNF1A, HNF4A and either m.3243A > G or HNF1B in the fourth position [37, 38]. This data clearly shows GCK-MODY as the most prevalent subtype not only in the pediatric setting but also all age data sets [37,38,39]. Several factors may explain such geographical and population inter-variability, given that routine genetic testing for MODY within healthy individuals (such as pregnant women) is easily performed in the United States, leading to a faster diagnosis, namely of milder phenotypes such as GCK-MODY [35]. Moreover, the lack of uniformization in participants’ selection for genetic testing, even within the European cohorts, applying diverse protocols for individuals’ selection, may partly explain some differences observed. In addition, other rarer MODY subtypes (HNF4A, HNF1B, PDX1, INS, KCNJ11 and APPL1) were also found in our cohort.
Our work led to the identification of novel disease-causing variants in known MODY genes (28%). Genetic diagnosis has significant management implications both for the individual and their family, given that treatment, prognosis and follow-up are rather heterogeneous among each MODY subtype. Firstly, heterozygous GCK deficiency constitutes a “benign” condition, characterized only by mildly elevated glucose values which do not lead to a higher risk for both micro and macrovascular diabetes-related complications [40]. Therefore, GCK-MODY individuals do not need glucose-lowering treatment, increasing genetic diagnosis cost-effectiveness within this subtype. Secondly, HNF1A, HNF4A and KCNJ11 individuals are usually sulfonylureas-responsive and can often transition off insulin/less effective non-insulin anti-hyperglycemic agents to an easier and more targeted treatment once the diagnosis is made [8]. Lastly, the identification of rarer subtypes by NGS is fundamental to increasing awareness of these specific genes, especially considering their phenotypic variability and the challenge to establish a specific clinical and analytical pattern. Particularly, here we considered heterozygous PDX1 missense variants as causative for MODY and not only predisposing for type 2 diabetes. These individuals presented some form of pancreatic dysgenesis with severe depleted beta-cell function and early insulin dependency (Table 4). Two missense variants were identified on the index case (PDX1c.92C > A and c.492G > T), increasing the possibility of compound heterozygosity as an explanation for the phenotype presented [23]. Further family study of both variants to determine inheritance and co-segregation may fully clarify our findings.
This study proposes a two-step approach for monogenic diabetes genetic testing, which aims to both maximize its diagnostic capability and minimize the associated cost, namely the burden of reporting variants of uncertain significance. First, GCK and HNF1A must be assessed, according to clinical suspicion, by Sanger sequencing. Second, next-generation sequencing should be considered for negative individuals with high pre-test probability for monogenic diabetes, to identify pathogenic variants in rare genetic causes of diabetes or even to identify novel MODY-associated genes. Specific tools, such as Exeter’s MODY Probability calculator, have recently been validated in our population and may improve individuals’ selection for genetic testing [41, 42]. Neonatal hyperinsulinemic hypoglycemia or the presence of kidney and urinary tract abnormalities may justify an early targeted screening for MODY. Lastly, given its increasing frequency in the adult setting, m.3243 A > G should also be included in the screening [38].
We have found that over 20% and 40% of our population were diagnosed with diabetes over 35 and 25 years of age, respectively. Specifically, HNF1A/HNF4A-MODY individuals were diagnosed at a median age of 30 years. Our results are in agreement with the available literature, given that it is already known that only approximately 60% of HNF1A-positive individuals develop diabetes below 25 years of age and 80% below 35 years [43]. A later diagnosis may result from either an intrinsic later diabetes presentation or from insufficient access to health care services. Our data reinforce the need to consider a higher age cut-off when evaluating non-GCK suspected-monogenic diabetes individuals.
A strong point of our work is that here we present and characterize the largest cohort of Portuguese individuals with a genetic diagnosis of MODY. A previous Portuguese genetic study found MODY mutations in 23/46 (50%) of families with clinically suspected MODY, with a higher predominance of GCK-MODY; however, only HNF4A, GCK and HNF1A genes were evaluated [44]. Our two-step genetic testing strategy (targeted and massive parallel sequencing techniques) led to the identification of a higher number of mutations and allowed us the detection of variants in MODY rare subtypes, especially considering that those genes are not commonly studied for the genetic diagnosis of MODY at most medical centers. The identification of these variants may help to expand the knowledge and further characterize these atypical forms of monogenic diabetes. Moreover, several novel disease-causing variants were identified, described and characterized in our work, adding valuable genetic and clinical information for all MODY research communities. Lastly, our targeted cascade genetic testing approach within available family members allowed identifying and tailoring diabetes management among participants’ relatives.
This study has some limitations. First, its retrospective design should be acknowledged, with potential selection bias inflicted. Second, most of the participants evaluated were from northern Portugal where ethnic white caucasian is predominant; therefore, our results should not be generalized to non-caucasian populations. Lastly, we did not have the possibility to perform genetic testing procedures on the family members from all probands.
Conclusion
In summary, our two-step genetic testing approach (targeted sequencing and NGS) led to the identification of novel MODY variants, further increasing the spectrum of MODY-associated genes. Our study contribute to a more personalized treatment, prognostic and follow-up assessment of these individuals and their families.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
References
Zhang H, Colclough K, Gloyn AL (2021) Monogenic diabetes: a gateway to precision medicine in diabetes. J Clin Invest 131(3):e142244. https://doi.org/10.1172/JCI142244
Vaxillaire M, Froguel P (2016) Monogenic diabetes: implementation of translational genomic research towards precision medicine. J Diabetes 8(6):782–795. https://doi.org/10.1111/1753-0407.12446
Shields BM, Hicks S, Shepherd MH et al (2010) Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53:2504–2508. https://doi.org/10.1007/s00125-010-1799-4
Laver TW, Wakeling MN, Knox O et al (2022) Evaluation of evidence for pathogenicity demonstrates that BLK, KLF11, and PAX4 should not be included in diagnostic testing for MODY. Diabetes 71(5):1128–1136. https://doi.org/10.2337/db21-0844
Patel KA, Kettunen J, Laakso M et al (2017) Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat Commun 8(1):888
Hattersley A, Kashyap AP (2017) Precision diabetes: learning from monogenic diabetes. Diabetologia 60:769–777
Kleinberger JW, Pollin TI (2015) Undiagnosed MODY: time for action. Curr Diabetes Rep 15:110
Forlenza GP, Calhoun A, Beckman KB et al (2015) Next generation sequencing in endocrine practice. Mol Genet Metab 115(2–3):61–71
Ellard S, Lango Allen H, De Franco E et al (2013) Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia 56:1958–1963
Richards S, Aziz N, Bale S, ACMG Laboratory Quality Assurance Committee et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17(5):405–24. https://doi.org/10.1038/gim.2015.30
Karczewski KJ, Weisburd B, Thomas B, The Exome Aggregation Consortium et al (2017) The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 45(D1):D840–D845. https://doi.org/10.1093/nar/gkw971
Clarke L, Fairley S, Zheng-Bradley X et al (2017) The international genome sample resource (IGSR): a worldwide collection of genome variation incorporating the 1000 genomes project data. Nucleic Acids Res 45(D1):D854–D859. https://doi.org/10.1093/nar/gkw829
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Apperley L, Giri D, Houghton JAL et al (2019) A rare case of congenital hyperinsulinism (CHI) due to dual genetic aetiology involving HNF4A and ABCC8. J Pediatr Endocrinol Metab 32(3):301–304. https://doi.org/10.1515/jpem-2018-0389 (PMID: 30730840)
Pruhova S, Dusatkova P, Sumnik Z et al (2010) Glucokinase diabetes in 103 families from a country-based study in the Czech Republic: geographically restricted distribution of two prevalent GCK mutations. Pediatr Diabetes 11(8):529–535. https://doi.org/10.1111/j.1399-5448.2010.00646.x
Aloi C, Salina A, Minuto N et al (2017) Glucokinase mutations in pediatric patients with impaired fasting glucose. Acta Diabetol 54(10):913–923. https://doi.org/10.1007/s00592-017-1021-y (Epub 2017 Jul 19)
Osbak KK, Colclough K, Saint-Martin C et al (2009) Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 30(11):1512–1526
Stern E, Strihan C, Potievsky O et al (2007) Four novel mutations, including the first gross deletion in TCF1, identified in HNF-4alpha, GCK and TCF1 in patients with MODY in Israel. J Pediatr Endocrinol Metab 20(8):909–921. https://doi.org/10.1515/jpem.2007.20.8.909
Weinert LS, Silveiro SP, Giuffrida FM et al (2014) Three unreported glucokinase (GCK) missense mutations detected in the screening of thirty-two Brazilian kindreds for GCK and HNF1A-MODY. Diabetes Res Clin Pract 106:44–48
Cho EH, Min JW, Choi SS et al (2017) Identification of maturity-onset diabetes of the young caused by glucokinase mutations detected using whole-exome sequencing. Endocrinol Metab (Seoul) 32(2):296–301. https://doi.org/10.3803/EnM.2017.32.2.296 (Epub 2017 May 29)
Shields BM, Shepherd M, Hudson M, UNITED study team et al (2017) Population-based assessment of a biomarker-based screening pathway to aid diagnosis of monogenic diabetes in young-onset patients. Diabetes Care 40(8):1017–1025. https://doi.org/10.2337/dc17-0224
Colclough K, Ellard S, Hattersley A et al (2022) Syndromic monogenic diabetes genes should be tested in patients with a clinical suspicion of maturity-onset diabetes of the young. Diabetes 71(3):530–537. https://doi.org/10.2337/db21-0517
Pihoker C, Gilliam LK, Ellard S et al (2013) Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for diabetes in youth. J Clin Endocrinol Metab 98(10):4055–62. https://doi.org/10.1210/jc.2013-1279 (Epub 2013 Jun 14. PMID: 23771925; PMCID: PMC3790621)
Estalella I, Rica I, Perez de Nanclares G et al (2007) Mutations in GCK and HNF-1alpha explain the majority of cases with clinical diagnosis of MODY in Spain. Clin Endocrinol (Oxf) 67(4):538–46. https://doi.org/10.1111/j.1365-2265.2007.02921.x
Colclough K, Bellanne-Chantelot C, Saint-Martin C et al (2013) Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat 34(5):669–685. https://doi.org/10.1002/humu.22279
Bellanné-Chantelot C, Carette C, Riveline JP et al (2008) The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes 57(2):503–8. https://doi.org/10.2337/db07-0859
Schwitzgebel VM, Mamin A, Brun T et al (2003) Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab 88(9):4398–4406. https://doi.org/10.1210/jc.2003-030046 (PMID: 12970316)
Alvelos MI, Rodrigues M, Lobo L et al (2015) A novel mutation of the HNF1B gene associated with hypoplastic glomerulocystic kidney disease and neonatal renal failure: a case report and mutation update. Medicine (Baltimore) 94(7):e469. https://doi.org/10.1097/MD.0000000000000469
Edghill EL, Bingham C, Ellard S et al (2006) Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet 43(1):84–90. https://doi.org/10.1136/jmg.2005.032854 (Epub 2005 Jun 1. PMID: 15930087; PMCID: PMC2564507)
Patouni K, Cinek O, Pruhova S et al (2021) A case of digenic maturity onset diabetes of the young with heterozygous variants in both HNF1Α and HNF1Β genes. Eur J Med Genet 64(9):104264. https://doi.org/10.1016/j.ejmg.2021.104264 (Epub 2021 Jun 20. PMID: 34161864)
Miller DT, Adam MP, Aradhya S et al (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86(5):749–764. https://doi.org/10.1016/j.ajhg.2010.04.006 (PMID: 20466091; PMCID: PMC2869000)
Johansson BB, Irgens HU, Molnes J et al (2017) Targeted next-generation sequencing reveals MODY in up to 65% of antibody-negative diabetes cases listed in the Norwegian childhood diabetes registry. Diabetologia 60(4):625–635. https://doi.org/10.1007/s00125-016-4167-1 (Epub 2016 Dec 2)
Snider KE, Becker S, Boyajian L et al (2013) Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 98(2):E355-63. https://doi.org/10.1210/jc.2012-2169 (Epub 2012 Dec 28. PMID: 23275527; PMCID: PMC3565119)
Søvika O, Irgens HU, Molnes J et al (2013) Monogenic diabetes mellitus in Norway Norwegian. J Epidemiol 23(1):55–60
Breidbart E, Deng L, Lanzano P et al (2021) Frequency and characterization of mutations in genes in a large cohort of patients referred to MODY registry. J Pediatr Endocrinol Metab 34(5):633–638. https://doi.org/10.1515/jpem-2020-0501
Schober E, Rami B, Grabert M et al (2009) Phenotypical aspects of maturity-onset diabetes of the young (MODY diabetes) in comparison with type 2 diabetes mellitus (T2DM) in children and adolescents: experience from a large multicentre database. Diabet Med 26(5):466–473 ([PubMed: 19646184])
Saint-Martin C, Bouvet D, Bastide M et al (2022) Gene panel sequencing of patients with monogenic diabetes brings to light genes typically associated with syndromic presentations. Diabetes 71(3):578–584. https://doi.org/10.2337/db21-0520
Pang L, Colclough KC, Shepherd MH et al (2022) Improvements in awareness and testing have led to a threefold increase over 10 years in the identification of monogenic diabetes in the UK. Diabetes Care 45(3):642–649. https://doi.org/10.2337/dc21-2056
Delvecchio M, Mozzillo E, Salzano G et al (2017) Monogenic diabetes accounts for 6.3% of cases referred to 15 Italian pediatric diabetes centers during 2007 to 2012. J Clin Endocrinol Metab 102(6):1826–1834. https://doi.org/10.1210/jc.2016-2490
Steele AM, Shields BM, Wensley KJ et al (2014) Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 311(3):279–286. https://doi.org/10.1001/jama.2013.283980 (PMID: 24430320)
Shields BM, McDonald TJ, Ellard S et al (2012) The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 55(5):1265–1272. https://doi.org/10.1007/s00125-011-2418
Santos TDS, Monteiro SS, Fonseca L et al (2022) MODY probability calculator utility in individuals’ selection for genetic testing: its accuracy and performance. Endocrinol Diabetes Metab. https://doi.org/10.1002/edm2.332
Lachance CH (2016) Practical aspects of monogenic diabetes: a clinical point of view. Can J Diabetes 40(5):368–375. https://doi.org/10.1016/j.jcjd.2015.11.004
Alvelos MI, Gonçalves CI, Coutinho E et al (2020) Maturity-onset diabetes of the young (MODY) in Portugal: novel GCK, HNFA1 and HNFA4 mutations. J Clin Med 9(1):288. https://doi.org/10.3390/jcm9010288
Acknowledgements
Authors gratefully thank all the doctors at Centro Hospitalar Universitário do Porto for referring eligible participants to our study. The authors also thank the participants for their contribution to clinical research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
SSM, TSS and LF designed the study. FL, IR, EP and SR performed the Sanger sequencing and MLPA studies. SG and MEV-M performed the NGS studies. SSM, TSS, LF, GA, AML and DBD acquired the data. SSM, TSS and LF interpreted the data. SSM and TSS drafted the original version of the manuscript and all authors revised it critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Ethical approval
This study was conducted according to the principles of the Declaration of Helsinki and was approved by the local Ethics Committee of Centro Hospitalar Universitário do Porto.
Informed consent
All participants or their guardians gave informed consent to genetic testing, according to national regulations.
Consent to participate
Consent to participate was waived by the Ethics Committee due to the retrospective nature of the study and full data anonymization.
Additional information
Managed by Fabrizio Barbetti.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Santos Monteiro, S., da Silva Santos, T., Fonseca, L. et al. Maturity-onset diabetes of the young in a large Portuguese cohort. Acta Diabetol 60, 83–91 (2023). https://doi.org/10.1007/s00592-022-01980-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00592-022-01980-2