
Abstract
Purpose
To screen for maturity-onset diabetes of the young (MODY) variants in subjects with an early age of onset and positive family history of diabetes mellitus.
Methods
60 subjects with onset of diabetes between 3 and 30 years of age and parental history (onset < 35 years) of diabetes were recruited after excluding autoimmune, pancreatic and syndromic forms of diabetes. Detailed pedigree chart and clinical data were recorded. MODY genetic testing (MODY 1–13) was performed and variant classification was done adhering to the ACMG guidelines.
Results
Baseline characteristics of subjects were as follows: mean age of onset of diabetes 19.9 ± 7 years, mean duration of diabetes 6.3 ± 6.8 years, BMI 23.3 ± 3 kg/m2 and C-peptide 1.56 ± 1.06 nmol/l. Four out of sixty (6.6%) were positive for variants classifiable as pathogenic/likely pathogenic: one patient with HNF4Ac.691C > T, (p.Arg231Trp), two with HNF 1A c.746C > A(p.Ser249Ter) and c.1340C > T(p.Pro447Leu), and one with ABCC8 c.4544C > T (p.Thr1515Met). MODY 1 and MODY 3 variants were documented in the paediatric age group (< 18 years).
Conclusion
A genetic diagnosis of MODY could be confirmed in only 6.6% (4/60) of patients clinically classifiable as MODY. This is less than that reported in clinically diagnosed MODY subjects of European descent. Newly published population data and more stringent criteria for assessment of pathogenicity and younger age of onset of type 2 diabetes in Indians could have contributed to the lower genetic confirmation rate. Apart from variants in the classical genes (HNF1A, HNF4A), a likely pathogenic variant in a non-classical gene (ABCC8) was noted in this study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is a high prevalence of young onset diabetes in India [1]. The prevalence of type 2 diabetes in youth in India varies from 25 to 40% in different studies [2, 3]. Maturity-onset diabetes of the young (MODY) is an important subset of monogenic diabetes with an estimated global prevalence of 1–4% in subjects with age of diabetes onset below 30 years [4]. 3.1% of subjects are clinically classified as MODY in the registry for youth onset diabetes in India (YDR) [3]. Apart from the obvious opportunity for genetic counselling, the diagnosis of MODY has therapeutic implications which include choosing not to treat mild hyperglycaemia (MODY 2) and appropriate selection of therapeutic agent (sulfonylurea in MODY 1 and MODY 3 and insulin in MODY 5) [5,6,7]. Normal BMI, strong family history of young onset diabetes, absent islet cell autoantibodies, absence of ketosis and responsiveness to oral hypoglycaemic agents are clinical pointers toward MODY. The original clinical criterion for a diagnosis of MODY is now known to have lower sensitivity than previously assumed [8, 9]. Proper categorisation of young onset diabetes is challenging in Indian subjects due to lower antibody positivity in type 1 diabetes, higher prevalence of fibro-calculous pancreatic diabetes and a lower BMI at diagnosis in patients with young onset type 2 diabetes [2, 10, 11]. MODY calculators and the biomarker approach with its limitations help in screening patients, but have not been well-validated in the Indian population [12, 13].
Genetic testing by Sanger sequencing alone is being increasingly replaced by multigene NGS panels [14, 15]. The last 10 years has seen significant advances in this direction [4]. Currently, at least 14 genetic loci have been associated with autosomal dominant MODY and the list is growing [4, 16, 17]. Interpretation of genetic variants detected by the aforementioned methods is sometimes challenging. American College of Medical Genetics guidelines 2015 and, ACGS best practice guidelines 2019, and the UK framework are used to arrive at the correct conclusions [18, 19]. Studies from different countries suggest that 10–33% of patients who are clinically categorised as MODY harbour disease-causing variants at the genetic loci studied [20,21,22,23,24,25, 29]. However, many of these variants which were earlier thought to be pathogenic are now classified as benign or variants of unknown significance (VUS). This has been made possible by large population-based genetic data sets published in the last 5 years [26, 27]. The current study describes our attempt at deciphering MODY genetics in the state of Kerala, South India.
Materials and methods
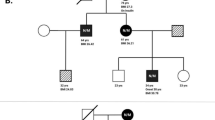
60 patients who had onset of diabetes between 3 and 30 years of age and a positive family history of DM (at least one parent/sibling with diabetes with an age of onset ≤ 35 years of age) were included in the study (Fig. 1). 17 patients were aged below 18 years. Patients with islet cell autoimmunity (positive auto antibodies to antigens-GAD 65, IA-2), clinical or radiological evidence of pancreatitis (absence of abdominal pain, steatorrhea, pancreatic calcification in ultrasound and X-rays), history of diabetic keto-acidosis (DKA) and identifiable forms of syndromic diabetes (lipodystrophy, Klinefelter syndrome, H syndrome, Wolfram syndrome (DIDMOAD), Thiamine-responsive megaloblastic anemia (TRMA), obesity syndromes) were excluded. Insulin use at enrolment was not considered as an exclusion criteria. C-peptide measurement was performed when random blood glucose was above 8 mmol/l. Oral hypoglycaemic agents were discontinued for 24 h prior to C-peptide testing. The family was informed of the need for genetic analysis of the parents or the sibling in the event of a positive result. Informed written consent was obtained from all participants. This study was approved by the Institutional Ethical committee (IRB No: IEC-AIMS-2017-ENDO-426 dated 23.11.2017). A detailed pedigree chart (Fig. 2) and the clinical data were recorded. MODY genetic testing was done using Targeted Next-Generation Sequencing at Christian medical college, Vellore for a comprehensive panel of 13 MODY genes (HNF1A, HNF4A, GCK, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, ABCC8, KCNJ11) as per a previously published protocol [28]. In short, the targeted MODY genes were amplified using multiplex PCR followed by a library preparation which involved fragmentation of long PCR amplicons, barcoded adaptor ligation and size selection. Equimolar libraries were then further utilised for template preparation using Ion one touch OT2 emulsion PCR and enrichment using Ion ES. Sequencing was done on the Ion Torrent PGM using the Ion PGM 200 Sequencing Kit (Ion Torrent, Life Technologies), 316 chips (multiplex 8–10 samples).

Study flow

Pedigree chart of variant positive patients
Bioinformatic analysis
The generated sequencing data were mapped to the human genome reference hg19. The Torrent suit software with v5 (Life Technologies) was used for all analysis. The coverage analysis was calculated using Torrent Coverage Analysis, and potential pathogenic variants were identified using the Torrent Variant Caller and DNA STAR software (DNASTAR, Madison, WI, USA). The Human Gene Mutation Database (HGMD®Professional 2019.4), was utilised to classify the identified variants as reported or novel. Furthermore, the population sequencing database GnomAD was explored to validate the novel variants identified adhering to the latest guidelines by the American College of Medical Genetics. All novel variants were evaluated for sequence conservation and the likelihood of pathogenicity evaluated using Mutation taster, Mutation Assessor, PROVEAN, FATH MM, FATHMM-MKL, META SVM, METAR, Sorting Intolerant From Tolerant (SIFT) and LRT. Final variant classification was based on ACMG guidelines 2015 [18]. Variants with pathogenic/likely pathogenic variant categorisation were taken as clinically significant and positive. Sanger sequencing was performed to confirm all identified mutations and rare variants were recorded.
Biochemical analysis
This was performed including plasma glucose (fasting/postprandial), HbA1C, stimulated C-peptide, alanine transaminase and fasting lipids (cholesterol, triglyceride, LDL and HDL cholesterol). Biochemical tests were done using a Cobas C 8000 auto analyser from Roche diagnostics (Germany). Glycosylated haemoglobin was measured by the ion exchange high-performance liquid chromatography (HPLC) method (Bio-Rad 2 Variant II turbo glycated haemoglobin (HbA1c) analyser; CV 0.69%). GAD65 antibodies were quantitatively measured using ELISA (Euroimmune kit CV 4.7%).
Statistical tools
Statistical analysis was done using IBM SPSS software (Version 21.0, Chicago, IL, USA). For descriptive statistics, categorical variables were expressed as numbers and percentages.
Continuous variables were expressed using mean, median and standard deviation (SD).
Results
The baseline characteristics of the study population are summarised in Table 1. In total, NGS analysis came out with 29 variants in 27 individuals. Variant segregation data were recorded when family genetic data were available. The mean read depth of these samples was > 300X with > 99% with 20× coverage. More importantly, the variants were confirmed by Sanger sequencing. The assessment of pathogenicity was made by application of ACMG 2015 guidelines and confirmed with the Clingen pathogenicity calculator and Varsome. Genome aggregation database (GnomAD)-based population allele count and frequency were recorded for each variant.
Pathogenic or likely pathogenic variants
Four out of sixty (6.6%) were positive for variants classifiable as positive (P/LP): one patient with HNF4A c.691C > T, (p.Arg231Trp), two with HNF 1Ac.746C > A, (p.Ser249Ter) and c.1340C > T, (p.Pro447Leu), and one with ABCC8 c.4544C > T, p.Thr1515Met). All the three variants in MODY 1 and MODY 3 genes were documented in paediatric age group (< 18 years). Three out of 17 paediatric patients (17.6%) had P/LP variants. Genetic characteristics of these patients are depicted in Tables 2 and 3.
Patient M1 (ABCC8:c.4544C > T(p.Thr1515Met) is a 35-year-old male with onset of diabetes at 20 years of age and on treatment with insulin. He had a BMI of 21.2 kg/m2 and C-peptide level of 0.65 nmol/l. Non-proliferative diabetic retinopathy had been documented at 31 years of age. His mother and brother both developed diabetes at around 33 years of age. Near-identical gene variants have been described previously in association with both congenital hyper-insulinism and MODY [29, 30]. He was planned for a trial of sulfonylurea; however, modest stimulated C-peptide, long-standing diabetes and poor follow-up dissuaded us.
Patient M2 is a boy with diabetes from 13 years of age carries a nonsense mutation HNF 1Ac.746C > A,
9p.Ser249Ter). He had a good response to sulfonylurea therapy and insulin could be stopped.
Patient M3 is a 12-year-old girl with diabetes from the age of 11 years carried a digenic cis variant HNF1 c.1340C > T (p.Pro447Leu /ABCC8 c.2152G > A (p.Gly718Ser). This MODY 3 variant is reported to be pathogenic [17, 31]. She had been maintaining good glycaemic control with very low dose sulfonylurea. Her mother, who has the same variant, developed diabetes at 15 years of age and has been maintaining adequate glycaemic control with a once daily dose of glimepiride 0.5 mg.
M4 is an 11-year-old boy on insulin since diagnosis one year prior to presentation. He had poor glycaemic control on insulin which improved with sulfonyl therapy. He carried the HNF4c.691C > T p.R231W variant. His mother had onset of diabetes at the age of 21 years and harboured the same variant. This variant has been reported in HGMD [32].
Variants classifiable as VUS and benign
Besides the pathogenic and likely pathogenic variants, other variants classifiable as VUS or benign were identified in classical genes (MODY 1 to MODY 3) and in non-classical genes like PAX4, Neuro D1, BLK1, PDX1, KLF 11 and CEL. Five patients had PDX1 c.670G > A(p.Glu224Lys) and one individual had PDX1 c.97C > A(p.Pro33Thr), which are currently considered as benign or VUS [33, 34]. The details of these variants are depicted in Table 4.
Discussion
The current study looked at the genetic confirmation rate in young subjects with multigenerational diabetes and reported rates which are lower than those reported previously. The genetically proven MODY detection rate based on the four common MODY types (HNF1A, HNF4A, HNF1B and GCK) has been shown to be lower in south Asians (SA) in a large population database from the UK [20]. Only 12.6% were genetically proven compared to 29.1% in the white Caucasian (WC) population. The detection rate in children was, however, similar (26.7% in SA vs 32.6% in WC). In the absence of variant description, it is unclear whether all the variants found in this study would be considered as pathogenic based on ACMG criteria. The genetic confirmation rates in recent studies from France, Ukraine, Greece and Turkey vary from 16 to 33% [22, 24, 25, 29].
Pathogenic/likely pathogenic changes were seen only in 6.6% of patients in the current study, despite using an NGS panel looking at 13 genes. Two out of nine patients (22.2%) aged less than 13 years were genetically proven to have MODY in our study, which is similar to the paediatric detection rate in a UK-based study [20]. All the variants in children were in classical genes (either MODY 1 or MODY 3).
Several Indian studies in the past that looked at MODY genetics were limited in nature, by virtue of the number of MODY genes screened [34,35,36]. There are two Indian studies, both from the neighbouring state of Tamil Nadu, which looked at the MODY genetic profile. Both studies had an inclusion strategy very similar to the current study, except that the study by Chapla et al. recruited subjects with an age of onset of diabetes less than 35 years compared to 30 years in the study by Mohan et al. In the study by Chapla et al., 56 patients clinically diagnosed to have MODY underwent NGS with a detection rate of 19.6% [21]. However, many of the variants considered pathogenic at that time are reclassified as benign/VUS now in light of ACMG2015 guidelines and new population data (PDX1 c.670G > A), HNF1A c1501G > T, NEUROD1 c723C > G) [33]. This study used an NGS panel incorporating 10 genes which did not include ABCC8. A more recent study (2018) from Chennai (Mohan et al.) included 152 subjects with a MODY diagnosis (age less than 30 years and satisfying Fajans’ clinical criteria) with a reported genetic confirmation rate of 15% [37]. 7.2% of these were noted in HNF1A and 3.3% were in ABCC8. There was one patient each with pathogenic variants in GCK and HNF1B genes and the rest were in non-classical genes like BLK, CEL, KLF11, PDX1 and KCNJ11 and newer gene variants (RFX6, WFS1, AKT2, NKX6-1) which are being recognised as contributing to early-onset diabetes in the heterozygous state. It should be stressed that, despite being a larger study both in terms of number of patients involved and the gene variants screened, the pathogenicity detection rate was lower than that reported in the first study. The role of KLF 11, PAX4 and BLK as MODY genes has been either disputed or refuted in recent years [38].
The variant in subject M1 (ABCC8NM_000352.5:c.4544C > T(p.Thr1515Met) was interesting (https://www.ncbi.nlm.nih.gov/snp/?term=rs769989185 has details of alternate transcripts). The likely pathogenic variant of ABCC8 p.Met1514Thr was reported in a recent study from Greece [29]. This is the same variant found in our study with an alternate transcript. Heterozygous variants (ABCC8c.4543A > G p.T1515A variant and ABCC8 c.4547C > T p.Thr1516Met) were previously reported as pathogenic in babies with congenital hyper-insulinism [30]. No known history of hypoglycaemia prior to the onset of diabetes was noted in this patient. However it has been reported that the parents of patients with congenital hyper-insulinism, who have a genetic variant in the heterozygous state, develop early-onset diabetes without a history of preceding manifestations of congenital hyper-insulinism [39]. Congenital hyper-insulinism is also known to evolve into a state of early-onset diabetes, both in the heterozygous and homozygous state [39, 40]. 8% of patients with a clinical diagnosis of MODY, but negative for MODY 1, MODY 2 and MODY 3 gene variants, were found to have pathogenic variants in the ABCC8 gene in a study from the UK [41]. A study from Singapore has also documented pathogenic variants in the ABCC8 gene, suggesting that among the non-classical genes; this is perhaps the most frequent one that contributes to the MODY spectrum. MODY due to ABCC8 variants is known to respond to sulfonylurea therapy, thereby underscoring the therapeutic implications of a precise genetic diagnosis [42]. Non-classical genes, especially ABCC8, have been reported as possible contributors to MODY pathogenesis in studies from Italy and Brazil [42, 43].
Assigning pathogenicity to gene variants is challenging. Many of the gene variants reported as pathogenic in the past are now known to be tolerated variants without phenotypic manifestations [26]. Patient M5 exemplifies this dilemma. This individual had a novel variant inABCC8 (p.V1165M) which had a likely pathogenic variant output in the variant classifier Varsome. However, the allele count of 18 (allele frequency 0.000588) for the South Asian population in the GnomAD database makes this conclusion questionable. Five patients had PDX c.670G > A(p.Glu224Lys), which was earlier thought to be pathogenic but is currently considered to be benign/VUS in view of its high prevalence in controls [33].
This study has several limitations. The limited number of analysed samples makes it impossible to derive prevalence of pathogenic or likely pathogenic positivity rates in subjects clinically diagnosed as MODY. The NGS methodology used in our study could have missed copy number variations [44], intronic and promoter variants. A backup MLPA strategy could have offset these short coming, at least partially [45]. The main strength of the study is the strict adherence to current guidelines for variant classification.
The question which naturally arises is the genetic make-up of the patients without pathogenic variants. Several explanations can be postulated, including an early-onset type 2 diabetes, intronic variants or copy number variations [44] (large deletions and duplications) which may be missed with the currently used sequencing technology or presence of as yet undiscovered genes (MODY X).
Conclusion
The detection rate of MODY-related pathogenic or likely pathogenic gene variants, by careful application of ACMG 2015 guidelines, was lower in this cohort of patients with a clinical diagnosis of MODY compared to the western literature. The contribution from some of the non-classical MODY genes, especially ABCC8, was evident in this study.
Data availability
Yes.
Code availability
NA.
Abbreviations
- MODY:
-
Maturity-onset diabetes of the young
- DKA:
-
Diabetic ketoacidosis
References
Tandon N, Anjana RM, Mohan V, Kaur T, Afshin A, Ong K, Mukhopadhyay S, Thomas N, Bhatia E, Krishnan A, Mathur P, India State-Level Disease Burden Initiative Diabetes Collaborators (2016) The increasing burden of diabetes and variations among the states of India: the Global Burden of Disease Study 1990–2016. Lancet Glob Health. https://doi.org/10.1016/S2214-109X(18)30387-5
Sahoo SK, Zaidi G, Vipin VP, Chapla A, Thomas N, Yu L, Asthana P, Bhatia E (2019) Heterogeneity in the aetiology of diabetes mellitus in young adults: a prospective study from north India. Indian J Med Res 149(4):479. https://doi.org/10.4103/ijmr.IJMR_1004_17
Praveen PA, Madhu SV, Mohan V, Das S, Kakati S, Shah N, Chaddha M, Bhadada SK, Das AK, Shukla DK, Kaur T (2016) Registry of youth onset diabetes in India (YDR) rationale, recruitment, and current status. J Diabetes Sci Technol 10(5):1034–1041. https://doi.org/10.1177/1932296816645121
Misra S, Owen KR (2018) Genetics of monogenic diabetes: present clinical challenges. Curr DiabRep 18(12):1–11. https://doi.org/10.1007/s11892-018-1111-4
Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT (2014) Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 311(3):279–286. https://doi.org/10.1001/jama.2013.283980
Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT (2003) No deterioration in glycemic control in HNF-1α maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care 26(11):3191–3192. https://doi.org/10.2337/diacare.26.11.3191-a
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT (2003) Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362(9392):1275–1281. https://doi.org/10.1042/cs104019p
Tattersall RB, Fajans SS (1975) A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes 24(1):44–53. https://doi.org/10.2337/diab.24.1.44
Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S (2010) Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53(12):2504–2508. https://doi.org/10.1007/s00125-010-1799-4
DiMeglio LA, Evans-Molina C, Oram RA (2018) Type 1 diabetes. The Lancet 391(10138):2449–2462. https://doi.org/10.1016/S0140-6736(18)31320-5
Sattar N, Gill JM (2015) Type 2 diabetes in migrant south Asians: mechanisms, mitigation, and management. Lancet Diabetes Endocrinol 3(12):1004–1016. https://doi.org/10.1016/S2213-8587(15)00326-5
Njølstad PR, Molven A (2012) To test, or not to test: time for a MODY calculator? Diabetologia 55(5):1231–1234. https://doi.org/10.1007/s00125-012-2514-4
Juszczak A, Pavić T, Vučković F, Bennett AJ, Shah N, Medvidović EP, Groves CJ, Šekerija M, Chandler K, Burrows C, Putarek NR (2019) Plasma fucosylated glycans and C-reactive protein as biomarkers of HNF1A-MODY in young adult–onset nonautoimmune diabetes. Diabetes Care 42(1):17–26. https://doi.org/10.2337/dc18-0422
Ellard S, Allen HL, De Franco E, Flanagan SE, Hysenaj G, Colclough K, Houghton JA, Shepherd M, Hattersley AT, Weedon MN, Caswell R (2013) Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia 56(9):1958–1963. https://doi.org/10.1007/s00125-013-2962-5
Owen KR (2013) Monogenic diabetes: old and new approaches to diagnosis. Clin Med 13(3):278. https://doi.org/10.7861/clinmedicine.13-3-278
Anık A, Çatlı G, Abacı A, Böber E (2015) Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab 28(3–4):251–263. https://doi.org/10.1515/jpem-2014-0384
Bansal V, Gassenhuber J, Phillips T, Oliveira G, Harbaugh R, Villarasa N, Topol EJ, Seufferlein T, Boehm BO (2017) Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med 15(1):1–4. https://doi.org/10.1186/s12916-017-0977-3
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–423. https://doi.org/10.1038/gim.2015.30
Ellard S, Baple EL, Berry I et al (2019) ACGS best practice guidelines for variant classification 2019. Assoc Clin Genet Sci 1–32. https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/. Accessed Mar 2021
Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, Greenbaum CJ, Imperatore G, Lawrence JM, Marcovina SM, Mayer-Davis E (2013) Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab 98(10):4055–4062
Chapla A, Mruthyunjaya MD, Asha HS et al (2015) Maturity onset diabetes of the young in India-A distinctive mutation pattern identified through targeted next-generation sequencing. Clin Endocrinol (Oxf) 82(4):533–542. https://doi.org/10.1111/cen.12541
Globa E, Zelinska N, Elblova L, Dusatkova P, Cinek O, Lebl J, Colclough K, Ellard S, Pruhova S (2017) MODY in Ukraine: genes, clinical phenotypes and treatment. J Pediatr Endocrinol Metab 30(10):1095–1103. https://doi.org/10.1515/jpem-2017-0075
Ang SF, Lim SC, Tan CS et al (2016) A preliminary study to evaluate the strategy of combining clinical criteria and next generation sequencing (NGS) for the identification of monogenic diabetes among multi-ethnic Asians. Diabetes Res Clin Pract 119:13–22. https://doi.org/10.1016/j.diabres.2016.06.008
Donath X, Saint-Martin C, Dubois-Laforgue D, Rajasingham R, Mifsud F, Ciangura C, Timsit J, Bellanné-Chantelot C (2019) Monogenic diabetes study group of the société francophone du diabète. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: a cross-sectional analysis. BMC Med. https://doi.org/10.1186/s12916-019-1363-0
Özdemir TR, Klrblylk Ö, Dündar BN et al (2018) Targeted next generation sequencing in patients with maturity-onset diabetes of the young (MODY). J Pediatr Endocrinol Metab 31(12):1295–1304. https://doi.org/10.1515/jpem-2018-0184
Flannick J, Johansson S, Njølstad PR (2016) Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes. Nat Rev Endocrinol 12(7):394–406. https://doi.org/10.1038/nrendo.2016.50
Lek M, Karczewski KJ, Minikel EV et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. https://doi.org/10.1038/nature19057
Mruthyunjaya MD, Chapla A, Shyamasunder AH et al (2017) Comprehensive maturity onset diabetes of the young (MODY) gene screening in pregnant women with diabetes in India. PLoS ONE 12(1):1–15. https://doi.org/10.1371/journal.pone.0168656
Tatsi EB, Kanaka-Gantenbein C, Scorilas A, Chrousos GP, Sertedaki A (2020) Next generation sequencing targeted gene panel in Greek MODY patients increases diagnostic accuracy. Pediatr Diabetes 21(1):28–39. https://doi.org/10.1111/pedi.12931 (Epub 2019 Nov 10 PMID: 31604004)
Bennett JT, Vasta V, Zhang M, Narayanan J, Gerrits P, Hahn SH (2015) Molecular genetic testing of patients with monogenic diabetes and hyperinsulinism. Mol Genet Metab 114(3):451–458. https://doi.org/10.1016/j.ymgme.2014.12.304
Yamagata K, Oda N, Kaisaki PJ et al (1996) Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature 384(6608):455–458. https://doi.org/10.1038/384455a0
Harries LW, Locke JM, Shields B, Hanley NA, Hanley KP, Steele A, Njølstad PR, Ellard S, Hattersley AT (2008) The diabetic phenotype in HNF4A mutation carriers is moderated by the expression of HNF4A isoforms from the P1 promoter during fetal development. Diabetes 57(6):1745–1752
National Center for Biotechnology Information (2020) ClinVar; [VCV000008863.3], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000008863.3. Accessed 10 May 2020. PDX1 E224K Clin var
PDX1P33T (2020) National Center for Biotechnology Information. ClinVar; [VCV000036414.5], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000036414.5. Accessed 10 May 2020
Radha V, Ek J, Anuradha S, Hansen T, Pedersen O, Mohan V (2009) Identification of novel variants in the hepatocyte nuclear factor-1alpha gene in South Indian patients with maturity onset diabetes of young. J Clin Endocrinol Metab 94(6):1959–1965. https://doi.org/10.1210/jc.2008-2371
Anuradha S, Radha V, Mohan V (2011) Association of novel variants in the hepatocyte nuclear factor 4A gene with maturity onset diabetes of the young and early onset type 2 diabetes. Clin Genet 80(6):541–549. https://doi.org/10.1111/j.1399-0004.2010.01577.x
Mohan V, Radha V, Nguyen TT et al (2018) Comprehensive genomic analysis identifies pathogenic variants in maturity-onset diabetes of the young (MODY) patients in South India. BMC Med Genet 19(1):1–10. https://doi.org/10.1186/s12881-018-0528-6
Clinical Genome Resource (2021). https://search.clinicalgenome.org/kb/gene-validity/CGGV:assertion_b1e38a49-7c12-4514-a2a1-109e04da146f-2020-02-12T170000.000Z, https://search.clinicalgenome.org/kb/genes/HGNC:8618, https://search.clinicalgenome.org/kb/genes/HGNC:1057. Accessed 3 Oct 2021
Işık E, Demirbilek H, Houghton JA, Ellard S, Flanagan SE, Hussain K (2019) Congenital hyperinsulinism and evolution to sulfonylurea-responsive diabetes later in life due to a novel homozygous p.L171F ABCC8 mutation. J Clin Res Pediatr Endocrinol 11(1):82–87. https://doi.org/10.4274/jcrpe.galenos.2018.2018.0077
Gussinyer M, Clemente M, Cebrián R, Yeste D, Albisu M, Carrascosa A (2008) Glucose intolerance and diabetes are observed in the long-term follow-up of nonpancreatectomized patients with persistent hyperinsulinemic hypoglycemia of infancy due to mutations in the ABCC8 gene. Diabetes Care 31(6):1257–1259. https://doi.org/10.2337/dc07-2059
Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, Hattersley AT, Ellard S (2012) Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia 55(1):123–127. https://doi.org/10.1007/s00125-011-2319-x
Pezzilli S, Ludovico O, Biagini T et al (2018) Insights from molecular characterization of adult patients of families with multigenerational diabetes. Diabetes 67(1):137–145. https://doi.org/10.2337/db17-0867
de Santana LS, Caetano LA, Costa-Riquetto AD et al (2019) Targeted sequencing identifies novel variants in common and rare MODY genes. Mol Genet Genomic Med. https://doi.org/10.1002/mgg3.962
Berberich AJ, Huot C, Cao H, McIntyre AD, Robinson JF, Wang J, Hegele RA (2019) Copy number variation in GCK in patients with maturity-onset diabetes of the young. J Clin Endocrinol Metab 104(8):3428–3436. https://doi.org/10.1210/jc.2018-02574
Komazec J, Zdravkovic V, Sajic S et al (2019) The importance of combined NGS and MLPA genetic tests for differential diagnosis of maturity onset diabetes of the young. Endokrynol Pol 70(1):28–36. https://doi.org/10.5603/EP.a2018.0064
Acknowledgements
We thank the RSSDI for the research grant and the staff of the Department of Endocrinology, Christian Medical College Vellore for helping us with the genetic testing.
Funding
This work was supported by Research Society of the Study of Diabetes in India.
Author information
Authors and Affiliations
Contributions
PPV: Conceptualization, Methodology, Software GEETHALAKSHMI SAMPATHKUMAR.: Data curation, Writing- Original draft preparation. UM, AS, NA: Visualization, Investigation. NB: Supervision: AC: Software, Validation: HK, VN, NT: Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest.
Research involving human participants and/or animals
This study was approved by the Institutional Ethical committee (IRB No: IEC-AIMS-2017-ENDO-426 dated 23.11.2017).
Consent to participate
Yes obtained. Informed consent was obtained from all individual participants and legal guardians included in the study.
Consent for publication
Yes obtained. The authors affirm that human research participants provided informed consent for publication of the data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sampathkumar, G., Valiyaparambil, P.P., Kumar, H. et al. Low genetic confirmation rate in South Indian subjects with a clinical diagnosis of maturity-onset diabetes of the young (MODY) who underwent targeted next-generation sequencing for 13 genes. J Endocrinol Invest 45, 607–615 (2022). https://doi.org/10.1007/s40618-021-01698-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-021-01698-y